

ABSTRACT
Primordial germ cells (PGCs) are embryonic founders of germ cells that ultimately differentiate into oocytes and spermatogonia. Embryonic proliferation of PGCs starting from E11.5 ensures the presence of germ cells in adulthood, especially in female mammals whose total number of oocytes declines after this initial proliferation period. To better understand mechanisms underlying PGC proliferation in female mice, we constructed a proteome profile of female mouse gonads at E11.5. Subsequent KEGG pathway analysis of the 3,662 proteins profiled showed significant enrichment of pathways involved in fatty acid degradation. Further, the number of PGCs found in in vitro cultured fetal gonads significantly decreased with application of etomoxir, an inhibitor of the key rate-limiting enzyme of fatty acid degradation carnitine acyltransferase I (CPT1). Decrease in PGCs was further determined to be the result of reduced proliferation rather than apoptosis. The inhibition of fatty acid degradation by etomoxir has the potential to activate the Ca2+/CamKII/5′-adenosine monophosphate-activated protein kinase (AMPK) pathway; while as an upstream activator, activated AMPK can function as activator of p53 to induce cell cycle arrest. Thus, we detected the expressional level of AMPK, phosphorylated AMPK (P-AMPK), phosphorylated p53 (P-p53) and cyclin-dependent kinase inhibitor 1 (p21) by Western blots, the results showed increased expression of them after treatment with etomoxir, suggested the activation of p53 pathway was the reason for reduced proliferation of PGCs. Finally, the involvement of p53-dependent G1 cell cycle arrest in defective proliferation of PGCs was verified by rescue experiments. Our results demonstrate that fatty acid degradation plays an important role in proliferation of female PGCs via the p53-dependent cell cycle regulation.
KEYWORDS: carnitine acyltransferase I A (CPT1A), fatty acid degradation, Primordial germ cells (PGCs), proliferation, p53-depedent G1 arrest
Introduction
Germ cells are specialized cells with the ability to differentiate into gametes, also known as oocytes and spermatogonia. During mammalian reproduction, these cells fuse to create a zygote capable of fetal development and containing both parents' genetic information. In mice, germ cell lineages are distinguished from somatic lineages at embryonic day (E) 6.5. Primordial germ cells (PGCs), which are derived from posterior proximal epiblast cells, appear at E7.5 under induction of BMP and other complex signaling.1,2 PGCs then migrate through the hindgut and body wall until finally arriving at the genital ridge (GR) between E10.5–E11.5,3 depending on mouse strain.4 Once PGCs colonize the GR,3 they proliferate rapidly until E13.5. During this significant period of PGC reserve formation, the GR simultaneously completes sex differentiation. In male mice, germ cells arrest during the G0 phase of mitosis at E13.5. In females, germ cells begin meiosis, but arrest at the diplotene stage of prophase I around E17.5.5
PGCs arrive at the GR and proliferate primarily between E11.5–E13.5. Abnormalities in proliferation may cause an insufficient number or depletion of germ cells after birth. In female mammals, reduced proliferation of germ cells has been associated with premature ovarian failure (POF).6 PGC prolifertation is dynamically regulated, but the mechanisms underlying progression of PGCs are not fully understood. To examine early development of PGCs after they colonize the GR, we constructed a proteome profile of female mouse gonads at E11.5. Based on KEGG pathway analysis of 3,662 identified proteins, fatty acid degradation pathways were highly enriched.
In this pathway, fatty acids are taken apart to produce adenosine triphosphate (ATP), the main energy source for animals. Fatty acid degradation is especially important for high energy-consuming tissues, such as skeletal and cardiac muscle. Deficiencies in fatty acid degradation can cause a range of disorders, including liver and cardiac dysfunction.7 However, the relationship between development of PGCs and fatty acid degradation remains largely uncharacterized. Fatty acid degradation includes 3 steps: lipolysis and release from adipose tissue, activation and transport into mitochondria, and β-oxidation. The second step of fatty acid degradation is critical. Carnitine acyltransferase I (CPT1), a mitochondrial transmembrane enzyme enabling fatty acid entry into the mitochondria, controls the rate-limiting step for the entire pathway.8 Therefore, in this study we chose CPT1 as the target protein to explore interdependency between fatty acid degradation and proliferation of PGCs, with the intent of identifying underlining mechanisms of PGC development.
Results
Significant enrichment of KEGG pathway for fatty acid degradation
E11.5 is the crucial point for development of PGCs, which have just colonized the GR and commenced rapid proliferation. At this point, proteomics technology was used to construct a proteome profile of female mouse gonads at E11.5, with a total of 3,662 proteins identified through 4 experiments (data not shown). Functional enrichment analysis made use of the KEGG pathway database; data from total protein were clustered using the analysis toolkit WebGestalt (http://bioinfo.vanderbilt.edu/ webgestalt/),9 to identify important pathways and biological processes. Fatty acid metabolism was significantly enriched, including fatty acid elongation in mitochondria, fatty acid degradation and the biosynthesis of unsaturated fatty acid, which yielded enrichment ratios of 15.99, 9 and 7.68, respectively (Fig. S1). Additionally, the tricarboxylic acid (TCA) cycle, responsible for transforming metabolites of fatty acid degradation into ATP, was also enriched at a ratio of 14.96. Of the 48 proteins involved in fatty acid degradation, 27 were identified in our proteome profile. A schematic of fatty acid degradation protein enrichment is shown in Figure S2, with red font indicating identified proteins. Next, a panel of 13 genes associated with each biochemical reaction was randomly selected for detection of expression in female GR from E11.5–E14.5. According to quantitative real-time PCR (qPCR) analysis, expression levels for most of these genes peaked at E12.5 and reached minimum at E14.5 (Fig. S3).
CPT1A is the primary CPT1 isoform expressed in genital ridge
CPT1A, an isoform of CPT1, was identified during our analysis. CPT1 is the rate-limiting enzyme responsible for regulating entry of fatty acids into mitochondria; repressing activity of CPT1 has the potential to inhibit fatty acid degradation. While other CPT1 isoforms include CPT1B and CPT1C, Cpt1a was identified as the primary isoform expressed in the GR (Fig. S4). qPCR analysis revealed mRNA expression levels of Cpt1a were high from E11.5–E13.5 compared with E14.5, peaking at E12.5. CPT1A protein expression levels were also quantified by Western blot at E11.5, E12.5 and E13.5, using anti-CPT1A antibody. High expression levels of CPT1A were observed at E12.5 and E13.5 (Fig. 1).
Figure 1.
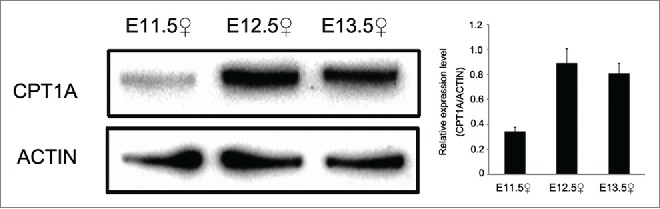
Expression level of protein of CPT1A between E11.5 and E13.5. Western blot analysis of CPT1A protein expression in genital ridge at E11.5 E12.5 and E13.5. Expression of CPT1A increased with embryonic development. ACTIN was used as a control. Densitometry of Western blotting was analyzed with Image-J software (NIH). Results are shown as mean ± SD.
Inhibition of fatty acid degradation leads to the decrease of PGCs
To explore effects of fatty acid degradation on development of PGCs, in vitro GR cultures were supplemented with etomoxir, a nonreversible inhibitor of CPT1,10 at doses of 2 μM, 10 μM and 50 μM. GR tissue was isolated at E11.5 and cultured for 48 h in medium containing etomoxir before transfer into normal medium for one day. GR cultured in normal medium served as a negative control. To quantify PGCs, immunofluorescence staining and confocal microscopy was performed for DDX4, a marker of germ cells (Fig. 2A). A slight decrease of PGCs was observed in 2 μM etomoxir-treated cultures, but no statistical difference existed when compared with controls (p > 0.05). However, in 10 μM and 50 μM etomoxir-treated cultures, PGCs decreased by 56.1% and 71.5% (P < 0.01; n = 3) compared with untreated cultures, respectively, in a concentration-dependent manner (Fig. 2B).
Figure 2.
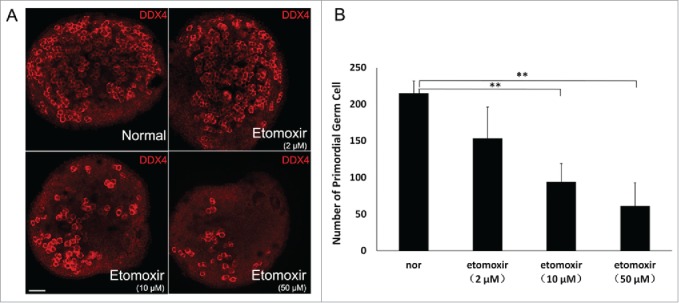
Inhibition of CPT1 led to decreased number of primordial germ cells (PGCs). (A) PGC number in E14.5 genital ridges cultured in normal medium containing various concentrations of etomoxir estimated by detection of DDX4-positive PGCs immunofluorescence staining. Bar =50 μm. (B) Average number of DDX4-positive PGCs in E14.5 genital ridge per 105 μm2. PGCs were counted in 3 sections from the middle part of individual genital ridges over the course of 3 independent experiments. Number of PGCs progressively decreased with inhibition of CPT1. (**), P < 0.01.
Decrease of PGCs in etomoxir-treated group resulted from reduced proliferation, not apoptosis
To ascertain mechanisms underlying an observed reduction in PGCs, we first assessed the apoptotic status of cells by using immunofluorescence staining to detect expression levels of cleaved caspase-3. A few cells exhibiting colocalized signals for cleaved caspase-3 and DDX4 were observed in all 4 groups (Fig. S5A), though this was determined to be statistically insignificant (Fig. S5B).
As apoptosis was not observed in etomoxir-treated PGCs and direct cytopathic effects of etomoxir had been excluded, we evaluated expression of proliferating cell nuclear antigen (PCNA) to determine the reason for reduced numbers of PGCs. The percentage of PCNA-positive cells in cultures treated with 2 μM, 10 μM and 50 μM etomoxir was 80%, 79% and 80%, respectively, compared with 82% in control cultures (Fig. 3A).
Figure 3.
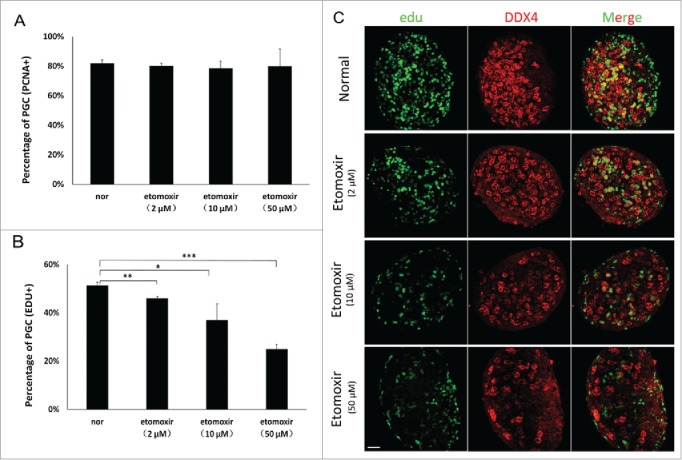
Reduced number of PGCs resulted from the proliferation defect. (A) Percentage of PCNA-positive PGCs in treated groups was similar to controls. Percentages in control and treated groups were 82±2 .3%, 80±1 .8%, 79±4 .3% and 80±11 .7%, respectively. (B) Edu was incorporated into PGCs. Edu-positive cells were counted in 3 sections from the middle part of individual genital ridges. The percentage of Edu-positive PGCs progressively decreased. (C) PGCs incorporating Edu were quantified using immunofluorescence staining; number of Edu-positive cells were determined in 3 independent experiments. Bar=50 μm.
GR was then cultured with varying doses of etomoxir and 50 μM Edu for 24 h (equal to E12.5), representing the peak stage of PGC proliferation. The ratio of Edu-positive PGCs was reduced in a concentration-dependent manner with increasing doses of etomoxir. Regarding percentage of Edu-positive PGCs, significant differences existed between control cultures (51.45 ± 1.2%) and cultures treated with etomoxir at doses of 2 μM (46.08 ± 0.9%, p < 0.01), 10 μM (37.09 ± 6.7%, p < 0.05) and 50 μM (25.04 ± 2.1%, p < 0.01), (n = 3) (Fig. 3B and C). In summary, etomoxir treatment had no effect on the percentage of PCNA-positive PGCs in vitro, but significantly decreased the percentage of Edu-positive PGCs.
Activation of p53-dependent G1 arrest in etomoxir-treated PGCs could be rescue by inhibition of p53
The expression level of AMPK, p53 and their phosphorylated forms in control and the etomoxir-treated groups was examined by Western blot. Results indicated expression levels of AMPK, p53 and their phosphorylated forms increased with escalating doses of etomoxir (Fig. 4A). Considering phosphorylation of p53 induces p21, an important G1 cell cycle phase inhibitor, expression of p21 was also evaluated by Western blot; expression levels of p21 also increased in a dose-dependent manner (Fig. 4A).
Figure 4.
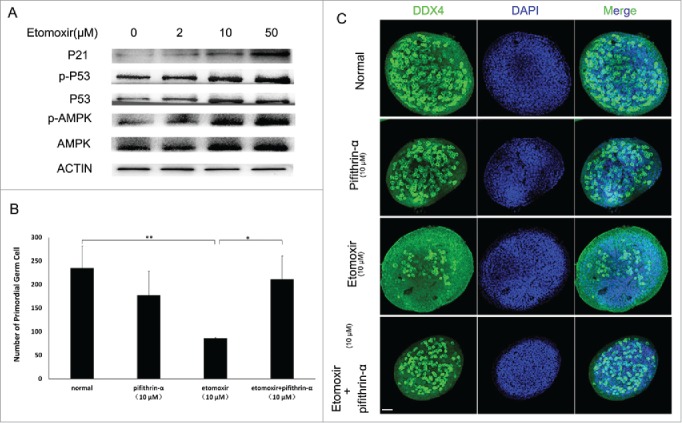
Verification of p53 activation dependent G1 arrest. (A) Western blot analysis showed expression of P-p53 and p21 increased with inhibition of fatty acid degradation. ACTIN was used as a control. (B and C). Quantity of PGCs was determined. Compared with controls, total number of PGCs significantly decreased after addition of CPT1 inhibitor. Total number of PGCs was restored to control levels by simultaneous addtion of CPT1 and p53 inhibitors into normal medium.
To confirm p53-dependent G1 arrest is required for a reduction in PGCs, GRs were cultured in 10 μM etomoxir supplemented with 10 μM of pifithrin-α, a p53 inhibitor, for 48 h. No statistical difference existed between the number of PGCs in control and pifithrin-α-treated cultures (235 ± 47 and 178 ± 50, respectively; p = 0.224). For positive controls, a decrease in PGCs was observed in 10 μM etomoxir-treated cultures (86 ± 1.6; p < 0.01), similar to previous experiments. Combined use of 10 μM etomoxir and 10 μM pifithrin-α rescued the effect of etomoxir on GR cultures by restoring the number of PGCs (211 ± 50; p = 0.58) to that of control cultures (Fig. 4B and 4C).
Discussion
PGCs are the first germ cell population established during development and are immediate precursors for both oocytes and spermatogonia. To comprehensively understand key proteins controlling PGC development, GR proteins were profiled at E11.5. Using the WebGestalt tool, we observed proteins involved in fatty acid degradation were highly enriched in the GR, indicating that fatty acid metabolism plays an important role during PGC development. While fatty acid degradation has been proven to play a major role in energy production in various cell types, its role and importance in PGCs is undefined.
Expression of CPT1, a rate-limiting enzyme for fatty acid degradation, was examined at various time points during PGC development. Results indicate CPT1 is hardly expressed at E11.5, however, a sharp increase in CPT1 expression occurs at E12.5 and E13.5. It is generally accepted that female mammals are born with ovaries containing a fixed number of primordial follicles. The period during which PGC proliferation occurs is vital for proper PGC development and formation of sufficient oocyte reserves for adult females. The period of rapid PGC proliferation is accompanied by increased CPT1 expression. Previous studies indicate PGCs reach the GR around E11 in most mouse strains,11 when the total number of PGCs is limited to approximately 1,000. Subsequently, PGCs undergo 2 or 3 additional rounds of mitosis to rapidly increase the total number to 20,000 by E13.5.12 After birth, the number of primordial follicles decreases progressively without possibility for renewal.13 Any impairment of proliferation, such as dysfunction caused by a mutation in the germ cell deficient (gcd) gene, can cause a drastic reduction of PGCs that leads to infertility in both males and females. Male mice with the gcd mutation exhibit vacuolated testes similar to mice with azoospermia; whereas, the female phenotype resembles POF syndrome in humans.14 In this study, addition of 10 μM and 50 μM etomoxir, an inhibitor of CPT1, significantly decreased the number of PGCs compared with untreated controls. This suggests fatty acid provides a vital energy source for PGC development.
The mechanism by which inhibition of CPT1 affects PGC development was also explored. First, apoptosis was evaluated by the presence of cleaved caspase-3 and DDX4 in cells, with results indicating colocalization in only a few germ cells. The number of caspase-3-positive cells was comparable between control and etomoxir-treated groups. The majority of cells in G0 phase did not express PCNA, a marker for cell proliferation; however, PCNA expression increased sharply during late G1 phase and reached peak levels in S-phase cells. During G2-M, cells demonstrated a reduced level of PCNA relative to S-phase cells.15 The results of double immunofluorescence staining for PCNA and Edu, a marker for DNA replication,16 indicated etomoxir treatment had no effect on percentage of PCNA-positive PGCs, but percentage of Edu-positive PGCs significantly decreased. Together, these data suggest inhibition of fatty acid degradation triggers G1-S cell cycle arrest, but not apoptosis, to decrease PGC proliferation. Although few studies have reported the association between PGC proliferation and fatty acid degradation, Tirado-Vélez and colleagues demonstrated inhibition of β-oxidation and de novo fatty acid synthesis reduces proliferation of human myeloma cells.17
AMPK, whose activation is regulated by ADP/ATP ratio,18 acts as a power switch to protect against energy deprivation in cells. Previous studies have demonstrated inhibition of fatty acid degradation by etomoxir activates the Ca2+/CamKII/AMPK pathway.19 Further, activation of AMPK has the potential to induce cell cycle arrest though p53 activation.20,21 p53 is a tumor suppressor protein exhibiting many anti-cancer mechanisms. First, p53 can arrest cell growth through regulation of the G1-S transition, while phosphorylated p53 (P-p53) induces p21 to block cell cycle progression out of G1. Second, p53 can initiate apoptosis through transcriptional activation. It is worth emphasizing that p53 initiation of either cell cycle arrest or apoptosis depends on cell type, cellular context and transcriptional activation of cancer genes.22 In this study, we detected increasing expression of P-p53 and p21, without changes to cleaved caspase-3 expression. We hypothesized that when fatty acid degradation is inhibited, activation of the p53 pathway triggers cell cycle arrest within PGCs, but not apoptosis. Further, addition of pifithrin-α to inactivate p53 in cultures successfully rescued decreases in PGCs induced by etomoxir treatment. Our results confirm inhibition of fatty acid degradation is capable of reducing PGC proliferation through regulation of the p53 pathway.
Based on our observations, we propose a model clarifying the mechanism of G1/S arrest during fatty acid metabolism, as shown in Figure 5. Etomoxir-induced inhibition of fatty acid degradation leads to phosphorylation of AMPK. Catalyzed by P-AMPK, P-p53 promotes transcriptional activation of p21, the key element of G1/S arrest. Thus, CPT1 is absolutely required for the development of PGCs and fertility. CPT1 deficiency is an autosomal recessive disorder of hepatic mitochondrial long-chain fatty acid oxidation. Patients generally experience a life-threatening episode (hypoketoitic hypoglycemia, liver cytolysis, and hepatomegaly) in early childhood;23 thus, the fertility of adult patients with CPT1 deficiency has been of little concern. Our research suggests more attention should be given to the reproductive capacity of patients with CPT1A deficiency and other inherited defects in mitochondrial fatty acid degradation. Recently, there is increasing evidence that maternal malnutrition during fetal development adversely affects the long-term health of her offspring, including reduced fertility;24,25 however, underlying mechanisms remain undetermined. Notably, a marked reduction in CPT1 expression was observed in the offspring of rats provided with a malnutrition diet during pregnancy.26 The vital role of fatty acid metabolism during proliferation of PGCs may bring new clues into the relationship of maternal nutritional signals and reproductive function of female offspring in adulthood. Our study also provides new insight into the development of oocytes, further studies will be helpful to germ cell reserve formation.
Figure 5.
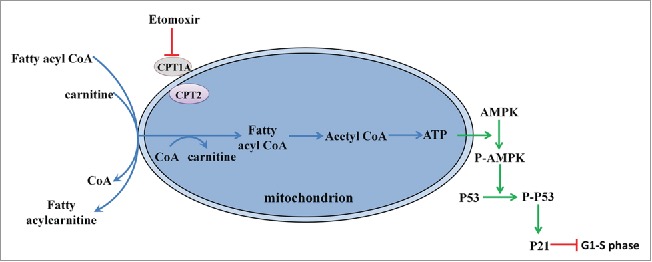
Working model based on our observations. Etomoxir inhibited fatty acid metabolism though the AMPK pathway, which catalyzed the phosphorylation of AMPK. P-p53, catalyzed by the P-AMPK, promoted the transcriptional activation of p21, the key element of G1/S arrest.
Materials and methods
Mice and ethics statement
All animal work was approved by the Ethics Committee of Nanjing Medical University, China. CD1 mice were kept within a strictly controlled environment of 20–25°C, 50–70% humidity, 12/12 h light/dark cycles, with free access to food and water.
Gonad collection and culture
Female CD1 mice (6–8 weeks old) were mated with males of the same strain in the afternoon. The following morning, females with mating plug were designated E0.5. The GR/mesonephric complex was isolated from embryos by dissection in physiological saline at room temperature. GR was separated from mesonephric tissue using a syringe, then cultured in DMEM/F-12 (Gibco, http://www.thermofisher.com/order/catalog/product/11330032?ICID=search-11330032 ) and MEM α (Gibco, http://www.thermofisher.com/order/catalog/product/12571063 ) containing 10% newborn calf serum (Gibco, http://www.thermofisher.com/order/catalog/product/16010159?ICID=search-16010159) and 50 μg/mL ampicillin at 37°C with 5% CO2. To inhibit function of cpt1 and p53, GRs were cultured in medium containing etomoxir (Sigma Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/e1905?lang=zhandregion=CN) and pifithrin-α (Santa Cruz Biotechnology, http://www.scbt.com/datasheet-45050.html) separately.
Sex appraisal
At E12.5, E13.5 and E14.5, GR of different sexes could be identified visually. However, at E11.5, no difference between male and female genital ridge is exhibited. Instead, PCR was used to perform sex appraisal. DNA was isolated from embryos and genotyping was performed using PCR primers to detect Sry, and β-actin.
Primer sequences are listed in Supplemental Table 1. PCR was performed under the following conditions: initial denaturation at 94°C for 6 min; 30 cycles of denaturation (94°C for 5 s), primer annealing (30 s at 60°C) and extension (45 s at 72°C); terminal elongation (10 min at 72°C); and a final hold at 4°C.
Immunofluorescence staining
GR was fixed in 4% paraformaldehyde for 2 h at room temperature. After washing in phosphate-buffered saline (PBS) 3 times for 15 min, tissue was incubated with 5% bovine serum albumin (BSA) in PBS for 2 h. Next, tissue was incubated with primary antibody at 4°C overnight. After washing in PBS 3 times for 15 min, secondary antibody was applied for 1 h at 37°C and nuclear stain Hoechst 33342 was applied. Images were captured on a Zeiss laser confocal microscope (LSM 710, Carl Zeiss; Oberkochen, Germany). Staining methods for paraffin-embedded genital ridge sections were similar; however, specimens had paraffin removed and were dehydrated before incuba-tion with 5% BSA in PBS. Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA) was used to quantify and normalize the number of PGCs. The detail information of the antibodies used is listed in Table S2.
Edu incorporation assay
Edu incorporation assay was performed using Cell-Light™ EdU Apollo®488 In Vitro Imaging Kit (Ribobio, http://www.ribobio.com/sitecn/productinfo_208231.html). Genital ridge was cultured in medium containing 50 μM Edu for 24 h prior to staining according to kit instructions. Images were captured on a Zeiss laser confocal microscope.
Real time PCR
In brief, total RNA was isolated using an RNeasy Plus Micro Kit (Qiagen, https://www.qiagen.com/us/products/catalog/sample-technologies/rna-sample-technologies/total-rna/rneasy-plus-micro-kit/), according to the manufacturer's instructions. Using Prime Script™ RT Reagent Kit (Takara, http://www.clontech.com/US/Products/Real-Time_qPCR_and_Reverse_Transcription/Reagents_and_Standards/Two-Step_RT-qPCR/PrimeScript_RT_Master_Mix?sitex=10020:22372:US), cDNAs were synthesized in 20 µl of reaction mixture according to manufacturer's instructions. qPCR was performed in the ABI step one plus Real-time PCR System. Primer sequences are listed in Table S1. Changes in gene expression were calculated as: fold change = 2-(ΔCT, Tg-ΔCT, control). The following PCR cycling was used: denaturation (6 min at 94°C);, 40 cycles of amplification (94°C for 5 s), annealing (60°C), extension (72°C for 30 s); 10 min at 72°C; and a final hold at 4°C.
Western blot analysis
Western blotting was performed as described previously.27,28 Genital ridge was cultured for 48 h and stored at −80°C. Tissue was lysed in ice cold RIPA lysis buffer and homogenized by motor cordless (749540-0000, Kimble). SDS-loading buffer was added to homogenates, where were then boiled for 5 min. First, proteins were separated by SDS-PAGE; second, they were transferred onto a polyvinylidene difluoride (PVDF) membrane. Next, the membrane was blocked with 5% BSA in Tris-buffered saline and tween (TBST) solution for 2 h at room temperature, then incubated overnight at 4°C with primary antibodies. After three 15-min washes in TBST buffer, the membrane was incubated at room temperature for 4 h with secondary antibodies. After washing with TBST, signals of targeted proteins were detected using the ChemiDoc XRS Imaging System (Bio-Rad, Hercules, CA, USA). Antibodies used during this process are listed in Table S2.
Statistics
All data were analyzed using mean and standard error of the mean. Data were evaluated by t-test statistical analysis or a non-parametric method to compare means between experimental and normal groups. Values of p < 0.05 were considered significant.
Supplementary Material
Abbreviations
- AMPK 5'
adenosine monophosphate-activated protein kinase
- ATP
adenosine triphosphate
- CPT1
carnitine acyltransferase I
- GR
genital ridge
- PGCs
Primordial germ cells
- P-p53
phosphorylated tumor protein p53
- p21
cyclin-dependent kinase inhibitor 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We gratefully acknowledge Liwen Bianji for editing the article.
Funding
This work was supported by grants from the National Basic Research Program of China (973 Program) (2012CB944704 and 2013CB911400) and the National Science Foundation of China (31171113).
References
- 1.Saga Y. Mouse germ cell development during embryogenesis. Curr Opin Genet Dev 2008; 18:337-41; PMID:18625315; http://dx.doi.org/ 10.1016/j.gde.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 2.Starz-Gaiano M, Lehmann R. Moving towards the next generation. Mech Dev 2001; 105:5-18; PMID:11429277; http://dx.doi.org/ 10.1016/S0925-4773(01)00392-6 [DOI] [PubMed] [Google Scholar]
- 3.Sosa BA, Demircioglu FE, Chen JZ, Ingram J, Ploegh HL, Schwartz TU. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife 2014; 3:e03239; PMID:25149450; http://dx.doi.org/ 10.7554/eLife.03239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molyneaux KA, Stallock J, Schaible K, Wylie C. Time-lapse analysis of living mouse germ cell migration. Dev Biol 2001; 240:488-98; PMID:11784078; http://dx.doi.org/ 10.1006/dbio.2001.0436 [DOI] [PubMed] [Google Scholar]
- 5.Menke DB, Koubova J, Page DC. Sexual differentiation of germ cells in XX mouse gonads occurs in an anterior-to-posterior wave. Dev Biol 2003; 262:303-12; PMID:14550793; http://dx.doi.org/ 10.1016/S0012-1606(03)00391-9 [DOI] [PubMed] [Google Scholar]
- 6.Duncan M, Cummings L, Chada K. Germ cell deficient (gcd) mouse as a model of premature ovarian failure. Biol Reprod 1993; 49:221-7; PMID:8373945; http://dx.doi.org/ 10.1095/biolreprod49.2.221 [DOI] [PubMed] [Google Scholar]
- 7.Olpin SE. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. Journal of inherited metabolic disease 2013; 36:645-58; PMID:23674167; http://dx.doi.org/ 10.1007/s10545-013-9611-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem 2011; 286:25655-62; PMID:21622568; http://dx.doi.org/ 10.1074/jbc.M111.228692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic acids research 2005; 33:W741-8; PMID:15980575; http://dx.doi.org/ 10.1093/nar/gki475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiorpes TC, Hoerr D, Ho W, Weaner LE, Inman MG, Tutwiler GF. Identification of 2-tetradecylglycidyl coenzyme A as the active form of methyl 2-tetradecylglycidate (methyl palmoxirate) and its characterization as an irreversible, active site-directed inhibitor of carnitine palmitoyltransferase A in isolated rat liver mitochondria. J Biol Chem 1984; 259:9750-5; PMID:6547720 [PubMed] [Google Scholar]
- 11.McLaren A. Primordial germ cells in the mouse. Dev Biol 2003; 262:1-15; PMID:14512014; http://dx.doi.org/ 10.1016/S0012-1606(03)00214-8 [DOI] [PubMed] [Google Scholar]
- 12.Pesce M, De Felici M. Purification of mouse primordial germ cells by MiniMACS magnetic separation system. Dev Biol 1995; 170:722-5; PMID:7649397; http://dx.doi.org/ 10.1006/dbio.1995.1250 [DOI] [PubMed] [Google Scholar]
- 13.De Felici M, Klinger FG, Farini D, Scaldaferri ML, Iona S, Lobascio M. Establishment of oocyte population in the fetal ovary: primordial germ cell proliferation and oocyte programmed cell death. Reprod Biomed Online 2005; 10:182-91; PMID:15823221; http://dx.doi.org/ 10.1016/S1472-6483(10)60939-X [DOI] [PubMed] [Google Scholar]
- 14.Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, Chada KK, Bishop CE. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet 2002; 11:3047-53; PMID:12417526; http://dx.doi.org/ 10.1093/hmg/11.24.3047 [DOI] [PubMed] [Google Scholar]
- 15.Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res 1986; 166:209-19; PMID:2874992; http://dx.doi.org/ 10.1016/0014-4827(86)90520-3 [DOI] [PubMed] [Google Scholar]
- 16.Zeng C, Pan F, Jones LA, Lim MM, Griffin EA, Sheline YI, Mintun MA, Holtzman DM, Mach RH. Evaluation of 5-ethynyl-2′-deoxyuridine staining as a sensitive and reliable method for studying cell proliferation in the adult nervous system. Brain Res 2010; 1319:21-32; PMID:20064490; http://dx.doi.org/ 10.1016/j.brainres.2009.12.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirado-Velez JM, Joumady I, Saez-Benito A, Cozar-Castellano I, Perdomo G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PloS one 2012; 7:e46484; PMID:23029529; http://dx.doi.org/ 10.1371/journal.pone.0046484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009; 458:1056-60; PMID:19262508; http://dx.doi.org/ 10.1038/nature07813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patella F, Schug ZT, Persi E, Neilson LJ, Erami Z, Avanzato D, Maione F, Hernandez-Fernaud JR, Mackay G, Zheng L, et al.. Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol Cell Proteomics 2015; 14:621-34; PMID:25573745; http://dx.doi.org/ 10.1074/mcp.M114.045575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009; 23:537-48; PMID:19270154; http://dx.doi.org/ 10.1101/gad.1756509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283-93; PMID:15866171; http://dx.doi.org/ 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- 22.Brown L, Boswell S, Raj L, Lee SW. Transcriptional targets of p53 that regulate cellular proliferation. Crit Rev Eukaryot Gene Expr 2007; 17:73-85; PMID:17361486; http://dx.doi.org/ 10.1615/CritRevEukarGeneExpr.v17.i1.50 [DOI] [PubMed] [Google Scholar]
- 23.Kompare M, Rizzo WB. Mitochondrial fatty-acid oxidation disorders. Seminars in pediatric neurology 2008; 15:140-9; PMID:18708005; http://dx.doi.org/ 10.1016/j.spen.2008.05.008 [DOI] [PubMed] [Google Scholar]
- 24.Dupont C, Cordier AG, Junien C, Mandon-Pepin B, Levy R, Chavatte-Palmer P. Maternal environment and the reproductive function of the offspring. Theriogenology 2012; 78:1405-14; PMID:22925651; http://dx.doi.org/ 10.1016/j.theriogenology.2012.06.016 [DOI] [PubMed] [Google Scholar]
- 25.Khorram O, Keen-Rinehart E, Chuang TD, Ross MG, Desai M. Maternal undernutrition induces premature reproductive senescence in adult female rat offspring. Fertility and sterility 2015; 103:291-8 e2; PMID:25439841; http://dx.doi.org/ 10.1016/j.fertnstert.2014.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang ZY, Dai YB, Wang HN, Wang MW. Supplementation of the maternal diet during pregnancy with chocolate and fructose interacts with the high-fat diet of the young to facilitate the onset of metabolic disorders in rat offspring. Clinical and experimental pharmacology & physiology 2013; 40:652-61; PMID:23819696; http://dx.doi.org/ 10.1111/1440-1681.12147 [DOI] [PubMed] [Google Scholar]
- 27.Huang XY, Guo XJ, Shen J, Wang YF, Chen L, Xie J, Wang NL, Wang FQ, Zhao C, Huo R, et al.. Construction of a proteome profile and functional analysis of the proteins involved in the initiation of mouse spermatogenesis. J Proteome Res 2008; 7:3435-46; PMID:18582094; http://dx.doi.org/ 10.1021/pr800179h [DOI] [PubMed] [Google Scholar]
- 28.Guo X, Shen J, Xia Z, Zhang R, Zhang P, Zhao C, Xing J, Chen L, Chen W, Lin M, et al.. Proteomic analysis of proteins involved in spermiogenesis in mouse. J Proteome Res 2010; 9:1246-56; PMID:20099899; http://dx.doi.org/ 10.1021/pr900735k [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.