

ABSTRACT
Apoptosis caused by deregulated MYC expression is a prototype example of intrinsic tumor suppression. However, it is still unclear how supraphysiological MYC expression levels engage specific sets of target genes to promote apoptosis. Recently, we demonstrated that repression of SRF target genes by MYC/MIZ1 complexes limits AKT-dependent survival signaling and contributes to apoptosis induction. Here we report that supraphysiological levels of MYC repress gene sets that include markers of basal-like breast cancer cells, but not luminal cancer cells, in a MIZ1-dependent manner. Furthermore, repressed genes are part of a conserved gene signature characterizing the basal subpopulation of both murine and human mammary gland. These repressed genes play a role in epithelium and mammary gland development and overlap with genes mediating cell adhesion and extracellular matrix organization. Strikingly, acute activation of oncogenic MYC in basal mammary epithelial cells is sufficient to induce luminal cell identity markers. We propose that supraphysiological MYC expression impacts on mammary epithelial cell identity by repressing lineage-specific target genes. Such abrupt cell identity switch could interfere with adhesion-dependent survival signaling and thus promote apoptosis in pre-malignant epithelial tissue.
KEYWORDS: apoptosis, lineage fate, MYC, MIZ1, mammary epithelial cells, repression
Introduction
Deregulated MYC expression is pervasive in multiple human tumor entities and is one of the first genetic alterations identified in human breast cancer.1
In a heterodimeric complex with MAX, MYC proteins bind E-boxes (5′-CANNTG-3′) in promoters to control gene expression and exert most of their biological functions, including growth control, cell cycle progression and differentiation.2 In some biological contexts, such as the mitogenic stimulation of resting B-cells, MYC enhances expression of all genes with an open chromatin structure.3-5 In contrast, deregulation of MYC activates and represses more restricted sets of target genes during tumorigenesis.6 For example, complex formation with MIZ1 (ZBTB17), which occurs predominantly at oncogenic MYC levels, mediates binding to low-affinity sites and shifts the direction of transcriptional responses at these “newly acquired” target genes toward repression.6,7 In addition, high levels of MYC can regulate genes by binding to distal enhancer elements.8
In addition to promoting cell growth and transformation, deregulated expression of MYC sensitizes cells to apoptosis in multiple settings.9-11 Consistently, impairment of apoptosis is a central feature during tumor progression and mutation or loss of apoptotic regulators accelerates MYC-induced tumorigenesis in different tissues in vivo.12-14
For example, mice expressing Myc under the control of either the whey acidic protein (WAP) or the mouse mammary tumor virus (MMTV) promoters develop mammary adenocarcinomas after long latencies, indicating that deregulation of Myc alone is insufficient to promote transformation in mammary epithelial cells without additional genetic lesions.15-17 In these models, Myc-dependent mammary tumorigenesis is most likely limited by the induction of apoptosis, since tumors display increased levels of apoptosis and overexpression of Bcl-2 in the MMTV-Myc background accelerates tumor development.18,19
Although several molecular factors involved in MYC-induced apoptosis have been identified, the mechanisms that enable cells to react to different levels of the transcription factor with the appropriate biological response had remained elusive. We have previously reported that MCF10A mammary epithelial cells engineered to express high levels of the 4-OHT inducible fusion protein MYC-ER undergo apoptosis. Importantly, a mutant form of MYC, MYCVD-ER, which is unable to bind to MIZ1, displays a strongly impaired response.7 Detailed analyses of MIZ1-dependent gene expression changes revealed that dose-dependent apoptosis induction by MYC correlated with repression of target genes of the serum response factor (SRF) and reduced AKT activity.7 These observations expand several other studies that had demonstrated a correlation between MYC-mediated transcriptional repression and the induction of apoptosis.20,21
Collectively, the available data suggest that oncogenic amounts of MYC, due to association with MIZ1 at these levels, lead to repression of genes that normally provide critical survival signals. Here, we aimed to gain more insight into the identity of these target genes and their significance for apoptosis induction in response to high levels of MYC.
Results
To elucidate how MIZ1-dependent repression contributes to MYC-induced apoptosis, we performed a gene set enrichment analysis (GSEA) using microarray expression data obtained from MCF10A cells expressing MYC-ER. We directly compared gene expression changes induced by 4-OHT-activated MYC-ER or the MYCVD-ER mutant form of MYC that cannot bind MIZ1.
MCF10As are non-tumorigenic mammary epithelial cells displaying a gene expression program characteristic of normal basal epithelial cells.22 Interestingly, the most prominently repressed gene set in response to MYC-ER was composed of genes that are upregulated in basal-like breast cancer cell lines relative to luminal cells (Fig. 1A). In addition, 3 other gene sets comparing basal- with luminal-specific gene expression patterns in tumor cell lines as well as normal mammary epithelial cells were identified with a false discovery rate of 0.000.(Fig. 1A).23-25
Figure 1.

(see previous page) The MYC/MIZ1 complex represses conserved genes of the basal cell lineage. (A) Example plots from a GSEA C2 analysis (curated gene sets) comparing MYC-ER- and MYCVD-ER-induced gene expression changes after 24 hours of 4-OHT treatment in MCF1A cells (100 nM). All represented gene sets are significantly repressed by MYC-ER relative to MYCVD-ER and indicate that MYC selectively represses marker genes of basal breast (cancer) cells in a MIZ1-dependent manner. ES = Enrichment score; NES = normalized enrichment score; FDR = false discovery rate. (B) qRT-PCR validating the GSEA analysis. Expression of KRT14, LAMC2 and ITGA6 in MCF10A cells was analyzed 24 hours after 4-OHT or ctr (EtOH) treatment. Data were normalized to B2M or RPS14 and plotted relative to MYC-ER ctr. Bars represent mean + SD from 3 independent biological replicates. Each gene is significantly repressed after activation of MYC-ER and less repressed by MYCVD-ER. p-values were calculated with Student's t-test (**: p < 0.01; ***: p < 0.001; ****: p < 0.0001). (C) Concept overlap graph of functional annotations generated with ConsensusPathDB (cpdb.molgen.mpg.de). All core enriched genes from the GSEA sets shown in A were merged into one list and used for a Gene Ontology analysis. Selected GO-terms of significantly over-represented biological processes are visualized. Edges between nodes represent shared genes between GO-terms. Numbers in each node indicate significance (–log10 q-value based on FDR) of the respective process. (D) Beeswarm boxplot depicting regulation of previously identified marker genes of the basal and luminal mammary epithelial lineage after activation of MYC-ER. Conserved gene signatures of basal (MaSC-enriched) and luminal (luminal progenitor and mature luminal) subpopulations were retrieved from.29 P-values were determined with a Wilcoxon rank sum test.
To validate repression of basal marker genes by MYC, we isolated RNA from pools of MYC-ER and MYCVD-ER MCF10A cells after 24 hours of 4-OHT treatment and tested expression of 3 genes: KRT14, LAMC2 and ITGA6. We observed an approximately 50 % reduction of relative mRNA expression for all 3 genes upon activation of MYC-ER (Fig. 1B). Importantly, all genes were significantly less repressed after activation of MYCVD-ER, suggesting the involvement of the partner protein MIZ1 (Fig. 1B).
We then combined all significantly repressed genes from the 4 sets shown in Figure 1A to generate a list of 339 unique lineage-related repressed MYC target genes. This list included genes encoding α-V and −6 integrins, laminin beta and gamma chains, as well as several basal keratins, such as K5 and K14. In order to explore their functional relationship, we performed an enrichment analysis of Gene Ontology categories and visualized over-represented terms in a concept overlap graph using ConsensusPathDB.26,27 Consistent with our previous observations, repressed genes were involved in epithelial development, proliferation and differentiation as well as several processes related to cellular adhesion, including wound healing, migration and extracellular matrix (ECM) organization (Fig. 1C and ref. 7).
We concluded that MYC represses a basal-like breast cancer gene signature in a MIZ1-dependent manner and that repressed genes are connected to key biological processes previously associated with basal/myoepithelial cells.28
Several MYC-repressed genes, including ACTA2, ITGA6, KRT14 and SNAI2, are not only part of the breast cancer signatures described above but also characteristic markers of the normal basal cell lineage in the mammary gland.28 To further confirm that MYC suppresses lineage-specific gene expression profiles associated with normal mammary epithelial subpopulations, we used datasets of conserved signature genes derived from functionally analogous mouse and human mammary epithelial cells.29 These signatures are composed of genes that are differentially expressed between purified populations of the 2 main epithelial cell types within the mammary gland: The basal/myoepithelial compartment, which is also enriched for mammary stem cells (MaSCs), and the luminal subset, which can be further divided into luminal progenitors and mature cells.30,31
We analyzed regulation of the basal or luminal gene signatures in response to high levels of MYC-ER in MCF10A cells. Whereas expression of genes characteristic for luminal cells was not significantly affected, expression of the basal gene signature was significantly downregulated after induction of MYC-ER (Fig. 1D).
Thus, we concluded that high levels of MYC lead to repression of a number of conserved genes involved in maintaining basal cell fate and lineage commitment.
MCF10A cells can be grown in 3D acinar cultures that more closely resemble the structural organization of the normal epithelial architecture within the mammary gland.32 Chronic MYC activation during development of acinar structures induces apoptosis (Fig. 2A).33 To test whether MYC-induced apoptosis correlates with disturbed lineage-identity under these culture conditions, we treated developing MCF10A acini with 4-OHT. In 2D monolayer culture, more than 80 % of MCF10A cells stain positive for the basal Keratin 14 (K14), while the remaining cells are Keratin 8 (K8, luminal) or double positive.34 We could observe similar frequencies under 3D culture conditions (Fig. 2B-C). Activation of high levels of MYC disturbed acinar morphogenesis (compare size of acini in Fig. 2B). Surprisingly, we could score K8-positive acini as early as 24 h after MYC induction and after 7 d of treatment, more than 80 % of structures contained K8- or double positive cells (Fig. 2C). In line with these findings, we observed a selection for decreased expression of basal Keratin 14 and a concomitant increase in luminal Keratin 8 expression in MCF10A cells surviving constitutive overexpression of high MYC levels in regular 2D culture (Fig. 2D). Taken together, these data suggest that high levels of MYC lead to a cell fate “switch” toward the luminal mammary epithelial lineage.
Figure 2.

High levels of MYC lead to a cell fate switch in MCF10A cells. (A) Myc activation induces apoptosis in developing acini. Representative pictures of control and 4-OHT treated MCF10A cells after 8 d in 3D culture (7 d 4-OHT). Cleavage of Caspase-3 indicates apoptosis. E-Cadherin staining was performed to visualize acinar structure. Nuclei are counterstained with Hoechst. Scale bar = 10 µm. (B) Myc activation induces a basal-to-luminal cell fate switch in 3D culture. Developing acini were treated with 4-OHT (1–7 days) and analyzed for expression of Keratin 14 and 8. Representative images are shown. Scale bar = 10 µm. (C) Quantification of B. Mean + SD of n = 3 independent experiments. 25–75 acini per group were analyzed. P-values were calculated with a Student's t-test (*: p < 0.05; **: p < 0.01; ***: p < 0.001). (D) Immunoblot documenting expression of K14 (basal Keratin 14) and K8 (luminal Keratin 8) in MCF10A cells after constitutive MYC overexpression. VINCULIN was used as loading control.
In summary, we present here evidence that high levels of MYC repress marker genes characteristic of basal mammary epithelial cells in a MIZ1-dependent manner. As several of these genes have previously been suggested to be essential determinants of basal lineage commitment and cell fate,28,29 we propose that MIZ1-dependent repression corrupts critical signals that are required to maintain survival of this cell type. This mechanism may play important roles in vivo, limiting the cancerous spread of cells with potentially oncogenic levels of MYC (Fig. 3).
Figure 3.
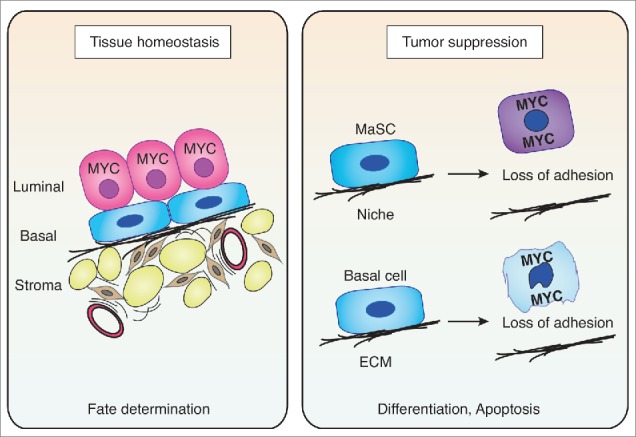
Model summarizing possible consequences of MYC expression/deregulation in the mammary gland. Left panel: During normal development, endogenous MYC is required to suppress basal gene expression programs (in a complex with MIZ1), leading to a confinement of MYC expression to the luminal compartment of the mammary gland during tissue homeostasis. Right panel: Deregulated MYC expression in either mammary stem cells (MaSC) or basal cells leads to MIZ1-dependent repression of adhesion genes and causes disruption of critical cell – niche or cell - ECM interactions and consequently, promotes apoptosis. Therefore, the MYC/MIZ1-induced cell identity switch could act as a critical fail-safe mechanism to prevent tumorigenesis within the basal/stem cell compartment.
Discussion
A number of observations suggest that high levels of MYC are detrimental to epithelial cells, which is considered as a failsafe mechanism to prevent accumulation of cancerous cells in the body. For example, whereas both low and high MYC protein levels induce transcription of the tumor suppressor ARF, only high levels lead to accumulation of ARF protein by blocking ULF-mediated ARF turnover.35 Furthermore, it has been suggested that due to an invasion of low-affinity sites in promoters and enhancers, high levels of MYC can regulate additional gene expression programs, including those involved in apoptosis.6,8 Last, complex formation with other transcriptional regulators such as MIZ1 can modulate transcriptional responses upon MYC overexpression.6,7 It is likely that these quantitative changes in gene expression patterns represent essential molecular cues that allow differentiation between physiological and oncogenic levels of MYC and, ultimately, crossing of the apoptotic “threshold.”36
We demonstrated previously that association with MIZ1 and subsequent repression of target genes of the SRF transcription factor are required to induce apoptosis in response to oncogenic MYC levels.7 Here, we show that MYC/MIZ1-mediated repression targets specific cell fate determining gene modules. In line with our previous findings, these genes are predominantly involved in biological processes such as adhesion and cytoskeletal organization (Fig. 1C and ref. 7). One particularly interesting conserved pathway with an important role in both basal and stem cell-enriched subpopulations of murine and human mammary epithelial cells is integrin signaling.29 In addition, based on differential expression relative to other subpopulations, extracellular matrix and cytoskeleton modules have emerged as key operational networks in basal/myoepithelial cells.28 In the same study, it was suggested that MYC is a crucial factor for mammary cell fate decisions, in particular for controlling the balance between basal and luminal differentiation.28 Interestingly, overexpression of MYC in basal mammary epithelial cells does not only repress basal marker genes but also promotes a cell fate shift toward the luminal lineage (Fig. 2B-C). It is possible that this abrupt and “unlicensed” switch in cell identity causes apoptosis of basal epithelial cells in vivo, due to MYC/MIZ1-mediated repression of critical, basal-specific adhesion genes and subsequent loss of contact with the ECM (Fig. 3, right panel). Thus, we propose that MYC/MIZ1-dependent transcriptional repression contributes to intrinsic tumor suppression mechanisms partly by switching epithelial cell identity.
MYC/MIZ1-dependent transcriptional repression could also inhibit tumorigenesis originating from adult mammary stem cells, which are believed to be located in the basal compartment.31
Interestingly, ectopic expression of MYC in the basal layer of the murine epidermis causes depletion of stem cells and increased differentiation.39,40 Furthermore, this exit from the stem cell compartment has been linked to repression of adhesion genes and the disruption of critical interactions between stem cells and their specified microenvironment or “niche.”41,42 Most notably, repression of these genes has already been shown to depend on association with MIZ1.43 Thus, it is tempting to speculate that MYC/MIZ1-dependent repression of the gene expression programs reported here could simultaneously represent an inbuilt fail-safe mechanism to prevent expansion of mammary stem cells with oncogenic MYC. (Fig. 3, right panel).31
Last, we also wish to point out that MYC is normally highly expressed in luminal epithelial cells of both murine and human mammary gland and this expression is important for luminal progenitor proliferation and survival (Fig. 3, left panel).37,38 Levels of MYC that are supraphysiological in the basal cells studied here might therefore be tolerated in luminal cells. It is tempting to speculate hat rare premalignant cells could survive the MYC-induced cell identity switch and these cancerous cells with new luminal identity will eventually benefit from MYC/MIZ1 pro-survival functions. From this angle, the MYC-induced cell fate switch could also be considered as a tumor survival mechanism. To summarize, it is conceivable that MYC/MIZ1-dependent modulation of mammary epithelial identity has a paramount influence on mammary tumor development, being able to either suppress or promote the tumorigenesis depending on the context and the stage of the pre-malignant or malignant lesion.
Despite a variety of tumor-suppressive mechanisms, deregulated MYC expression is frequently observed in human breast cancer and most commonly associated with the basal/triple-negative subtype.44,45 As this type of cancer has been proposed to originate in luminal progenitor cells, this could reflect a “memory” of the physiological expression pattern of MYC in this compartment.46 Alternatively, as the majority of these tumors have lost expression of the p53 tumor suppressor, MYC/MIZ1-dependent gene repression might be tolerated because apoptosis can be escaped.7 Further studies will determine whether the potential MYC-induced cell fate switch observed here bears similarities to recent discoveries of oncogene-induced cell fate conversions in lineage restricted subpopulations and whether it contributes to tumor heterogeneity in human breast cancer.47,48
Materials and methods
Tissue culture and lentiviral transduction
2D and 3D organotypic culture of MCF10A and MCF10A MycERtm cells was performed as previously described.7,33 Stable overexpression of MYC or MYC-ER fusion proteins was achieved by transduction with lentiviral pRRL-SFFV vectors and packaging plasmids psPAX2 and pMD2.G (Didier Trono) in the presence of 8 μg/ml protamine sulfate.
Cells were either treated with 100 nM 4-OHT to induce expression of MYC-ER, or ethanol as solvent control.
Gene expression, GSEA and Gene Ontology analysis
Total RNA was extracted with peqGOLD TriFast Reagent (PEQLAB). First-strand cDNA synthesis was performed with M-MLV Reverse Transcriptase (Invitrogen) and random hexamer primers (Roche). Gene expression was analyzed by qRT-PCR on an Agilent MX3000P platform in technical triplicates using ABsolute SYBR Green Mix (Thermo Scientific) and the following primers (5′−3′): ccattgaggacctgaggaac and caatctgcagaaggacattgg (KRT14); ctcagcccaacgactagacc and tcacctgttgattcccaaga (LAMC2); tttgaagatgggccttatgaa and ccctgagtccaaagaaaaacc (ITGA6). Data were quantified with the comparative CT method using B2M (5′gtgctcgcgctactctctc3′ and 5′gtcaacttcaatgtcggat3′) or RPS14 (5′ggcagaccgagatgaatcctca3′ and 5′caggtccaggggtcttggtcc3′) as reference for normalization. Results from 3 independent biological experiments were combined to calculate relative expression and determine significance. The microarray analysis is described in ref. Seven.
To analyze differentially repressed gene sets between MYC-ER and MYCVD-ER, M-values from the microarray analysis were used for a gene set enrichment analysis (GSEA) of curated gene sets (“C2”). The two different phenotypes were compared with n = 1000 permutations and permutation type “gene_set.”
To analyze which functional annotation categories are over-represented among differentially repressed genes, we used all core enriched genes (significantly repressed by MYC-ER relative to MYCVD-ER) from the 4 curated gene sets shown in Fig. 1A. The resulting list of 339 unique genes was analyzed with the web interface of ConsensusPathDB (http://consensuspathdb.org) and selected Gene Ontology categories were visualized in a concept overlap graph.27
Analysis of conserved lineage signature genes
To determine the regulation of previously identified conserved lineage signature genes by MYC, we used Supplementary Tables 1, 2 and 3 published by Lim et al.29 Upregulated genes in MaSC-enriched (“Basal”), or luminal progenitor and mature luminal subsets (“Luminal”) were merged with significantly regulated genes after MYC-ER induction in MCF10A cells (p < 0.05). The overlap between both lists (153 basal (31 %) and 67 (38 %) luminal genes, respectively) was displayed in a beeswarm boxplot using the R environment. Significance was determined with a 2-sided Wilcoxon rank sum test.
Immunofluorescence (IF) staining of 3D MCF10A cultures
Acinar structures were immunostained according to established protocols.49 Briefly, acini were fixed with 2 % PFA for 15 min and permeabilized with 0.25 % Triton X-100/PBS for 10 min and blocked with 10 % goat serum/PBS for 1 h. Primary antibodies were diluted in IF-buffer (0.1 % BSA/0.2 % Triton X-100/0.05 % Tween-20 in PBS) and 3D cultures were stained overnight. Following washes (3×15 min with IF-buffer), structures were incubated with secondary antibodies and washed again. Nuclei were counterstained with Hoechst 33258 (Life Technologies) and mounted with Immu-Mount reagent (Fisher Scientific). Imaging was performed on a Leica TCS CARS SP8 confocal microscope.
Antibodies
Anti-cleaved Caspase-3 (1:300, Asp175, #9661, Cell Signaling), anti-E-Cadherin (1:500, 36/E-Cadherin, BD Biosciences), anti-Keratin 8 (1:300, 1E8, Biolegend), anti-Keratin 14 (1:300, Poly19053, Biolegend) and appropriate Alexa Fluor® conjugated secondary antibodies (Life Technologies) were used for 3D-IF stainings. Anti-Keratin 8 (M20, Acris Antibodies GmbH), anti-Keratin 14 (LL002, abcam) and anti-Vinculin (hVIN-1, Sigma) were used for immunoblot.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
MCF10A cells were a kind gift from M. Bentires-Alj (Friedrich Miescher Institute, Basel). We would like to thank all members of the Eilers and Klefström laboratories for constant fruitful discussions.
Funding
This work was supported by grants from the Deutsche Krebshilfe (109696), the Thyssen Foundation and the Deutsche Forschungsgemeinschaft (Ei 222/12–1) as well as the Academy of Finland, TEKES, Finnish Cancer Organizations and Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115188. HMH is supported by the Integrative Life Sciences (ILS) doctoral program, the Finnish Cancer Foundation, Orion-Farmos Foundation, Biomedicum Helsinki Foundation and Emil Aaltonen Foundation.
References
- 1.Escot C, Theillet C, Lidereau R, Spyratos F, Champeme MH, Gest J, Callahan R. Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc Natl Acad Sci U S A 1986; 83:4834-8; PMID:3014513; http://dx.doi.org/ 10.1073/pnas.83.13.4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dang C V. MYC on the path to cancer. Cell 2012; 149:22-35; PMID:22464321; http://dx.doi.org/ 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. Revisiting global gene expression analysis. Cell 2012; 151:476-82; http://dx.doi.org/ 10.1016/j.cell.2012.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151:56-67; PMID:23021215; http://dx.doi.org/ 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al.. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012; 151:68-79; PMID:23021216; http://dx.doi.org/ 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al.. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014; 511:483-7; PMID:25043018; http://dx.doi.org/ 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiese KE, Haikala HM, von Eyss B, Wolf E, Esnault C, Rosenwald A, Treisman R, Klefström J, Eilers M. Repression of SRF target genes is critical for Myc-dependent apoptosis of epithelial cells. EMBO J 2015; 34:1554-71; PMID:25896507; http://dx.doi.org/ 10.15252/embj.201490467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al.. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014; 511:488-92; PMID: 25043028 http://dx.doi.org/ 10.1038/nature13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991; 6:1915-22; PMID:1923514 [PubMed] [Google Scholar]
- 10.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992; 69:119-28; PMID:1555236; http://dx.doi.org/ 10.1016/0092-8674(92)90123-T [DOI] [PubMed] [Google Scholar]
- 11.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene 2008; 27:6462-72; PMID:18955973; http://dx.doi.org/ 10.1038/onc.2008.312 [DOI] [PubMed] [Google Scholar]
- 12.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999; 13:2658-69; PMID:10541552; http://dx.doi.org/ 10.1101/gad.13.20.2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002; 109:321-34; PMID:12015982; http://dx.doi.org/ 10.1016/S0092-8674(02)00738-9 [DOI] [PubMed] [Google Scholar]
- 14.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A 2004; 101:6164-9; PMID:15079075; http://dx.doi.org/ 10.1073/pnas.0401471101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 1984; 38:627-37; PMID:6488314; http://dx.doi.org/ 10.1016/0092-8674(84)90257-5 [DOI] [PubMed] [Google Scholar]
- 16.Schoenenberger CA, Andres AC, Groner B, van der Valk M, LeMeur M, Gerlinger P. Targeted c-myc gene expression in mammary glands of transgenic mice induces mammary tumours with constitutive milk protein gene transcription. EMBO J 1988; 7:169-75; PMID:2834201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandgren EP, Schroeder JA, Ting Hu Qui, Palmiter RD, Brinster RL, Lee DC. Inhibition of mammary gland involution is associated with transforming growth factor α but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res 1995; 55:3915-27; PMID:7641211 [PubMed] [Google Scholar]
- 18.Hundley JE, Koester SK, Troyer DA, Hilsenbeck SG, Barrington RE, Windle JJ. Differential regulation of cell cycle characteristics and apoptosis in MMTV-myc and MMTV-ras mouse mammary tumors. Cancer Res 1997; 57(4):600-3; PMID:9044833 [PubMed] [Google Scholar]
- 19.Jäger R, Herzer U, Schenkel J, Weiher H. Overexpression of Bcl−2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene 1997; 15:1787-95; http://dx.doi.org/ 10.1038/sj.onc.1201353 [DOI] [PubMed] [Google Scholar]
- 20.Conzen SD, Gottlob K, Kandel ES, Khanduri P, Wagner AJ, O'Leary M, Hay N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol Cell Biol 2000; 20:6008-18; PMID:10913183; http://dx.doi.org/ 10.1128/MCB.20.16.6008-6018.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oster SK, Mao DYL, Kennedy J, Penn LZ. Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene 2003; 22:1998-2010; PMID:12673205; http://dx.doi.org/ 10.1038/sj.onc.1206228 [DOI] [PubMed] [Google Scholar]
- 22.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al.. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515-27; PMID:17157791; http://dx.doi.org/ 10.1016/j.ccr.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JGM, Foekens JA, Martens JWM. Subtypes of breast cancer show preferential site of relapse. Cancer Res 2008; 68:3108-14; PMID:18451135; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5644 [DOI] [PubMed] [Google Scholar]
- 24.Huper G, Marks JR. Isogenic normal basal and luminal mammary epithelial cells isolated by a novel method show a differential response to ionizing radiation. Cancer Res 2007; 67:2990-3001; PMID:17409405; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4065 [DOI] [PubMed] [Google Scholar]
- 25.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, et al.. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006; 25:2273-84; PMID:16288205; http://dx.doi.org/ 10.1038/sj.onc.1209254 [DOI] [PubMed] [Google Scholar]
- 26.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res 2011; 39:D712–D717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamburov A, Stelzl U, Lehrach H, Herwig R The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res 2013; 41:D793–D800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kendrick H, Regan JL, Magnay F-A, Grigoriadis A, Mitsopoulos C, Zvelebil M, Smalley MJ. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genomics 2008; 9:591; PMID:19063729; http://dx.doi.org/ 10.1186/1471-2164-9-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim E, Wu D, Pal B, Bouras T, Asselin-Labat M-L, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res 2010; 12:R21; PMID:20346151; http://dx.doi.org/ 10.1186/bcr2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asselin-Labat M-L, Sutherland K D, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al.. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol 2007; 9:201-9; PMID:17187062; http://dx.doi.org/ 10.1038/ncb1530 [DOI] [PubMed] [Google Scholar]
- 31.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439:84-8; PMID:16397499; http://dx.doi.org/ 10.1038/nature04372 [DOI] [PubMed] [Google Scholar]
- 32.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer 2005; 5:675-88; PMID:16148884; http://dx.doi.org/ 10.1038/nrc1695 [DOI] [PubMed] [Google Scholar]
- 33.Partanen JI, Nieminen AI, Mäkelä TP, Klefstrom J. Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci U S A 2007; 104:14694-9; PMID:17766436; http://dx.doi.org/ 10.1073/pnas.0704677104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchwalter G, Hickey M, Cromer A, Selfors L, Gunawardane R, Frishman J, Jeselsohn R, Lim E, Chi D, Fu X, et al.. PDEF Promotes Luminal Differentiation and Acts as a Survival Factor for ER-Positive Breast Cancer Cells. Cancer Cell 2013; 23:753-67; PMID:23764000; http://dx.doi.org/ 10.1016/j.ccr.2013.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D, Kon N, Zhong J, Zhang P, Yu L, Gu W. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol Cell 2013; 51:46-56; PMID:23747016; http://dx.doi.org/ 10.1016/j.molcel.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct Thresholds Govern Myc's Biological Output In Vivo. Cancer Cell 2008; 14:447-57; PMID:19061836; http://dx.doi.org/ 10.1016/j.ccr.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoelzle T, Schwarb P, Trumpp A, Hynes NE. c-Myc affects mRNA translation, cell proliferation and progenitor cell function in the mammary gland. BMC Biol 2009; 7:63; PMID:19785743; http://dx.doi.org/ 10.1186/1741-7007-7-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Von Eyss B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, Sauer M, Rosenwald A, Evan GI, Kempa S, et al.. A MYC-driven change in mitochondrial dynamics limits YAP/TAZ function in mammary epithelial cells and breast cancer. Cancer Cell 28(6): 743–757. [DOI] [PubMed] [Google Scholar]
- 39.Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol 2001; 11:558-68; PMID:11369200; http://dx.doi.org/ 10.1016/S0960-9822(01)00154-3 [DOI] [PubMed] [Google Scholar]
- 40.Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 2001; 28:165-8; PMID:11381265; http://dx.doi.org/ 10.1038/88889 [DOI] [PubMed] [Google Scholar]
- 41.Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development 2003; 130:2793-808; PMID:12736221; http://dx.doi.org/ 10.1242/dev.00462 [DOI] [PubMed] [Google Scholar]
- 42.Watt FM, Frye M, Benitah SA. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat Rev Cancer 2008; 8:234-42; PMID:18292777; http://dx.doi.org/ 10.1038/nrc2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gebhardt A, Frye M, Herold S, Benitah SA, Braun K, Samans B, Watt FM, Elsässer HP, Eilers M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol 2006; 172:139-49; PMID:16391002; http://dx.doi.org/ 10.1083/jcb.200506057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hynes NE, Stoelzle T. Key signalling nodes in mammary gland development and cancer: Myc. Breast Cancer Res 2009; 11:210; PMID:19849814; http://dx.doi.org/ 10.1186/bcr2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV., Smyth JW, Davis SE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012; 209:679-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, MacKay A, Grigoriadis A, Tutt A, Ashworth A, et al.. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010; 7:403-17; PMID:20804975; http://dx.doi.org/ 10.1016/j.stem.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 47.Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, Wuidart A, Bouvencourt G, Dubois C, Salmon I, et al.. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015; 525:119-23; PMID:26266985; http://dx.doi.org/ 10.1038/nature14665 [DOI] [PubMed] [Google Scholar]
- 48.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O, et al.. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015; 525:114-8; PMID:26266975; http://dx.doi.org/ 10.1038/nature14669 [DOI] [PubMed] [Google Scholar]
- 49.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30:256-68; PMID:12798140; http://dx.doi.org/ 10.1016/S1046-2023(03)00032-X [DOI] [PubMed] [Google Scholar]