

ABSTRACT
Doxorubicin and other anthracycline compounds exert their anti-cancer effects by causing DNA damage and initiating cell cycle arrest in cancer cells, followed by apoptosis. DNA damage generally activates a p53-mediated pathway to initiate apoptosis by increasing the level of the BH3-only protein, Puma. However, p53-mediated apoptosis in response to DNA damage has not yet been validated in prostate cancers. In the current study, we used LNCaP and PC3 prostate cancer cells, representing wild type p53 and a p53-null model, to determine if DNA damage activates p53-mediated apoptosis in prostate cancers. Our results revealed that PC3 cells were 4 to 8-fold less sensitive than LNCaP cells to doxorubicin-inuced apoptosis. We proved that the differential response of LNCaP and PC3 to doxorubicin was p53-independent by introducing wild-type or dominant negative p53 into PC3 or LNCaP cells, respectively. By comparing several apoptosis-related proteins in both cell lines, we found that Bcl-xl proteins were much more abundant in PC3 cells than in LNCaP cells. We further demonstrated that Bcl-xl protects LNCaP and PC3 cells from doxorubicin-induced apoptosis by using ABT-263, an inhibitor of Bcl-xl, as a single agent or in combination with doxorubicin to treat LNCaP or PC3 cells. Bcl-xl rather than p53, likely contributes to the differential response of LNCaP and PC3 to doxorubicin in apoptosis. Finally, co-immunoprecipitation and siRNA analysis revealed that a BH3-only protein, Bim, is involved in doxorubicin-induced apoptosis by directly counteracting Bcl-xl.
KEYWORDS: apoptosis, Bcl-xl, Bim, doxorubicin, LNCaP, PC3, p53
Introduction
The anthracycline doxorubicin is widely used for chemotherapy, with great efficacy in several solid and non-solid cancers.1 Several mechanisms including topoisomerase II poisoning,2 DNA-adduct formation,3,4 and oxidative stress5,6 have been proposed to explain doxorubicin-mediated cell death.1 Doxorubicin and other anthracycline compounds also directly intercalate into DNA, leading to torsional stress and nucleosome destabilization.7 Accordingly, the main effect of doxorubicin and other anthracyclines is to trigger a DNA damage response (DDR) and subsequently activates an apoptotic pathway to kill proliferative cells such as cancer cells.
The main mediators of DDR are the ataxia–telangiectasia mutated (ATM) and ataxia–telangiectasia and Rad3 related (ATR) kinases.8 The actions of ATM/ATR lead to cell cycle arrest, DNA repair, or programmed cell death through complicated but finely concerted pathways mainly regulated by modifications of their downstream targets.9,10,11 Among these pathways, the p53-mediated DDR responses have been intensively explored.11 P53 proteins are activated and stabilized through several post-translation modifications by ATM12,13 and ATM target genes such as checkpoint kinase 214 and homeodomain-interacting protein kinase 2.15 The activated form of p53 acts as a transcription factor to control the expression of genes at cell cycle checkpoints for DNA repair and the programmed cell death pathway.16,17
The expressions of p21, Puma and Noxa are regulated by p53 in DDR.18 P21 functions as a negative regulator to shut down cell cycle progression, resulting in cell cycle arrest.19 This arrest allows the cells to activate DNA repair. In contrast, both Puma and Noxa are members of the BH3-only protein family, responsible for the initiation of the apoptosis pathway.20 The threshold model has been proposed to address how p53 controls the decision between cell cycle arrest and apoptosis.21 The p53 transcription factor might only turn on p21 and DNA repair proteins after minor DNA damage, whereas serious DNA damage increases the levels of p53 protein leading to the accumulation of Puma and Noxa, thus overcoming an apoptotic threshold to initiate cell death.
There are at least 8 members of the BH3-only protein family in mammals, including Bad, Bik, Bid, Bmf, Hrk, Bim, Noxa and Puma.22 These BH3-only proteins contain a homologous BH3 domain that is required for interaction with Bcl-2 family proteins such as Bcl-2, Bcl-XL, Bcl-W, Mcl-1 and A1.23 Differential binding affinity has been demonstrated between various BH3-only proteins and anti-apoptotic Bcl-2 family proteins.24 For example, both Bim and Puma can bind to all anti-apoptotic family members with equal affinity, whereas the other members exhibit different binding affinities toward various anti-apoptotic proteins.24 Despite their obvious binding affinity to anti-apoptotic proteins, the mechanism by which BH3-only proteins interact with anti-apoptotic proteins to initiate apoptosis remains uncertain. Two alternative models, direct and indirect activation, have been proposed to depict how BH3-only proteins counteract their respective anti-apoptotic proteins to activate apoptosis.25 In the direct activation model, some BH3-only proteins including Bad, BMF, Noxa, Bik and HRH work as sensitizers to displace activators such as Bim, Bid or probably Puma from the respective anti-apoptotic member, Bcl2, Bcl-xl or Mcl-1. The free activator molecules activate Bax/Bak to form oligomers to release cytochrome c. In the indirect activation model, the respective BH3-only protein displaces Bax/Bak from the anti-apoptotic protein complex to form oligomer for releasing cytochrome c.
The DDR-activating p53-mediated apoptosis pathway is logically reasonable in either the direct or indirect activation model, and is widely reported in various cancer chemotherapies using DNA-targeting agents. Moreover, p53 itself has become an attractive target as a therapeutic strategy for cancer.18 The main concern is whether this pathway occurs in all cancers. Indeed, 2 previous studies have shown that patients with p53 mutant breast cancers have a better response than patients with wild type p53 breast cancers to anthracycline-based chemotherapy.26,27 One recent study further demonstrated that the role of p53 in MMTV-Wnt1 mammary tumors is to induce p21-dependent and -independent growth arrest and cellular senescence instead of apoptosis.28 Therefore, we asked if DDR-activating p53-mediated apoptosis appears in prostate cancers, using LNCaP and PC3 prostate cancer cells that represent the wild type p53 and p53-null models, respectively. Our results revealed that PC3 cells display about 4 to 8-fold less sensitivity to doxorubicin apoptosis than LNCaP cells. This differential response of apoptosis between LNCaP and PC3 is p53-independent, but Bcl-xl-dependent. We found that Bim counteracts Bcl-xl to control doxorubicin-induced apoptosis through the indirect activator model.
Results
LNCaP cells showed 4- to 8-fold greater sensitivity than PC3 cells to doxorubicin treatment in apoptosis
Doxorubicin causes severe DNA damage to activate p53-mediated DDR. LNCaP carries the wild type p53 genes, which is easily induced by doxorubicin. In contrast, PC3 is a p53-null cell line and cannot produce p53 proteins. Thus, we compared the apoptotic response between LNCaP and PC3 cells in response to doxorubicin. The results indicated that doxorubicin induces apoptosis in LNCaP cells at relatively low concentrations, about 0.125-0.25 μM, based on the minimum concentration of doxorubicin needed to activate caspase 3 (Figure 1A). Doxorubicin can also activate caspase 3 in PC3 cells, although the concentration required is about 4- to 8-fold higher, about 1 μM in PC3, compared to LNCaP (Figure 1A).
Figure 1.

LNCaP cells were more sensitive in response to doxorubicin than PC3 cells. (A) LNCaP cells showed 4- to 8- fold greater sensitivity than that of PC3 cells to doxorubicin treatment in apoptosis. LNCaP and PC3 cells were cultured and treated with various concentrations of doxorubicin as indicated for 48 hr. Immunoblot analysis of cell lysates were used to detect the activated form of caspase 3 and PARP with cleavage PARP. Experiments were repeated 3 times and representative results are shown. Doxo: doxorubicin. Caspase 3(a): the activated form of caspase 3. (B) Doxorubicin induced significant cell death in LNCaP cells, but not in PC3 cells. Flow cytometric analyses of PC3 or LNCaP cells treated by doxorubicin for 24 and 48 hours. Analyses were in triplicate and representative histograms are shown. X- and Y-axes represent DNA content and cell numbers, respectively.
We further used flow cytometry to examine the doxorubicin effect on cell cycle progression and cell death in both LNCaP and PC3 cells. The results revealed that doxorubicin causes cell arrest at G1 and G2 phase, and induces cell death, the appearance of sub-G1, in dose-dependent manner for LNCaP cells (Figure 1B). In contrast, doxorubicin significantly induces cell cycle arrest at intra-S and G2 phase and the most of cells remain alive even in 5 μM of doxorubicin for PC3 cells (Figure 1B).
Doxorubicin-induced apoptosis is p53-independent
Since the role of p53 in apoptosis is to activate the expression of Puma, we first examined if p53 affects Puma expression in response to doxorubicin treatment. Our results disclosed that Puma expression is not affected by p53 in LNCaP cells, and its expression can be also observed in PC3 cells (Figure 2A). We further asked if p53 is involved in doxorubicin-induced apoptosis by over-expression of wild type p53 in PC3 or overexpression of dominant negative p53 in LNCaP cells. Interestingly, the results revealed that neither the over-expression of p53 in PC3 cells nor the knockdown of p53 with dominant negative p53 in LNCaP affected doxorubicin-induced apoptosis (Figure 2B and C). Clearly, p53 did not participate in the doxorubicin-induced apoptotic pathway in either the LNCaP or PC3 prostate cancer model.
Figure 2.
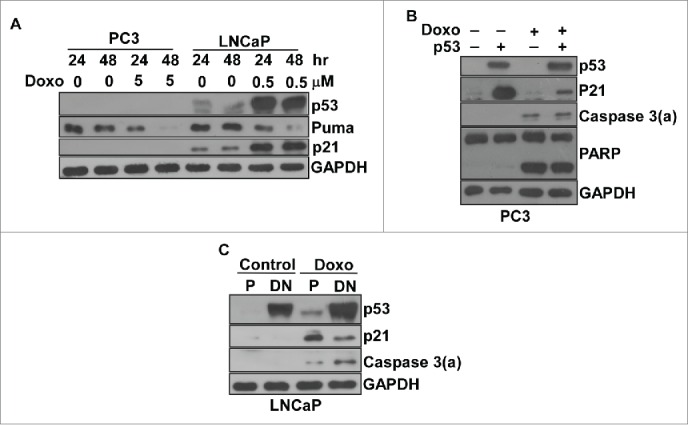
Doxorubicin-induced apoptosis was p53-independent in prostate cancer cells. (A) The expression of p53, puma and p21 in LNCaP and PC3 cells with/without doxorubicin. Immunoblot analysis of cell lysates from LNCaP or PC3 cells treated by doxorubicin over time as indicated. (B) The overexpression of wild type p53 did not affect doxorubicin-induced apoptosis in PC3 cells. PC3 cells were transiently transfected with pCMV-p53 or empty vector for 24 hr. Cells were treated with/without 3 μM doxorubicin for 48 hr, and then subjected to immunoblot analysis for the detection of p53, the activated form of caspase 3 and PARP with cleavage PARP. (C) The overexpression of dominant negative p53 did not affect doxorubicin-induced apoptosis in LNCaP cells. LNCaP cells were transfected with pCMV-p53mt135 to establish stably transfected LNCaP cells expressing dominant negative p53. The stably transfected LNCaP cells were treated with/without 0.5 μM doxorubicin for 48 hr, and then subjected to immunoblot analysis to detect p53 and the activated form of caspase 3. All experiments were repeated 3 times and representative results are shown. P, parental cells; DN, dominant negative.
Bcl-xl determines the differential sensitivity of LNCaP and PC3 cells to doxorubicin
Since p53 did not govern doxorubicin-induced apoptosis in prostate cancers, we asked what factor did lead to the 4 to 8-fold difference between LNCaP and PC3 cells in response to doxorubicin. We previously demonstrated that the level of Bcl-xl in PC3 cells is about 5-fold higher than in LNCaP cells.29 In the current study, we confirmed this result (Figure 3A). To determine if Bcl-xl also contributes to doxorubicin-induced apoptosis, we used ABT-263,30 an inhibitor of Bcl-xl, alone or in combination with doxorubicin to treat PC3 and LNCaP cells. The results indicated that ABT-263 with doxorubicin has significant synergistic effects triggering apoptosis compared to either ABT-263 or doxorubicin alone (Figure 3B). The minimum concentration of doxorubicin used in combination with 1 μM ABT-263 to trigger apoptosis, judged by the activation of caspase 3 for LNCaP cells and PC3 cells, is 0.25 μM and 1 μM respectively, about a 4-fold difference (Figure 3B). Thus, the low sensitivity of PC3 cells to doxorubicin was mainly due to their high level of Bcl-xl. Another anti-apoptotic protein, Bcl2, can only be detected in PC3 cells, not in LNCaP cells (Figure 3A); however, this protein was not involved in the ABT-263-induced apoptotic pathway.29
Figure 3.
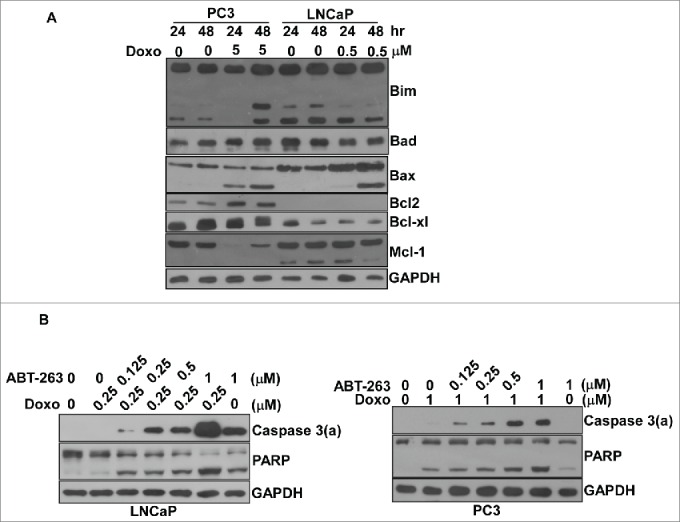
Bcl-xl played an essential role protecting LNCaP and PC3 cells against doxorubicin-induced apoptosis. (A) The expression of Bcl2 family proteins including Bim, Bad, Bax, Bcl2, Bcl-xl and Mcl2 with/without doxorubicin in LNCaP and PC3 cells. Immunoblot analysis of cell lysates from LNCaP or PC3 cells treated by doxorubicin over time as indicated. Experiments were repeated 3 times and representative results are shown. (B) The combination of doxorubicin with various concentrations of ABT-263 had a synergistic effect on apoptosis in both LNCaP and PC3 cells. Cell lysates from LNCaP or PC3 cells treated by doxorubicin in combination with ABT-263 or ABT-263 alone for 48 hr at various concentrations were subjected to immunoblot analysis. Experiments were repeated 3 times and representative results are shown.
Bim antagonizes Bcl-xl to initiate apoptosis through an indirect activation model
Since Bcl-xl is the anti-apoptotic protein protecting LNCaP and PC3 from apoptosis, we wanted to know which BH3-only protein could counteract Bcl-xl in response to doxorubicin treatment. The results from the Bcl-xl antibody co-immunoprecipitation indicated that Bim, not Puma, can interact with Bcl-xl (Figure 4A). The treatment of 0.5 μM doxorubicin decreased the interaction of Bcl-xl with Bim in LNCaP (Figure 4A). This consequence might be due to that the Bim proteins extracted from the LNCaP cells in sub-G1 phase (Figure 1B) loss the binding capacity to Bcl-xl. Bim siRNA knockdown further proved that Bim is the BH3-only protein that can antagonize Bcl-xl activity in response to doxorubicin-induced apoptosis in LNCaP cells (Figure 4B).
Figure 4.
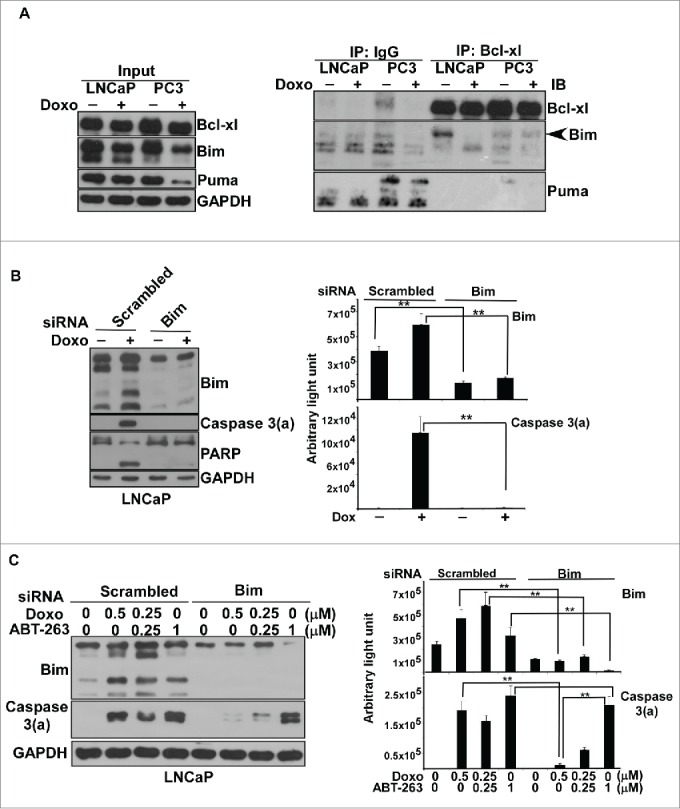
Bim interacted with Bcl-xl in doxorubicin-induced apoptosis in prostate cancers. (A) Coimmunoprecipitation of Bcl-xl or the control IgG with Bim. Cell lysates from LNCaP or PC3 cells with/without doxorubicin treatment, 0.5 μM for LNCaP and 3 μM for PC3 for 48 hr, were incubated with antibody against Bcl-xl or IgG, and then precipitated with magnetic beads for further immunoblot analysis. Antibodies against Puma and Bim were used for immunoblotting. The Bim co-precipitated with Bcl-xl was indicated by arrow head. Experiments were repeated 3 times and representative results are shown. (B) Bim knockdown reduced doxorubicin-induced apoptosis in LNCaP. Bim and scrambled RNAi knockdown cell lysates from LNCaP cells with/without 0.5 μM doxorubicin treatment were subjected to immunoblotting analysis. Experiments were repeated 3 times and representative results are shown. The respective immunoblot image of Bim and caspase 3(a) normalized to GAPDH was quantitated and shown at the right. (**) statistical significance between 2 readouts. (C) Doxorubicin-induced apoptosis is Bim-dependent, whereas ABT-263-induced apoptosis is Bim-independent. Bim and scrambled RNAi knockdown cell lysates from LNCaP cells with/without doxorubicin, ABT-263 or doxorubicin plus ABT-263 treatment were subjected to immunoblot analysis. Experiments were repeated 3 times and representative results are shown. The respective immunoblot image of Bim and caspase 3(a) normalized to GAPDH was quantitated and shown at the right. (**) statistical significance between 2 readouts.
The above results indicated that doxorubicin-induced apoptosis is Bim-dependent in prostate cancer cells. Next, we wanted to know if the action of Bim occurred through the direct activator-derepressor or indirect activator model for doxorubicin-induced apoptosis. To answer that, we examined whether ABT-263 action was Bim-dependent or Bim-independent. ABT-263 is a BH3 mimetic inhibitor of Bcl-xl. Based on the indirect activator model, ABT-263, just like BH3-only proteins such as Bim, binds to Bcl-xl to disrupt the interaction of Bcl-xl with Bak/Bax, triggering mitochondria-mediated apoptosis. In contrast, ABT-263, like the sensitizer of BH3-only proteins, binds to Bcl-xl to disrupt the interaction of Bcl-xl with the activator of BH3-only proteins such as Bim, releasing Bim to activate Bak/Bax for initiation of apoptosis by following the direct activator-derepressor model. We speculated that if ABT-263-induced apoptosis is Bim-dependent, doxorubicin-induced apoptosis would follow the direct activator-derepressor model. On the other hand, if ABT-263-induced apoptosis was Bim-independent, doxorubicin-induced apoptosis would follow the indirect activator model.
Thus, we explored the Bim knockdown effect on the levels of the activated form of caspase 3 in response to doxorubicin or ABT-263 treatment. The results demonstrated that Bim knockdown significantly decreases the activation form of caspase 3 for doxorubicin treatment (Figure 4C). In contrast, the decrease of Bim in ABT-263 treatment did not apparently reduce the activation form of caspase 3 (Figure 4C). Thus, we concluded that doxorubicin-induced apoptosis is Bim-dependent, but ABT-263-induced apoptosis is Bim-independent.
In sum, the Bim activated by doxorubicin-induced DNA damage might directly interrupt the interaction of Bcl-xl with pro-apoptotic proteins, Bak/Bax, to activate mitochondria-driven apoptosis.
Discussion
Puma is a BH3-only protein regarded as a p53 target gene involved in DNA damage-induced apoptosis. The current study showed that Puma is not the target of p53 for LNCaP and PC3 cells. A previous study demonstrated that Puma is the target gene of Foxo3 instead of p53 in prostate cancer cell lines.31 We were still not sure if Puma is regulated by Foxo3 in LNCaP or PC3 cells, but our results are consistent with the previous finding that Puma is not the target gene of p53. However, our results are consistent with previous findings that show the up-regulation of p21 by p53 overexpression in PC3 or LNCaP cells with doxorubicin treatment. Therefore, we concluded that p53 might be a crucial factor for cycle arrest and DNA repair, but not for apoptosis in response to doxorubicin treatment in prostate cancer cells.
The concentration of doxorubicin needed to induce apoptosis in PC3 cells was about 4 to 8-fold higher than that in LNCaP cells. This outcome was mainly caused by over-expression of Bcl-xl, not loss-of-function of p53 in PC3 cells. Overexpression of Bcl-xl also contributes to the apoptosis-resistant phenotype of PC3 cells in response to paclitaxel, in comparison with LNCaP cells.29 Examination of clinical samples indicated that overexpression of Bcl-xl in high-grade prostate carcinoma is associated with a hormone-refractory phenotype.32 Thus, disclosure of the mechanisms of Bcl-xl overexpression might allow us to find other therapeutic targets to control prostate cancer progression. One study has demonstrated that the expression of Bcl-xl is upregulated by the signal transducer and activator of transcription-6 (STAT6) in prostate cancers.33 However, the levels of STAT6 are not correlated with the expressions of Bcl-xl in 3 prostate cancer cell lines, Du145, LNCaP and PC3.34,35 Among these 3 cells, Du145 contains the highest levels of STAT6 whereas the levels of STAT6 in LNCaP and PC3 cells are low.34 In contrast, the expression of Bcl-xl in PC3 cells is much higher than in Du145 and LNCaP cells.35 Thus, the regulators responsible for the overexpression of Bcl-xl in prostate cancers remain controversial, and PC3 cells might represent the best cell model to study the mechanism of Bcl-xl overexpression in prostate cancers.
This study combined with our previous publication29 suggests that Bcl-xl has a protective role against paclitaxel-induced and doxorubicin-induced apoptosis in LNCaP and PC3 cells. We may ask what BH3-only protein counteracts Bcl-xl for paclitaxel-induced or doxorubicin-induced apoptosis. The current study demonstrated that Bim represses Bcl-xl function for doxorubicin-induced apoptosis. However, we still do not know which BH3-only protein participates in paclitaxel-induced apoptosis. We used RNAi to knock down Bim in paclitaxel-treated LNCaP cells, which revealed that Bim is not involved in paclitaxel-induced apoptosis.29 We also observed that the levels of Bim significantly decrease in LNCaP and PC3 cells in response to paclitaxel treatment,29 providing more evidence that Bim has no function in paclitaxel-induced apoptosis in prostate cancer cells. Interestingly, Bim's functionality in doxorubicin-induced apoptosis but not in paclitaxel-induced apoptosis further supports the indirect activator model rather than the direct activator-derepressor model for how Bim inhibits Bcl-xl function in doxorubicin-induced apoptosis in prostate cancer cells (Figure 4). If the direct activator-derepressor model is true in prostate cancers, Bim should activate Bak/Bax in both doxorubicin and paclitaxel-induced apoptosis. Our findings are inconsistent with those of Kutuk et al,36 who demonstrated that displacement of Bim by Bmf and Puma mediates paclitaxel-induced apoptosis in breast cancer cells, representing a typical example of the direct activator-derepressor model. Our explanation is that different cancer models might display variation in the apoptotic machinery in response to the chemotherapy agents.
In sum, we demonstrated that DDR-activating p53-mediated apoptosis pathway does not occur in prostate cancers. Doxorubicin sensitivity in prostate cancer cell models is determined by the level of the anti-apoptotic protein Bcl-xl, not by p53. It might be necessary to reassess the paradigm for the role of p53 in apoptosis in all other cancer types, since this scenario is being challenged by more and more studies.26,27,28,31 In addition, we showed that Bim is involved in doxorubicin-induced apoptosis but not in paclitaxel-induced apoptosis, and that the respective effect of doxorubicin and ABT-263 are Bim-dependent and Bim-independent. The indirect activator, rather than the direct activator-derepressor model, is suitable to explain the role of BH3-only proteins in prostate cancer apoptosis.
Materials and methods
Compounds and plasmids- Doxorubicin (Merck Millipore), ABT-263 (Selleck) and G-418 sulfate (Affymetrix) were purchased from commercial sources as indicated. The plasmids corresponding to the expression of wild type p53 and dominant negative p53, pCMV-p53 and pCMV-p53mt135 (Clontech), were purchased from commercial sources as indicated.
Cell culture, doxorubicin and ABT-263 treatment
PC3 and LNCaP cells were purchased from the Bioresource Collection and Research Center (BCRC) in Taiwan. The cells were seeded at 4 × 105 −6 × 105 cells per petri dish (10 cm) in RPMI 1640 with 10%' fetal bovine serum and grown at 37°C under 5%' CO2. When cell growth was up to 80%' confluence, the cell medium was replaced with fresh medium. Then the cells were incubated with various concentrations of doxorubicin, ABT-263 or doxorubicin in combination with ABT-263 for 24 or 48 hours. After treatment the cells were harvested, washed with PBS and spun down.
Flow cytometric cell cycle analysis
PC3 and LNCaP cells were treated by various concentrations of doxorubicin for 24 and 48 hours. The cells were harvested by trypsinization, centrifuged, washed with PBS and collected by centrifugation. The cells were fixed with ice-cold 70%' ethanol for 30 min, washed with PBS and centrifuged to remove supernatant. The cells were re-suspended in PBS containing 0.05%' Triton X-100 and RNase A (40 μg/mL) and incubated at 37oC for 1 hr, and propidium iodide (PI) was added to the cell suspension to a final concentration of 50 μg/mL for another 1 hr incubation. The cells were harvested by centrifugation, washed with PBS and centrifuged to remove supernatant. Finally, the cells were re-suspended by PBS and analyzed by BD LSR II Flow Cytometer (BD Biosciences) with BD FACSDiva™ Software (BD Biosciences).
Generation of the stable transfectants of LNCaP cells expressing dominant negative p53- pCMV-p53mt135 plasmid DNAs were transfected into LNCaP cells by electroporation. The transfected cells were grown in a medium containing 500 μg/ml G-418 sulfate for 3 weeks, and then the resistant colonies, regarded as the stable clones, were picked for validation. The stable clones were further verified by immunoblotting analysis of cell lysates using an anti-p53 antibody.
Transient transfection of wild type p53 into PC3 cells
At about 70%' confluence, PC3 cells in 10-cm plates were transiently transfected with 3 μg of pCMV-p53 using transfection reagents, FuGENE 6 (Promega), according to the manufacturer's instructions for 24 h. Then the transfected cells were incubated with the fresh medium with/without doxorubicin for 48 h, and cells were harvested for further analysis.
Immunoblotting
The harvested cells were lysed in RIPA buffer (25 mM Tris-HCl pH7.6, 150 mM NaCl, 1%' NP40 1 mM DTT, 0.1%' NP-40, 1%' sodium deoxycholate, 0.1%' SDS) containing proteinase and phosphatase inhibitors by sonication. Protein concentration was determined by a BCA Protein Assay Kit (Pierce). About 60 μg of protein per well was subjected to SDS-PAGE. After electrophoresis, the proteins were transferred to a nitrocellulose membrane. The transferred membranes were blocked in 5%' (w/v) nonfat dry milk or 5%' (w/v) BSA in TBS (0.5 M NaCl, 20 mM Tris-HCl, pH 7.4) with 0.1%' (v/v) Tween 20 and probed for the first antibody, followed by incubation with a secondary antibody conjugated with horseradish peroxidase (anti-rabbit, Cell Signaling; anti-mouse, Jackson ImmunoResearch) with visualization by ECL (Merck Millipore) with photographic film development. The first antibodies used in this study were as follows: anti-GAPDH (Cell Signaling, #5174) anti-Bcl-xl (Abcam, ab32370), Anti-Bax (Cell Signaling, #5023), anti-Bcl2 (Cell Signaling, #9941), anti-Bim (Cell Signaling, #2933), anti-caspase 3(a) (Cell Signaling, #9661), anti-Mcl-1 (Cell Signaling, #5453), anti-PARP (Cell Signaling, #9542), anti-Bad (Cell Signaling, #9239), anti-p21 (Abcam, ab109199), anti-Puma (Cell Signaling, #4976) and anti-p53 (Santa Cruz Biotechnology, Sc-126). Immunoblot images were quantitated by Image Studio Lite (LI-COR Biosciences).
Coimmunoprecipitation
Total cell lysates of doxorubicin-treated or control LNCaP or PC3 cells were prepared in Chaps buffer (5 mM MgCl2, 137 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1%' Chaps, 20 mM Tris–HCl (pH 7.5)) and protease inhibitors. About 500 μg of proteins were immunoprecipitated with Bcl-xl antibody (Abcam) or the control IgG at 4oC for 2 h. The control IgG for coimmunoprecipitation was rabbit IgG (Cell Signaling, #4096). Immunoprecipitates were captured by 50 μl of the slurry of protein A-Magnetic beads in Chaps buffer at 4oC overnight. Immunoprecipitates were then recovered by magnetic stand and washed 3 times in 1%' Chaps buffer. Immunoprecipitates were eluted by SDS-PAGE sample buffer, subjected to SDS-PAGE and then immunoblotting.
Bim knock down by siRNA
LNCaP cells were grown up to 60%' confluence. Bim (Hs_BCL2L11_5 HP Validated siRNA; Qiagen) siRNA was pre-incubated in 0.5 ml culture medium without serum before transfection, and then 40 μl INTERFERin transfection reagent (Polyplus) was added to the same culture medium, mixed by vortex and incubated for 10 min at room temperature to allow the formation of transfection complexes. The siRNA complex was added drop-wise to LNCaP cells on 10 cm dish culture. After 24 hours, LNCaP cells were incubated with various concentrations of doxorubicin or ABT-263 paclitaxel and harvested after 48 hours.
Statistical analysis
A paired t-test was used to show the statistical significance of the results using SigmaPlot 10. **P < 0.01 was considered significant.
Abbreviations
- DDR
DNA damage response
- ATM
ataxia–telangiectasia mutated
- ATR
ataxia–telangiectasia and Rad3 related
- STAT6
signal transducer and activator of transcription-6
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We want to thank Gary Mawyer for critical editing of this manuscript. We also thank the Center for Resources, Research and Development of Kaohsiung Medical University for providing the flow cytometer for cell cycle analysis.
Author contributions
Conceived and designed the experiments: CW. Performed the experiments: MCY, RWL, SBH, SYH, WJC. Analyzed the data: CW and YRH. Wrote the paper: CW, SW and YRH. All authors read and approved the final manuscript.
Funding
This research was partly supported by Grant 103-2815-C-037-012-B (to CW) from the Ministry of Science and Technology of Taiwan, Grant KMU-M103005 (to CW) and KMU-KMUH Co-Project of Key Research," grant No. KMU-DK105004 (toYRH) from Kaohsiung Medical University.
References
- 1.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta 2014; 1845(1):84-9; PMID:24361676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, Altman RB. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011; 21(7):440-6; PMID:21048526; http://dx.doi.org/ 10.1097/FPC.0b013e32833ffb56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forrest RA, Swift LP, Rephaeli A, Nudelman A, Kimura K, Phillips DR, Cutts SM. Activation of DNA damage response pathways as a consequence of anthracycline-DNA adduct formation. Biochem Pharmacol 2012; 83(12):1602-12; PMID:22414726; http://dx.doi.org/ 10.1016/j.bcp.2012.02.026 [DOI] [PubMed] [Google Scholar]
- 4.Coldwell KE, Cutts SM, Ognibene TJ, Henderson PT, Phillips DR. Detection of Adriamycin-DNA adducts by accelerator mass spectrometry at clinically relevant Adriamycin concentrations. Nucleic Acids Res 2008; 36(16):e100; PMID:18632763; http://dx.doi.org/ 10.1093/nar/gkn439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strigun A, Wahrheit J, Niklas J, Heinzle E, Noor F. Doxorubicin increases oxidative metabolism in HL-1 cardiomyocytes as shown by 13C metabolic flux analysis. Toxicol Sci 2012; 125(2):595-606; PMID:22048646; http://dx.doi.org/ 10.1093/toxsci/kfr298 [DOI] [PubMed] [Google Scholar]
- 6.Myers C. The role of iron in doxorubicin-induced cardiomyopathy. Semin Oncol 1998; 25(4 Suppl 10):10-4; PMID:9768818 [PubMed] [Google Scholar]
- 7.Yang F, Kemp CJ, Henikoff S. Doxorubicin enhances nucleosome turnover around promoters. Curr Biol 2013; 23(9):782-7; PMID:23602475; http://dx.doi.org/ 10.1016/j.cub.2013.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 2015; 149:124-38; PMID:25512053; http://dx.doi.org/ 10.1016/j.pharmthera.2014.12.001 [DOI] [PubMed] [Google Scholar]
- 9.Cremona CA, Behrens A. ATM signalling and cancer. Oncogene 2014; 33(26):3351-60; PMID:23851492; http://dx.doi.org/ 10.1038/onc.2013.275 [DOI] [PubMed] [Google Scholar]
- 10.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16(1):2-9; PMID:24366029; http://dx.doi.org/ 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14(4):197-210; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 12.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998; 281(5383):1677-9; PMID:9733515; http://dx.doi.org/ 10.1126/science.281.5383.1677 [DOI] [PubMed] [Google Scholar]
- 13.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281(5383):1674-7; PMID:9733514; http://dx.doi.org/ 10.1126/science.281.5383.1674 [DOI] [PubMed] [Google Scholar]
- 14.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73-112; PMID:21034966; http://dx.doi.org/ 10.1016/B978-0-12-380888-2.00003-0 [DOI] [PubMed] [Google Scholar]
- 15.Hofmann TG, Glas C, Bitomsky N. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays 2013; 35(1):55-64; PMID:23169233; http://dx.doi.org/ 10.1002/bies.201200060 [DOI] [PubMed] [Google Scholar]
- 16.Sullivan KD, Gallant-Behm CL, Henry RE, Fraikin JL, Espinosa JM. The p53 circuit board. Biochim Biophys Acta 2012; 1825(2):229-44; PMID:22333261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi M, Shi J, Jung SH, Chen X, Cho KH. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci Signal 2012; 5(251):ra83; PMID:23169817; http://dx.doi.org/ 10.1126/scisignal.2003363 [DOI] [PubMed] [Google Scholar]
- 18.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13(3):217-36; PMID:24577402; http://dx.doi.org/ 10.1038/nrd4236 [DOI] [PubMed] [Google Scholar]
- 19.Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res 2010; 704(1-3):12-20; PMID:20096807; http://dx.doi.org/ 10.1016/j.mrrev.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 20.Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, Dewson G, Michalak EM, Vandenberg CJ, Smyth GK, et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 2010; 116(24):5256-67; PMID:20829369; http://dx.doi.org/ 10.1182/blood-2010-04-280818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ 2013; 20(4):576-88; PMID:23306555; http://dx.doi.org/ 10.1038/cdd.2012.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doerflinger M, Glab JA, Puthalakath H. BH3-only proteins: a 20-year stock-take. FEBS J 2015; 282(6):1006-16; PMID:25565426; http://dx.doi.org/ 10.1111/febs.13190 [DOI] [PubMed] [Google Scholar]
- 23.van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res 2006; 16(2):203-13; PMID:16474435; http://dx.doi.org/ 10.1038/sj.cr.7310028 [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17(3):393-403; PMID:15694340; http://dx.doi.org/ 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- 25.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11(9):621-32; PMID:20683470; http://dx.doi.org/ 10.1038/nrm2952 [DOI] [PubMed] [Google Scholar]
- 26.Bertheau P, Turpin E, Rickman DS, Espie M, de Reynies A, Feugeas JP, Plassa LF, Soliman H, Varna M, de Roquancourt A, et al. Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin-cyclophosphamide regimen. PLoS Med 2007; 4(3):e90; PMID:17388661; http://dx.doi.org/ 10.1371/journal.pmed.0040090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertheau P, Plassa F, Espie M, Turpin E, de Roquancourt A, Marty M, Lerebours F, Beuzard Y, Janin A, de Thé H. Effect of mutated TP53 on response of advanced breast cancers to high-dose chemotherapy. Lancet 2002; 360(9336):852-4; PMID:12243922; http://dx.doi.org/ 10.1016/S0140-6736(02)09969-5 [DOI] [PubMed] [Google Scholar]
- 28.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012; 21(6):793-806; PMID:22698404; http://dx.doi.org/ 10.1016/j.ccr.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Huang SB, Yang MC, Lin YT, Chu IH, Shen YN, Chiu YH, Hung SH, Kang L, Hong YR, et al. Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PloS One 2015; 10(3):e0120913; PMID:25811469; http://dx.doi.org/ 10.1371/journal.pone.0120913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68(9):3421-8; PMID:18451170; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5836 [DOI] [PubMed] [Google Scholar]
- 31.Dey P, Strom A, Gustafsson JA. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene 2014; 33(33):4213-25; PMID:24077289; http://dx.doi.org/ 10.1038/onc.2013.384 [DOI] [PubMed] [Google Scholar]
- 32.Castilla C, Congregado B, Chinchon D, Torrubia FJ, Japon MA, Saez C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology 2006; 147(10):4960-7; PMID:16794010; http://dx.doi.org/ 10.1210/en.2006-0502 [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Tao T, Xu B, Chen S, Liu C, Zhang L, Lu K, Huang Y, Jiang L, Zhang X, et al. MiR-361-5p acts as a tumor suppressor in prostate cancer by targeting signal transducer and activator of transcription-6(STAT6). Biochem Biophys Res Commun 2014; 445(1):151-6; PMID:24491557; http://dx.doi.org/ 10.1016/j.bbrc.2014.01.140 [DOI] [PubMed] [Google Scholar]
- 34.Das S, Roth CP, Wasson LM, Vishwanatha JK. Signal transducer and activator of transcription-6 (STAT6) is a constitutively expressed survival factor in human prostate cancer. Prostate 2007; 67(14):1550-64; PMID:17705178; http://dx.doi.org/ 10.1002/pros.20640 [DOI] [PubMed] [Google Scholar]
- 35.Parrondo R, de Las Pozas A, Reiner T, Perez-Stable C. ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 2013; 1:e144; PMID:24058878; http://dx.doi.org/ 10.7717/peerj.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kutuk O, Letai A. Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ 2010; 17(10):1624-35; PMID:20431602; http://dx.doi.org/ 10.1038/cdd.2010.41 [DOI] [PMC free article] [PubMed] [Google Scholar]