

ABSTRACT
The replicative DNA polymerases insert ribonucleotides into DNA at a frequency of approximately 1/6500 nucleotides replicated. The rNMP residues make the DNA backbone more susceptible to hydrolysis and can also distort the helix, impeding the transcription and replication machineries. rNMPs in DNA are efficiently removed by RNaseH2 by a process called ribonucleotides excision repair (RER). In the absence of functional RNaseH2, rNMPs are subject to cleavage by Topoisomerase I, followed by further processing to result in deletion mutations due to slippage in simple DNA repeats. The topoisomerase I-mediated cleavage at rNMPs results in DNA ends that cannot be ligated by DNA ligase I, a 5′OH end and a 2′–3′ cyclic phosphate end. In the budding yeast, the mutation level in RNaseH2 deficient cells is kept low via the action of the Srs2 helicase and the Exo1 nuclease, which collaborate to process the Top1-induced nick with subsequent non-mutagenic gap filling. We have surveyed other helicases and nucleases for a possible role in reducing mutagenesis at Top1 nicks at rNMPs and have uncovered a novel role for the RecQ family helicase Sgs1 in this process.
KEYWORDS: DNA helicase, ribonucleotides, RNaseH2, rNMPs, Sgs1
Introduction
We previously uncovered a role for the Srs2 DNA helicase and Exo1 nuclease in reducing mutations arising from Top1 cleavage at misinserted rNMP residues that are not removed by RNaseH2.1 In the absence of RNaseH2, Top1 cleavage at rNMPs at simple repeats, mononucleotide runs, and short dinucleotide repeats results in −1 or −2 deletions at these sites. In addition to mutations, misinserted rNMPs also stimulate homologous recombination and other events that cause genome instability. Additional loss of either the Srs2 helicase or the Exo1 nuclease results in a synergistic increase in mutations, in particular the −1 and −2 deletions at the same hotspots that are observed in cells deleted for the RNH202 gene encoding one of the 3 subunits of RNaseH2. In vitro, Srs2 can unwind from a Top1 generated nick at a rUTP residue, and Exo1 nuclease can digest from the nick, both from the 5′OH end. Srs2 associates with Exo1 and stimulates its nuclease activity.1
There are several additional nucleases and helicases that have been implicated in action at the replication fork and in maintenance of genome stability. rNMP residues that remain within DNA are likely recognized during the subsequent DNA replication cycle. Thus, we wished to examine these nucleases and helicases to see if they also could act on Top1 generated nicks at rNMPs to suppress mutagenesis and genome instability, and to see if we could identify factors that might act at the 2′–3′ cyclic phosphate end of the nick. We used several assays for this study: slippage mutagenesis rates, interaction of candidate helicases with Exo1 and stimulation of Exo1 nuclease activity in vitro. Additionally, we determined the spectrum of mutation events at the CAN1 gene when both RNH202 and the test gene were deleted. From these studies, we conclude that only Exo1 collaborates with Srs2, and that the Sgs1 helicase has a role in mutation prevention, possibly through unwinding from the 2′–3′ cyclic phosphate end and prevention of further processing of this end by Top1, or acting at an earlier stage in DNA replication to allow tolerance of rNMPs.
Results
DNA repair nucleases
We previously identified Exo1 as being important for preventing mutations arising from rNMPs in DNA and furnished evidence that it collaborates with Srs2 to process the 5′ end of a nick induced by Top1 cleavage. To identify other nucleases that could be involved in processing rNMP-induced DNA nicks, we surveyed 7 additional nucleases that have a known role in DNA replication and/or repair in rnh202 cells, using a dinucleotide repeat hotspot reporter for slippage mutation that is suited to assessing rNMP-mediated mutagenesis 1,2 (Fig. 1). In some cases, we studied point mutations that eliminate the activity of the nuclease but otherwise leave the protein intact, as the double deletion of RNH202 and the nucleases is lethal. We also examined the Polδ proofreading-defective allele, pol3-01.3 Some of the mutants increased the basal level of slippage mutations. In the case of rad27Δ, which affects the processing of RNA primers in Okazaki fragments, the increase in slippage mutations is expected based on the fact that there are more rNMPs remaining in the DNA of mutant cells.4 Accordingly, the double mutant rnh202Δ rad27Δ displayed a synergistic increase in slippage mutations, indicative of overlap in function in removing rNMPs from DNA. The pol3-01 mutant gave an increase in mutation slippage in an RNaseH2-proficient strain, confirming the role of the Pol3 editing function in mutation prevention. The mutation rate in rhn202Δ pol3-01 cells is lower than that of rnh202Δ alone, with statistical significance (P=0.02). This is in line with the biochemical finding that the proofreading activity of Pol3 does not edit incorporated ribonucleotides,5 and one would expect a synergistic increase in mutation rate in rnh202Δ pol3-01 otherwise. To gain more insights into the influence of pol3-01 in this reporter one would need to conduct sequence analysis of the dinucleotide slippage segregants.
Figure 1.

Effects of nuclease defects on mutation rates. Mutations rates using the dinucleotide slippage mutation reporter are shown with the fold increase over wild type RNH202. Median rates are plotted with error bars representing 95% confidence limits (n = 18).
None of the other nuclease mutants examined, including those that are involved in recombinational repair and the nucleolytic processing of DNA breaks, namely, sae2Δ, mre11-H125N and mus81-D414A D415A, increased the rnh202Δ mutation rate. The APN2- and YEN1-encoded products are involved in base excision repair (BER) 6 and the resolution of DNA intermediates generated during replication fork repair and homologous recombination,7 respectively.
It remains possible that redundancy in nucleases that can process rNMP-induced nicks will be revealed only through a more detailed analysis of combinatorial mutants. This is part of our ongoing effort.
Roles of Top1 and Tdp1 in cleavage and processingat an rNMP residue
During the Top1-mediated cleavage at an embedded rNMP, a transient covalent linkage between the enzyme and DNA target is formed. The Top1-T722A mutant of S. cerevisiae generates a long-lived enzyme-DNA conjugate, thus impairing the ligation step and increasing genome instability from processing embedded rNMPs.8 The Top1-mediated cleavage reaction generates a free cyclic 2′–3′ phosphate end from the transesterification of the 2′OH residue in the ribose.9 There is good evidence that Top1-mediated nicks at rNMP residues act as the initiating DNA lesion in slippage mutation induction.1,2,10,11 To determine the linkage of the ligation step to mutations, we used the dinucleotide slippage reporter to examine the effect of the top1-T722A allele on mutation rates. As shown in Figure 2A, top1-T722A behaves identically to the null allele, thus showing that both rNMP cleavage and DNA ligation are needed to generate the necessary lesion for mutation induction.
Figure 2.
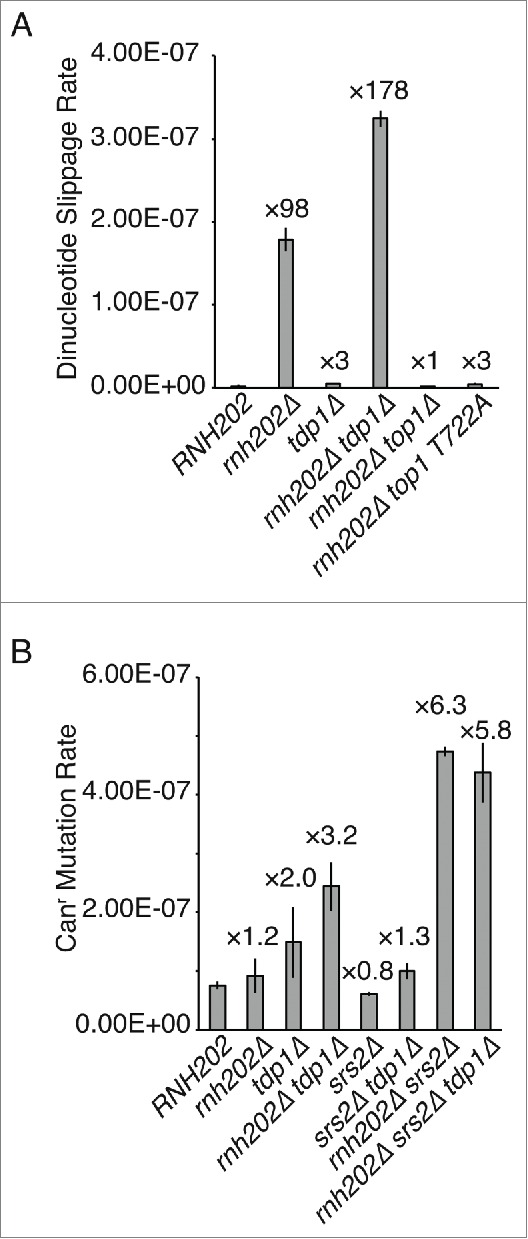
Effects of TDP1 and TOP1 mutations on mutation rates in the absence of RNase H2. (A) Dinucleotide slippage mutation rates are shown with the fold increase over wild type RNH202. Median rates are plotted with error bars representing 95% confidence limits (n = 18). (B) Spontaneous mutation rates at the CAN1 locus are shown with the fold increase over wild type RNH202. Median rates are plotted with error bars representing 95% confidence limits (n = 18).
It has been proposed that, following Top1 cleavage at a rNMP residue to form a cyclic 2′–3′ phosphate end, a second cleavage 2 nucleotides from this end removes the cyclic phosphate, leading to a −2 deletion upon realignment with the intact DNA strand and ligation to the 5′-OH end.2 This reaction has been reconstituted in vitro.12,13 These studies also revealed that the second Top1 cleavage 2 nucleotides from the cyclic 2′–3′ phosphate led to a covalent Top1-cleavage complex (Top1-cc).13 The bound Top1 could be removed by realignment and ligation to the 5′-OH, or via proteolysis and action of tyrosyl-DNA phosphodiesterase (Tdp1)14 and other enzymes.13 Here, we examined the role of Tdp1 in mutation formation. If Tdp1 indeed aided in the removal of the covalently attached Top1 residue at rNMPs, then loss of Tdp1 function would affect mutation rates. To assess the effect of Tdp1 loss, we determined mutation rates at the CAN1 locus, which reveals both base substitution and slippage mutations. The basal level of mutation in the tdp1Δ mutant was at the wild type level (Fig. 2B), and when combined with rnh202Δ, the rate appeared additive but not synergistic. Additionally, tdp1Δ did not alter the mutation rate of the rnh202Δ srs2Δ double mutant, indicating that the mutation-inducing lesion occurs in the absence of Tdp1 can be processed to form mutations. As there are several types of mutations at CAN1, and we expect Tdp1 to act only on Top1-cc, we also employed the slippage specific reporter. In this case, the effect of tdp1Δ was readily apparent, leading to an increase in slippage mutations specifically caused by the absence of RNaseH2 (Fig. 2A). We interpret these results as a channeling of some Top1-cc intermediates into an error-free repair mode dependent on TDP1 and others into the error-prone mode by slippage and ligation. In the absence of Tdp1, more Top1-cc intermediates are diverted to the error-prone repair mode with a concomitant increase in slippage mutation.
DNA repair helicases
We have previously shown that Srs2 prevents mutations from occurring in the absence of RNaseH2 and that it stimulates the activity of Exo1 in this process. To determine whether additional DNA helicases can act at rNMP-induced nicks, we examined the action of Mph1 and Sgs1. Mph1, a 3′–5′ helicase does not enhance rnh202Δ slippage mutation rates nor does it unwind a Top1-induced nick substrate or stimulate Exo1 nuclease.1 In contrast, we found that Sgs1, a 3′–5′ helicase of the RecQ family, can unwind DNA in the 3′–5′ direction from the Top1-induced nick (Fig. 3). Interestingly, Sgs1 also physically interacts with Exo1 although it does not enhance the activity of the latter (Fig. 3).
Figure 3.

Sgs1 unwinds Topo-I-induced nick and interacts Exo1, but does not stimulate Exo1 catalyzed cleavage. (A) DNA substrates containing a Topo-I-induced nick and labeled at the 3′ end were incubated with Srs2, Mph1 or Sgs1. The reactions were analyzed on a polyacrylamide gel. HD: Heat denatured. (B) Affinity pull-down through the (His)6 tag on Sgs1 was used to analyze its interaction with Exo1. The supernatant (S), wash (W) and SDS eluate (E) fractions were analyzed by 7.5% SDS–polyacrylamide gel electrophoresis. Sgs1 and Exo1, both FLAG tagged, were detected by western blot analysis with an anti-FLAG-M2 monoclonal antibody (Sigma). (C) Exonucleolytic cleavage of a Topo-I-induced nick by Exo1 was examined without and with Srs2, Mph1 or Sgs1.
Effect of the sgs1Δ mutation on rNMP-induced mutagenesis
In light of the biochemical activity of Sgs1 at a Top1-induced nick, we examined the effect of SGS1 deletion on mutagenesis. We first determined mutation rates using the dinucleotide slippage reporter and found that the sgs1Δ mutation resulted in a synergistic increase in mutation rate with rnh202Δ (Fig. 4A). Consistent with the idea that the slippage events in the dinucleotide reporter arise as a result of Top1 cleavage at rNMP residues, the rnh202Δ sgs1Δ slippage events were mostly eliminated by the top1Δ mutation, being reduced from 140-fold increase over wildtype to 8X increase. A synergistic interaction between sgs1Δ and rnh203Δ (which has another subunit of RNaseH2 deleted) on gross chromosome rearrangement (GCR) rates has been reported,15 however in this published study the synergistic increase was independent of Top1.
Figure 4.
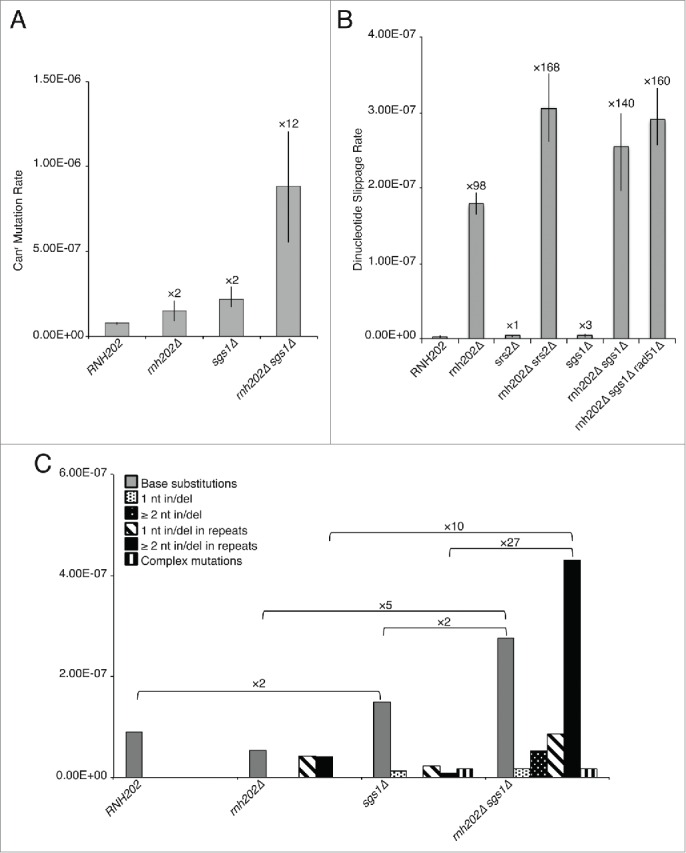
Effect of SGS1 mutation in a rnh202 context on mutation. (A) Spontaneous mutation rates at the CAN1 locus are shown with the fold increase over wild type RNH202. Median rates are plotted with error bars representing 95% confidence limits (n = 18). (B) Dinucleotide slippage mutation rates are shown with the fold increase over wild type RNH202. Median rates are plotted with error bars representing 95% confidence limits (n = 18). (C) Mutation spectra of Canr mutants obtained in (A) with fold increases indicated.
We next examined the sgs1Δ srs2Δ double mutant to determine if Sgs1 acts in the same mutation avoidance pathway as Srs2. Since this combination is lethal unless RAD51 or one of the genes encoding accessory factors of Rad51 is also defective, we had to perform these experiments in a rad51Δ background. The quadruple mutant rnh202Δ sgs1Δ srs2Δ rad51Δ showed no increase in mutation rate over that observed for rnh202Δ sgs1Δ rad51Δ or rnh202Δ srs2Δ rad51Δ, leading us to conclude that Sgs1 and Srs2 act in the same pathway to prevent mutations arising as a consequence of embedded rNMPs (Fig. 4A). The rad51Δ mutation had no impact on mutation rate in wild type, rnh202Δ, srs2Δ and sgs1Δ strains and only a slight impact in the rnh202Δ srs2Δ mutant, indicating that some of the initial lesions arising in the double mutant may be channeled into recombinational repair that requires Rad51 protein.
As the dinucleotide slippage reporter measures only one type of mutation at one sequence, we examined mutations at the CAN1 locus as an independent measure for mutagenesis. Here, we saw a strong synergistic increase dependent on loss of Sgs1 in an rnh202Δ context (Fig. 4B). To determine the spectrum of events resulting in functional loss of CAN1, over 50 independent isolates from rnh202Δ sgs1Δ were sequenced at CAN1 and compared to those obtained from wild type, rnh202Δ, and sgs1Δ. This analysis showed that new classes of mutations occur in the rnh202Δ sgs1Δ strain, most notably an increase in slippage mutations at dinucleotide repeats and mononucleotide runs as well small deletions and insertions not associated with any DNA repeat. The distribution of Canr events in the rnh202 sgs1 strain was significantly different from those observed in the rnh202 strain (P<0.002) (Fig. 4C). Among these was the increase in base substitution events. The triple mutant rnh202Δ sgs1Δ top1Δ showed a partial reduction in the Canr rate, consistent with loss of the slippage events at dinucleotide repeats but with other types of mutation events that are Top1-independent persisting.
Discussion
We previously reported that Exo1 acts with and is stimulated by Srs2 to promote error-free gap filling repair at rNTPs in simple DNA repeats.1 Here, we have surveyed several additional nucleases that function in DNA damage repair and recombination and find that only Fen1/Rad27 prevents mutations at dinucleotide repeats. As Fen1 was previously shown to function in RER and to remove RNA primers from Okazaki fragments, its loss is expected to increase the load of rNTPs in DNA to be acted upon by a mutagenic repair pathway. The other nucleases examined have no role in preventing error-prone repair, but we cannot yet exclude the possibility of redundancy. This could be addressed by analysis of mutants ablated for more than one of these nucleases. However, as these nucleases function in different DNA repair pathways, such an endeavor would require specific measures to eliminate off-target effects.
As originally proposed in the model for Top1 cleavage at rNMPs in DNA,2,10 we have shown that the ligation activity of Top1 is needed to induce slippage mutations. In vitro studies have provided evidence for a role of Tdp1 in the ribonucleotide-induced lesions by processing the Top1-cc. Consistent with this finding, the use of the dinucleotide slippage reporter has revealed a role of Tdp1 in error-free repair in vivo.13 It has been suggested that in vivo either ubiquitin-mediated proteolysis or the action of the Wss1 protease acts on the Top1-cc to create an intermediate that is amenable to further processing by Tdp1.16 This hypothesis is supported by the synthetic lethal genetic interaction between wss1 and tdp1.17
Our studies have revealed an unexpected role for the Sgs1 helicase in mutation avoidance stemming from the Top1-mediated cleavage at rNMPs in DNA. Unlike Srs2, Sgs1 does not stimulate the Exo1 activity, and the loss of Sgs1 results in a much greater increase in mutations at the CAN1 locus than what was observed in the srs2 mutant. Moreover, slippage mutations in simple repeats and base substitution mutations were observed, but only the −2 slippage mutations in repeats are dependent on Top1 activity. This too is in contrast to the spectrum of mutations observed with srs2Δ in rhn202Δ cells. Genetically, both Srs2 and Sgs1 seem to act in the same pathway to avoid slippage mutations. We suggest that Sgs1 acts at a step in rNMP repair or tolerance different from that dependent on Srs2. Sgs1 may act during DNA replication when a DNA polymerase encounters an rNMP residue in the template strand. In an RNaseH2 proficient strain, the RER process would remove the rNMP residue. In the absence of RNaseH2, Sgs1 may promote accurate bypass of the rNMP residue. In the absence of Sgs1, some of the rNMP residues are bypassed inaccurately, accounting for the increase in base substitution mutations, while others are subject to cleavage by Top1 and error-free repair via Srs2-Exo1.
Materials and methods
Determination of mutation rates
Mutation rates for the (AG)4 slippage reporter and Canr were determined by the Lea and Coulson median method as previously described.1,2,18 For the (AG)4 slippage reporter, single colonies were inoculated into 5 mL YPD and grown for 2 d at 30°C, washed and resuspended in 1 mL H2O. Cells were plated onto SC-lysine medium to select for slippage mutants and onto SC medium for total cell number. Plates were incubated at 30°C for 3 d At least 18 independent cultures with a minimum of 2 different isolates per genotype were used to determine rates with 95% confidence intervals.
For mutation rates at the CAN1 locus, CAN1+ strains were grown in 5 mL YPD overnight, washed and resuspended in 1 mL H2O. Cells were plated onto SC-arginine + 60 μg/ml canavanine to select for can1 mutants and onto SC medium for total cell number. Plates were incubated at 30°C for 3–4 d At least 18 independent cultures with a minimum of 2 different isolates per genotype were used to determine rates with 95% confidence intervals. Independent Canr segregants were sequenced at the CAN1 locus to identify mutations and the to establish rates of each class of mutational event.
Biochemical assays: Srs2, Mph1, Sgs1 and Exo1 were expressed and purified as described.1,19-21 For helicase and nuclease assays, a duplex DNA substrate with a unique U residue was treated by topoisomerase I from calf thymus (Invitrogen) to create a nick at the ribonucleoside site.1 Helicase, nuclease, and affinity pull-down assays were conducted as before.1
Supplementary Material
Abbreviations
- RER
ribonucletide excision repair
- rNMP
ribonucleoside-monophosphate
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank S. Jinks-Robertson and R. Rothstein for strains and plasmids.
Funding
This work was supported by National Institutes of Health grants R01GM053738 (to H.L.K.), RO1GM57814 (to P.S.) and R00ES021441 (to H.N.).
References
- 1.Potenski CJ, Niu H, Sung P, Klein HL. Avoidance of ribonucleotide-induced mutations by RNase H2 and Srs2-Exo1 mechanisms. Nature 2014; 511:251-254; PMID:24896181; http://dx.doi.org/ 10.1038/nature13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 2011; 332:1561-1564; PMID:21700875; http://dx.doi.org/ 10.1126/science.1205016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison A, Sugino A. The 3′–>5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet 1994; 242:289-296; PMID:8107676; http://dx.doi.org/ 10.1007/BF00280418 [DOI] [PubMed] [Google Scholar]
- 4.Rydberg B, Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc Natl Acad Sci U S A 2002; 99:16654-16659; PMID:12475934; http://dx.doi.org/ 10.1073/pnas.262591699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase delta. DNA Repair (Amst) 2013; 12:121-127; PMID:23245697; http://dx.doi.org/ 10.1016/j.dnarep.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guillet M, Boiteux S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J 2002; 21:2833-2841; PMID:12032096; http://dx.doi.org/ 10.1093/emboj/21.11.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC. Identification of Holliday junction resolvases from humans and yeast. Nature 2008; 456:357-361; PMID:19020614; http://dx.doi.org/ 10.1038/nature07470 [DOI] [PubMed] [Google Scholar]
- 8.Megonigal MD, Fertala J, Bjornsti MA. Alterations in the catalytic activity of yeast DNA topoisomerase I result in cell cycle arrest and cell death. J Biol Chem 1997; 272:12801-12808; PMID:9139740; http://dx.doi.org/ 10.1074/jbc.272.19.12801 [DOI] [PubMed] [Google Scholar]
- 9.Sekiguchi J, Shuman S. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol Cell 1997; 1:89-97; PMID:9659906; http://dx.doi.org/ 10.1016/S1097-2765(00)80010-6 [DOI] [PubMed] [Google Scholar]
- 10.Cho JE, Kim N, Li YC, Jinks-Robertson S. Two distinct mechanisms of Topoisomerase 1-dependent mutagenesis in yeast. DNA Repair (Amst) 2013; 12:205-211; PMID:23305949; http://dx.doi.org/ 10.1016/j.dnarep.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol Cell 2013; 49:1010-1015; PMID:23375499; http://dx.doi.org/ 10.1016/j.molcel.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang SY, Ghosh S, Pommier Y. Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J Biol Chem 2015; 290:14068-14076; PMID:25887397; http://dx.doi.org/ 10.1074/jbc.M115.653345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sparks JL, Burgers PM. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J 2015; 34:1259-1269; PMID:25777529; http://dx.doi.org/ 10.15252/embj.201490868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 1999; 286:552-555; PMID:10521354; http://dx.doi.org/ 10.1126/science.286.5439.552 [DOI] [PubMed] [Google Scholar]
- 15.Allen-Soltero S, Martinez SL, Putnam CD, Kolodner RD. A saccharomyces cerevisiae RNase H2 interaction network functions to suppress genome instability. Mol Cell Biol 2014; 34:1521-1534; PMID:24550002; http://dx.doi.org/ 10.1128/MCB.00960-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 2014; 158:327-338; PMID:24998930; http://dx.doi.org/ 10.1016/j.cell.2014.04.053 [DOI] [PubMed] [Google Scholar]
- 17.Stingele J, Habermann B, Jentsch S. DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem Sci 2015; 40:67-71; PMID:25496645; http://dx.doi.org/ 10.1016/j.tibs.2014.10.012 [DOI] [PubMed] [Google Scholar]
- 18.Spell RM, Jinks-Robertson S. Determination of mitotic recombination rates by fluctuation analysis in Saccharomyces cerevisiae. Methods Mol Biol 2004; 262:3-12; PMID:14769952 [DOI] [PubMed] [Google Scholar]
- 19.Krasner DS, Daley JM, Sung P, Niu H. Interplay between Ku and Replication Protein A in the Restriction of Exo1-mediated DNA Break End Resection. J Biol Chem 2015; 290:18806-18816; PMID:26067273; http://dx.doi.org/ 10.1074/jbc.M115.660191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L. et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 2010; 467:108-111; PMID:20811460; http://dx.doi.org/ 10.1038/nature09318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue X, Choi K, Bonner J, Chiba T, Kwon Y, Xu Y, Sanchez H, Wyman C, Niu H, Zhao X. et al. Restriction of replication fork regression activities by a conserved SMC complex. Mol Cell 2014; 56:436-445; PMID:25439736; http://dx.doi.org/ 10.1016/j.molcel.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.