

Abstract
Purpose
Human γS-crystallin (CrygS) is an important component of the human eye lens nucleus and cortex. The mutation G57W in the molecule is reported to be associated with congenital cataract in children. We compare the conformational features and aggregation properties of the mutant protein G57W with the wild-type CrygS to understand how the structural changes in the mutant are related to the mechanism of opacification.
Methods
Wild-type and mutant proteins were cloned, expressed, and purified, and their structural properties were studied in solution. Conformational features and the structural stability of the proteins were compared in solution, using circular dichroism (CD) and fluorescence spectroscopic analysis, and the proteins’ tendencies to aggregate were compared using extrinsic spectral probes. In addition, we analyzed the proteins’ structural differences with extensive molecular modeling in silico.
Results
CD and intrinsic fluorescence analysis suggested the secondary and tertiary structures of the mutant are slightly altered. Experiments using extrinsic spectral probes revealed that the compact close-packed structure is loosened somewhat, and the mutant tends to self-aggregate. Denaturation (both thermal and chemical) studies indicate that the replacement of glycine (G) in position 57 by tryptophan (W) lowered the structural stability of the molecule. Further, the mutant had a tendency to precipitate and scatters light more easily than the wild-type.
Conclusions
The replacement of glycine at position 57 by the tryptophan residue in human γS-crystallin weakens the stability of the mutant molecule and causes the molecule to self-aggregate, thus generating light-scattering particles. This set of changes in the mutant offers a molecular insight into the mechanism of opacification.
Introduction
As the lens grows in the eye of the human fetus, starting with the embryonic nucleus, this nuclear region is found to be particularly rich in the proteins called the βγ-crystallins (concentrations >400 mg/ml). The human lens has three major γ-crystallins, namely, γC, γD, and γS. A detailed summary of the structural and functional features of the γ-crystallins of the human eye lens and the role they play in providing short-range order in packing and providing transparency, and appropriate refractive index gradient in the lens, has recently been published [1]. Mutations in these crystallins are expected to affect their packing in the lens and compromise transparency, giving rise to lens opacification or cataract. Such mutations, when inherited, can lead to congenital cataracts.
Two comprehensive reviews of the epidemiology and molecular genetics of congenital cataracts have recently been published [2,3]. A significant proportion of these cataracts (about 25%) are estimated to be hereditary [4]. Mutations in more than 22 genes are reported to be associated with congenital cataracts [2]. And among the human γ-crystallins, 28 mutations have been reported thus far to be associated with cataracts, seven in γC, 24 in γD, and four in γS; a complete list of the 28 mutations in these three genes and their associated cataract phenotypes were presented in our earlier publication [5].
A fifth one in human γS-crystallin (abbreviated as CrygS), G57W, has been recently reported in a three-generation Chinese family wherein a young boy and his mother were found to have pulverulent cataract in the center of the lens [6]. While reporting on this, the authors also pointed out how the glycine residue in position 57 in CrygS is conserved across species and surmised that replacing it with the tryptophan residue (W) could affect the structural stability of the whole molecule. We have now cloned, expressed, isolated, purified, and studied the wild-type (WT) CrygS and the G57W mutant proteins in solution and present our results here. In addition, we also performed molecular modeling of both proteins, which shows how the introduction of the bulkier aromatic side chain of W in place of glycine leads to notable changes in the intra- and intermolecular interactions in the mutant. In effect, the mutant is less compact than the WT, easier to unfold upon heating and upon the addition of chemical denaturants, and tends to generate light-scattering aggregates more readily.
Methods
The study adhered to the tenets of the Declaration of Helsinki and the ARVO statement on human subjects. All materials, chemicals, enzymes, media, and buffers were obtained from standard firms (e.g.,Sigma Aldrich, St.Louis, MO., Invitrogen, Carlsbad, CA., New England BioLabs, Ipswich, MA., HiMedia, Mumbai, India.) and were of the highest available purity. A human cadaveric eye lens was collected from the Ramayamma International Eye Bank, L.V. Prasad Eye Institute, after ethical and scientific approval was received from the Institutional Review Board. Total RNA was isolated from the lens using TRIzol reagent. The first strand was synthesized by reverse transcription PCR (RT–PCR) using oligo-dT primer and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). Human γS-crystallin (CrygS) cDNA was amplified from the first strand using the forward primer with the Nde1 restriction site and reverse primer with the HindIII restriction site. The WT Crygs cDNA thus amplified was cloned into a SmaI digested pBSSK+ vector using T4-DNA ligase. The recombinant clones were confirmed with PCR and restriction digestion. WT cDNA was released from the pBSSK+ vector by restriction digestion with the Nde1 and HindIII restriction enzymes. The released cDNA was ligated into the Nde1 and HindIII sites of the pET-21a (+) vector using T4-DNA ligase. The mutant clone pET-21a γS G57W was generated from the pET-21a γS WT template with PCR-based site-directed mutagenesis using Phusion DNA polymerase. The amplification conditions were as follows: an initial denaturation step at 98 °C for 30 s, followed by 16 cycles of denaturation, annealing, and extension at 98 °C (10 s), 55 °C (30 s), 72 °C (3 min), respectively, with a final extension step at 72 °C for 10 min. The PCR product was digested with Dpn1 for 1 h at 37 °C and transformed into DH5-α, and the plasmids were isolated. Table 1 gives the list of primers used for cloning and sequencing the wild-type and mutant molecules.
Table 1. List of primers used for cloning and sequencing.
| Clone | Primer sequence (5’-3’) |
|---|---|
| pET21a-γS |
F: GGGAGTTCCATATGTCTAAAACTGGAACC |
| |
R: CCGGAATTCTTACTCCACAATGCG |
| pET21a-γSG57W |
F: GAAAGGCCCAACTTTGCTTGGTACATGTACATCTTACCACAGGGAG |
| |
R: GGTAAGATGTACATGTACCAAGCAAAGTTGGGCCTTTCATAAACAGCCC |
| T7 |
F: TAATACGACTCACTATAGG |
| T7 |
R: TATGCTAGTTATTGCTCAG |
| BGH | R: TAGAAGGCACAGTCGAGG |
Overexpression of recombinant proteins
The recombinant constructs pET-21a γS WT and pET-21a γS G57W were transformed into Escherichia coli BL21 (DE3) pLys (S) cells. A single colony containing the recombinant construct was picked, inoculated into 15 ml of Luria-Bertoni (LB) medium (HiMedia), containing 50 μg/ml ampicillin, and 34 μg/ml chloramphenicol and grown for 8 h by shaking at 225 rpm, 37 °C. After 8 h, 10 ml of the culture was transferred into 1 l of LB medium containing 50 μg/ml ampicillin and 34 μg/ml chloramphenicol. The cultures were grown at 37 °C to an absorbance value of 0.6 at 600 nm. Protein synthesis was induced with the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, and the cultures were grown for an additional 3.5 h. Cells were pelleted down from the 1 l culture by centrifugation at 6,000 ×g for 10 min at 4 °C.
The cell pellets were suspended in 40 ml of lysis buffer containing 50 mM Tris hydroxymethyl aminomethane chloride (Tris-Cl; pH 7.3), 100 mM KCl, 1 mM EDTA, 1 mM phenyl methyl sulfonyl fluoride (PMSF), and 20 μg/ml aprotinin. The cell suspension was extensively sonicated for 30 cycles (15 s bursts of sonication, followed by 45 s rest cycles) at 35% amplitude at 4 °C using a high-intensity ultrasonic processor (Sonics Vibra Cell; Sonics & Materials Inc., Newton, MA).The cell lysate was centrifuged at 30,000 ×g for 20 min at 4 °C. The resulting supernatant and the pellet were checked for the presence of the recombinant protein on 14% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Wild-type γS and G57W were predominantly found in the soluble fraction.
Purification of γS-WT and G57W proteins
The supernatant was subjected to ammonium sulfate fractionation at 30% concentration. Ammonium sulfate was added pinch by pinch, and the solution was incubated on ice for 2 h and centrifuged at 30,000 ×g for 20 min at 4 °C. The pellet and the supernatant were checked for the presence of the recombinant protein using 14% SDS–PAGE. The proteins CrygS-WT and G57W were predominantly found in the supernatant. The supernatant was loaded onto a Phenyl-Sepharose column equilibrated with 50 mM Tris-Cl buffer (pH 7.3) containing 30% of ammonium sulfate and eluted using a gradient of 0–30% of ammonium sulfate in 50 mM Tris-Cl buffer (pH 7.3). Individual fractions were checked on 14% SDS–PAGE. Fractions containing the required protein were pooled, concentrated using an Amicon stirred ultrafiltration cell with 3 kDa cut-off membrane. The concentrated protein was further purified to homogeneity with gel filtration chromatography using a Sephadex G-75 column.
The purity and specificity of the proteins were assessed with SDS–PAGE. The concentration of each protein was measured using A280 in 4.5 M guanidinium chloride (GdnCl), using its molar extinction coefficient (εM) calculated from Expasy.
Spectroscopic analysis
Circular dichroism (CD) spectra were recorded using a spectropolarimeter (J-810; Jasco, Easton, MD) at room temperature (27 °C). Far-ultraviolet (UV) CD spectra were recorded in the region between 250 and 190 nm, using 2 mm path length quartz cells and the near-UV CD spectra in the 340–250 nm region were recorded with 1 cm path length quartz cells. CD was measured at every 0.5 nm between 190 and 340 nm with 2 s response time at 100 nm/s speed. At least three scans of each spectrum were averaged, and the baselines of the buffer alone were subtracted. The protein concentration used to determine the far-UV spectra was 0.2 mg/ml in 20 mM Tris-Cl buffer (pH 7.3), and for the near-UV spectra, the protein concentration was 0.5 mg/ml in 50 mM Tris-Cl buffer (pH 7.3).
Intrinsic fluorescence spectra were recorded at room temperature (27 °C) using a fluorescence spectrophotometer (F-2500; Hitachi, Yokohama, Japan), and the spectra were recorded in the 300 to 400 nm range using an excitation wavelength of 295 nm, with 5 nm excitation and emission slits. The protein concentrations used were 0.2 mg/ml in 50 mM Tris-Cl buffer (pH 7.3). At least three scans of each spectrum were averaged, and the baselines of the buffer alone were subtracted.
Extrinsic fluorescence spectra of proteins were recorded at room temperature (27 °C) using two surface hydrophobicity probes, namely, 4,4’-dianilino-1,1’-binaphthyl-5,5′-disulfonate (bis-ANS) [7] and 9-diethylamino-5H-benzo[alpha]phenoxazin-5-one (Nile Red) [8]. With bis-ANS, spectra were recorded in the range of 400 to 600 nm, using an excitation wavelength of 390 nm with 5 nm excitation and emission slits. With Nile Red, the excitation was at 540 nm, and the emission was recorded between 570 and 700 nm, using 10 nm slits. The protein concentrations used in each case were 0.2 mg/ml in 50 mM Tris-HCl buffer (pH 7.3). Stock solutions of bis-ANS and Nile Red were prepared in methanol, and the final alcohol concentration was maintained below 7% v/v when the reagents were mixed with the proteins. The bis-ANS and Nile Red concentrations were measured using extinction coefficients of 16.8 mM−1 cm−1 at 385 nm and 45 mM−1 cm−1 at 552 nm, respectively. To check for protein aggregation and the possibility of amyloid type aggregates, we used the dye Thioflavin T [9], with the protein concentration at 0.2 mg/ml and increasing dye concentration from 10 μM to 70 μM. The spectra were recorded between 470 and 570 nm, with an excitation at 444 nm, and the data were analyzed as in [10].
Equilibrium unfolding and refolding experiments were performed at 37 °C by diluting the purified proteins to 0.2 mg/ml in a series of 46 different concentration solutions in the range of 0 to 4.5 M GdnCl with increasing intervals of 0.1 M GdnCl in a buffer containing 50 mM Tris-HCl, 1 mM EDTA, and 5 mM dithiothreitol (DTT). The samples were incubated at 37 °C for 16 h. The procedure used here was the same as in our earlier papers [5,10]. Fluorescence emission spectra were recorded for each unfolding sample as described. Data were analyzed by plotting the GdnCl concentration for each sample versus the ratio of fluorescence intensities at 360 and 320 nm. This ratio of fluorescence intensities at these wavelengths was chosen for the analysis to simultaneously monitor changes in native and unfolded fractions. The unfolding data were analyzed to determine the transition midpoints and the free energy of unfolding, ∆G°, by fitting the data to Greene and Pace’s two-state model [11] using GraphPad Prism software. The model that best fit the data was selected based on a random distribution of residuals. The transition midpoints and ∆G° were calculated for all transitions from these fits. In all fluorescence experiments, the response time used was 0.08 s, the scan speed was 60 nm/s, with the photomultiplier tube voltage below 400 V. Time-dependent light-scattering measurements of WT and G57W were performed by monitoring the protein solution turbidity at 600 nm, using a spectrofluorimeter with 5 nm excitation and emission slits, at different temperatures with the protein concentration of 0.200 mg/ml in 50 mM Tris-Cl buffer (pH 7.3).
Modeling studies
Several models of the G57W mutant were generated using the crystal structure 2M3T as the template [12,13]. The best two models, as deduced by GROMACS energy terms, were picked for molecular dynamics simulations and the comparison of amino acid pair interaction energies [14,15]. The protein molecule was placed in a cubic water box with volume 900 nm3 that contained about 9,200 water molecules, and the charge of the system was neutralized with Na+ ions. The potential energy of the system was initially lowered using steepest gradient minimization. The system was equilibrated at a constant temperature, first in an isochoric (constant volume) setting and then in an isobaric (constant pressure) setting for 100 ps each. Molecular dynamics for 5 ns were then performed in the CHARMM27 force field in GROMACS, version 4.5.6 [16]. System stability (300 K, 1 bar) was maintained with Berendsen temperature coupling [17] and Parrinello-Rahman pressure coupling [18]. Energy estimates, as well as snapshots of molecular structures were collected from the last 100 ps of the simulation, where residues 52 and 57 were recorded as separate energy groups. Interaction energies between select amino acid pairs were estimated using the g_energy function in GROMACS, as sums of the short-range Lennard-Jones terms (for van der Waals interactions) and short-range coulomb terms (for electrostatic interactions). The cut-off distance for the neighbor list was set at 1.15 nm; the cut-off distance for the short-range van der Waals and coulomb was 0.9 nm [19].
Results
Human CrygS is known to be folded using four Greek key motifs [20], each motif constituting an interlocking set of four β-strands. The N-terminal half of the molecule has two such motifs, in sequences 1–40 and 42–83, respectively, while residues 88–128 and 129–171 form the two Greek key motifs in the C-terminal domain. The two domains fold on each other, leading to a compact, stable, and close-packed globular structure [1]. Spectroscopic studies on the protein bear this out. Figure 1A compares the circular dichroism spectra in the far-UV region (260–190 nm) of WT and G57W. The characteristic negative band around 218 nm is indicative of a significant amount of β-pleated sheet secondary structural conformation in the WT protein. The slight reduction in the 218 nm peak in the mutant appeared to suggest that introduction of W in position 57 in the second GK motif perturbs the conformation mildly.
Figure 1.
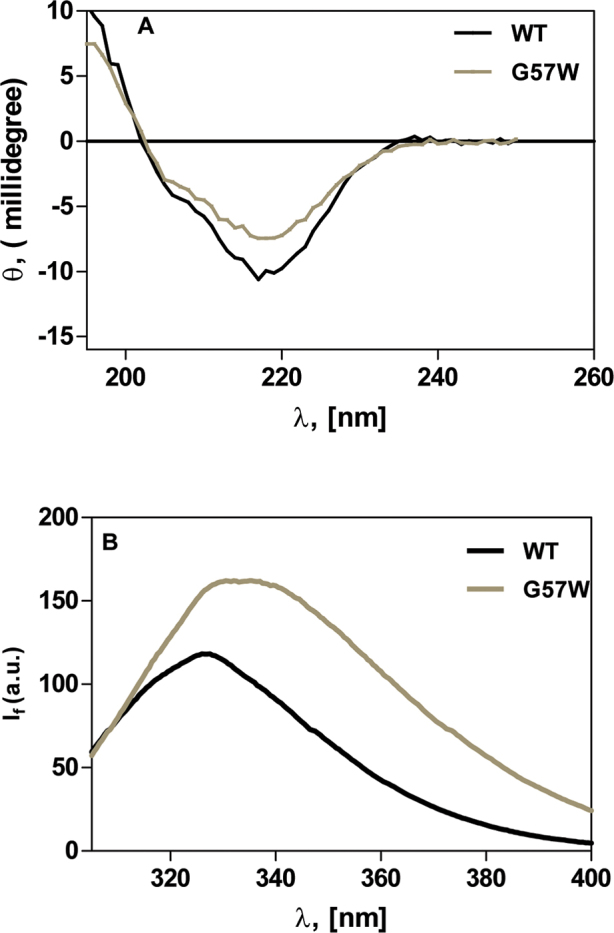
The G to W change at position 57 mildly affects the conformation of the protein. A: Far-ultraviolet (UV) circular dichroism (CD) spectra of wild-type CrygS and its mutant G57W. θ = ellipticity in millidegrees. The protein concentration in each case was 0.2 mg/ml in 10 mM sodium phosphate buffer (pH 7.3). The cell path length was 2 mm, and all spectra were recorded at 27 °C, corrected for the background buffer signal. Each spectrum is an average of three independent runs. B: Intrinsic fluorescence of wild-type and G57W CrygS. If = fluorescence emission intensity in arbitrary units; λexc = 295 nm. The protein concentrations used were 5 μM in 50 mM Tris-Cl, (pH 7.3). The cell path length was 3 mm, the excitation and emission slits were 5 nm, and the spectra were recorded at 27 °C.
Wild-type human CrygS has a total of 4 W, 14 Y, and 8 F residues in its sequence, and these aromatic side chains display intrinsic fluorescence emission in the 280–340 nm region of the near ultraviolet, upon excitation in the 255–300 nm region. Of these three fluorophores, the fluorescence emission quantum yield is in the order W > Y > F. And, among them, the emission wavelength (λf) and the intensity (If) of W are particularly sensitive to the polarity of the environment in which the fluorophore is placed. Monitoring the λf and If of the emission of the W residues in proteins thus offers an idea about the microenvironment that the residues experience. In a nonpolar microenvironment of the aromatic residues, the emission maximum occurs in the 320 nm region with a low quantum yield, but in a more polar microenvironment, it displays a red shifted emission band and an increase in the emission intensity. Figure 1B compares the emission profiles of the two molecules. When the W residues are excited using 295 nm, the λf maximum of emission of the W residues in the WT is centered at around 327 nm, with an If value of 118 arbitrary units. These suggest that the W residues in the WT are essentially located in a nonpolar environment. The mutant displays a slightly red-shifted (by 3.5 nm) band maximum at λf of 330.5 nm and a higher If of 162 arbitrary units, suggesting a slightly more polar microenvironment. We next performed fluorescence quenching experiments to investigate this microenvironment around the W residues, per [21], using the ionic quencher KI, which would quench the emission of fluorophores that are accessible to the surface of the proteins. The Stern-Volmer quenching constant KSV of the WT molecule, using KI as the quencher, was estimated to be 0.96 M−1. In contrast, the KSV for the mutant under the same conditions was higher, with a value of 1.62 M−1. In contrast to KI, the nonionic quencher molecule, acrylamide, can reach out to polar and nonpolar environments in the molecule. We found the KSV value for acrylamide quenching of the WT to be 0.45 M−1, while for the G57W mutant, it was 0.91 M−1. Thus, the KI and acrylamide quenching results suggest that the W residue(s) in the mutant are in a somewhat more polar microenvironment.
To gain a better idea of the solvent-accessible surface of the proteins, we next used the extrinsic fluorescence probe, bis-ANS, whose intensity increases upon binding to a surface (exposed) region of the macromolecule [7]. Figure 2A reveals that the If value of bis-ANS bound to the WT is about 8 arbitrary units, but when bound to an equivalent concentration of G57W, the If value increases to 60 arbitrary units, suggesting that the mutation brings about a greater amount of surface exposure in the protein. This was confirmed when we used the other (neutral) extrinsic probe, Nile Red [8]. Figure 2B reveals that this probe shows consistently higher emission intensities upon binding to the mutant than with the wild-type. We also studied a third surface-binding probe, Thioflavin T, which is used to monitor the aggregation (and amyloid-type) behavior of proteins it binds to [9]. The higher signal intensity it displays when bound to the mutant than to the wild-type (Figure 2C) suggests that G57W has a significant higher tendency to self-aggregate (though whether there is any amyloid type aggregation is not clear).
Figure 2.
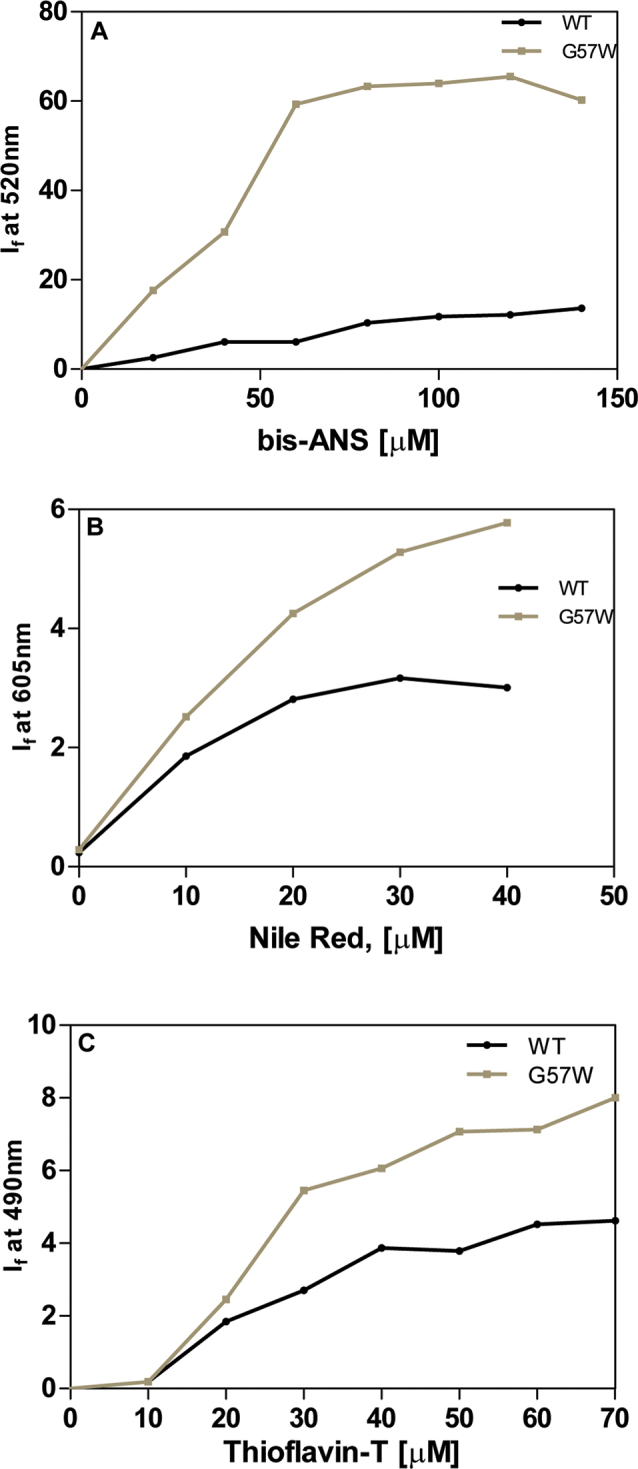
Mutant is more surface-accessible and readily self-aggregates. A: Surface exposure of residues in the wild-type and G57W CrygS, monitored using bis-ANS as the extrinsic probe. λexc = 390 nm. If at 490 nm of the probe was measured as a function of its increasing concentration. The cell path length was 3 mm, the excitation and emission slits were 5 nm, and the spectra were recorded at 27 °C. Each point is an average of three independent runs. B: Aggregation tendencies of the wild-type and mutant human γS-crystallin G57W, estimated using Nile Red as the extrinsic probe. λexc = 540 nm. If at 605 nm of the probe was measured as a function of its increasing concentration. The cell path length was 3 mm, the excitation and emission slits were 10 nm, and the spectra were recorded at 27 °C. Each point is an average of three independent runs. C: Aggregation behavior of the wild-type and mutant γS crystallin G57W, probed using Thioflavin-T. If values at 490 nm were measured as a function of the increasing concentration of the probe. The cell path length was 3 mm, the excitation and emission slits were 10 nm, and the spectra were recorded at 27 °C. Each point is an average of three independent runs.
Such an “opening up” is expected to tell on the stability of the molecule. To check this possibility, we compared the unfolding of the protein, first upon heating and then using a chemical denaturant such as GdnCl. Figure 3A shows that the native molecule CrygS unfolds upon heating, at 75.8 °C (349 K). In contrast, the mutant is thermally less stable, denaturing at 63 °C (336 K). Turning to chemical denaturation, Figure 3B shows that the wild-type molecule displays a sharp transition from the native to the denatured state, with a midpoint at 2.15 M GdnCl, while the mutant G57W displays a more gradual transition. Both curves were found to fit the Greene-Pace “two-state model” of denaturation (native ↔ denatured), with no intermediate states [11]. Fitting them to such a two-state model, we found the free energy of unfolding of WT was 10.97±0.34 kcal/mol and that of G57W was substantially lower, that is, 4.20±0.50 kcal/mol.
Figure 3.
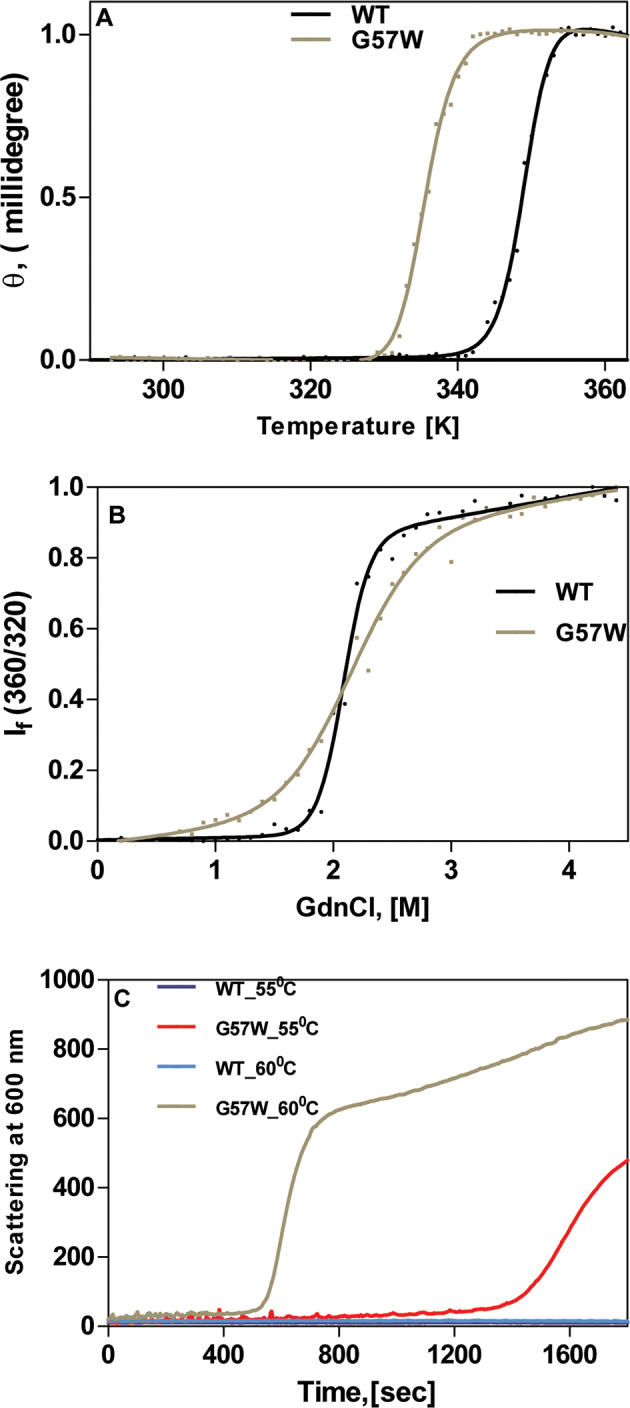
The mutant is less stable as seen by thermal and chemical denaturation and tends to aggregate producing light-scattering particles. A: The mutant G57W denatures at a lower temperature than the wild-type (WT) CrygS. The circular dichroism (CD) spectrum of each protein was measured at a wavelength of 222 nm, as a function of increasing temperature (290–360 K), and the data were fitted using the two-state model. The protein concentration in each case was 0.2 mg/ml in 10 mM sodium phosphate buffer (pH 7.3). The cell path length was 2 mm, and all spectra were corrected for background buffer signal. Each spectrum is an average of three independent runs. B: Guanidinium chloride–induced denaturation of wild-type and G57W CrygS. The relative emission intensity of the 360 nm band (of the denatured form) was compared to that of the 320 nm band (of the WT protein) and monitored as a function of denaturant concentration. λexc in each case was 295 nm. The protein concentration in each sample was fixed at 0.2 mg/ml in 50 mM Tris buffer, 1 mM EDTA, and 5 mM dithiothreitol (DTT). The cell path length was 3 mm, the excitation and emission slits were 5 nm, and the spectra were recorded at 27 °C. The spectrum shown is the average of three runs. C: Thermal aggregation of WT and G57W CrygS at two different temperatures. Light scattering was measured at 600 nm for 1800 s at 55 °C and 60 °C. The protein concentration was 0.2 mg/ml in 50 mM Tris buffer, the cell path length was 10 mm, the excitation and emission slits were 5 nm, and the spectra were recorded at 27 °C.
The tendency of the mutant to aggregate was also studied using temperature-dependent light scattering, as was performed previously with other mutant molecules [10,22]. Figure 3C shows that the wild-type molecule, upon standing at 55 °C (or even at 60 °C) for as long as 1,800 s (30 min), does not generate light-scattering aggregates, but the mutant molecule does after standing for about 1,400 s at 55 °C, and more readily, by 600 s at 60 °C.
Discussion
Spectral analysis of the two proteins offers some clues into the changes that occur to CrygS when Gly57 is replaced by the Trp residue. That G57 is a highly conserved residue across species has been noted [6]. Replacing it with the bulkier Trp is seen to affect the structural compactness of the protein. Although the secondary structure (backbone conformation) is only mildly altered (Figure 1A), the tertiary structure is seen to be affected more noticeably. That the intrinsic fluorescence emission of G57W is quenched by the ionic quencher KI to a higher extent than in the WT suggests that W57 is exposed to the surface. Results obtained using the three extrinsic reporters bis-ANS, Nile Red, and Thioflavin T (Figure 2A–C) confirmed this inference and showed that the microenvironment around this segment of the second Greek key motif (sequence 42–83, where W57 is located) in the mutant molecule is significantly more polar than in the case of WT. This appears to lead to intermolecular interactions and self-aggregation of the mutant. Results shown in Figure 3C confirm this point.
All these data, taken together, suggest that replacement of the highly conserved residue Gly57 by Trp leads to a “loosening up” of the compact conformation of CrygS. Exposing W57 and its immediate neighborhood in the second Greek key motif to the surface also appears to lead to a compromise in the stability of the mutant¸ as can be seen in Figure 3A,B. The mutant needs less than half of the energy to unfold (∆G° of 4.2 kcal/mol, cf. 11 kcal/mol for the WT). In these features, this mutant of CrygS behaves remarkably similar to the other mutant V42M (also a mutation in the second Greek key fold) of CrygS [22]. Interestingly, the three other reported mutants of CrygS that have been studied, namely, G18V [23-25], D26G [10], and S39C [1], all perturbing the folding of the first Greek key motif of the molecule, behave in much the same manner: They lose their compactness and expose residues to the surface, are weaker in stability, and self-aggregate to generate light-scattering particles. These studies highlight the vital role that the Greek key motifs play in maintaining the compact, globular packing, and structural stability of βγ-crystallins [5,26].
We analyze and discuss these results further using additional input from molecular modeling studies. Although Yang et al. [6] used the CLC Main Workbench software to look at the possible changes that might occur to the molecule upon substituting G57 by W, we took it further and supplemented molecular modeling with extended molecular dynamics simulations, based on the fact that the crystal structure of the C-terminal domain of the CrygS molecule and the solution state nuclear magnetic resonance (NMR) spectral analysis of the entire molecule have been published [24].
Figure 4A,B illustrate the space-filling models. Introduction of W in position 57 leads to a close side chain-side chain interaction between the polar (cationic) R residue in position 52 and the π-electron cloud of the indole ring of W 57, which are estimated to come in close proximity (about 5 Å). This is reflected in Figure 5 which shows that the inter-side chain interaction is about three times stronger in the mutant. In this connection, Gallivan and Dougherty [27], based on a study of about 593 proteins with dissimilar sequences in the Protein Data Bank, calculated that about 99% of all acceptable cation–π interactions in these proteins lie within a distance of 6 Å, that Arg is more likely than Lys to be in a cation–π interaction, and among the aromatics, a strong bias toward Trp is clear.
Figure 4.

Modeled structures of human CrygS and its G57W mutant. A: Space-filling rendering molecular model of wild-type CrygS. B: Similar space-filling model of the mutant G57W.
Figure 5.
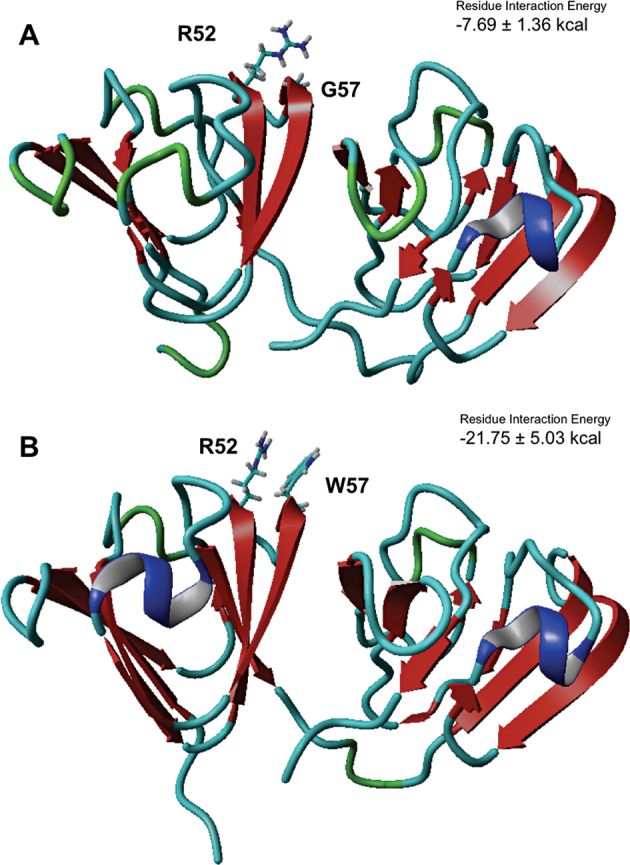
Comparison of the residue interactions between R52 and residue 57 in the WT and in the mutant. Ribbon diagrams of the wild-type (A) and mutant (B) models, viewed from the standard perspective, with the C-terminal domain on the right side. The calculated interaction energies between residues R52 and G57 or W57 are shown in the insets.
The combined solvent-accessible surface area of the two residues (R52 and W57) is estimated to be 244.6 Å2, considerably higher than the 122.23 Å2 for the R52/G57 pair in the wild-type protein. Figure 4B also suggests a slight readjustment in the packing of the N-terminal domain of the mutant in comparison to WT, and it appears that this might be reflected in the readjustment of residue Y70. Accommodating the bulky side chains of R52 and W57, however, has a disruptive effect on the loop that carries these residues; as a result, the distance between the alpha carbons of the two residues increases to 6.3 Å from 5.46 Å in the wild-type protein. (The other altered Cα-Cα distances are the coming together of residues R52 and D29, from 9.5 Å in the WT to 7.7 Å in the mutant, of R52 and S81, from 5.8 Å to 5.0 Å in the mutant, and the slight moving away of E51 and G/W57 from 5.2 Å in the WT to 5.64 Å in the mutant.) We also note that there is a salt bridge between residues E51 and R52 in the WT molecule; this salt bridge is disrupted in the G57W mutant due to a shift in the position of R52 to facilitate the cation–π interaction described. Salt bridges are often found across residues that are sequence-wise close and in the same secondary structure feature [28].
Although the N-terminal domain of the molecule is affected by the mutation, modeling studies and Figure 4A,B suggest no particularly notable change in the packing or the conformation of the C-terminal domain (see Figure 4A,B). Interestingly, all the mutations reported thus far in human CrygS are in the N-terminal domain (N-tD). The N-tD of CrygS has been found to be inherently less stable than its C-tD (upon thermal and chemical unfolding), the stability of the C-tD is comparable to that of the full-length molecule itself, and the domain interface presumably does not make a significant contribution to the overall stability of the full molecule [26]. Therefore, studying a naturally occurring or laboratory-made C-terminal mutant of CrygS, and comparing its packing and stability features with those of the wild-type molecule, would be interesting. All these results, taken together, indicate that a single point mutation in position 57 in the second Greek key motif of the N-terminal domain of CrygS leads to opening up of the compact packing of the molecule and a notable decrease in its stability.
Acknowledgments
We thank Mr. Srinivasu Karri of the Department of Biochemistry, School of Life Sciences, University of Hyderabad, India for his ready help and advice with some experiments and interpretation. IK thanks the Department of Biotechnology, India for a Research Associateship. This resesrch is supported in part by a grant from the Department of Biotechnology, India (grant number BT/MB/Indo-US/LEVR/2/2013.
References
- 1.Vendra VP, Khan I, Chandani S, Muniyandi A, Balasubramanian D. Gamma crystallins of the human eye lens. Biochim Biophys Acta. 2016;1860(1 Pt B):333–43. doi: 10.1016/j.bbagen.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Shiels A, Hejtmancik JF. Genetics of human cataract. Clin Genet. 2013;84:120–7. doi: 10.1111/cge.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi J, Yun J, Li Z-K, Xu C-T, Pan B-R. Epidemiology and molecular genetics of congenital cataracts. Int J Ophthalmol. 2011;4:422–32. doi: 10.3980/j.issn.2222-3959.2011.04.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckstein M, Vijayalakshmi P, Killedar M, Gilbert C, Foster A. Aetiology of childhood cataract in south India. Br J Ophthalmol. 1996;80:628–32. doi: 10.1136/bjo.80.7.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vendra VPR, Agarwal G, Chandani S, Talla V, Srinivasan N, Balasubramanian D. Structural integrity of the Greek key motif in βγ-Crystallins is vital for central eye lens transparency. PLoS One. 2013;8:e70336. doi: 10.1371/journal.pone.0070336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Li Q, Zhu S, Ma XA. G57W mutation of CRYGS associated with autosomal dominant pulverulent cataracts in a Chinese family. Ophthalmic Genet. 2015;36:281–3. doi: 10.3109/13816810.2013.865761. [DOI] [PubMed] [Google Scholar]
- 7.Rosen CG, Weber G. Dimer formation from 1-amino-8-naphthalenesulfonate catalyzed by bovine serum albumin, a new fluorescent molecule with exceptional binding properties. Biochemistry. 1969;8:3915–20. doi: 10.1021/bi00838a006. [DOI] [PubMed] [Google Scholar]
- 8.Sutter M, Oliveira S, Sanders NN, Lucas B, Van Hoek A, Hink MA, Visser AJ, De Smedt SC, Hennink WE, Jiskoot W. Sensitive spectroscopic detection of large and denatured protein aggregates in solution by use of fluorescent dye Nile Red. J Fluoresc. 2007;17:181–92. doi: 10.1007/s10895-007-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Vine, H III. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–84. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 10.Srinivasu K, Ramesh Babu K, Vendra VPR, Chandani S, Balasubramanian D. Structural analysis of the mutanat protein D26G of human γS-crystallin associated with Coppock cataract. Mol Vis. 2013;19:1231–7. [PMC free article] [PubMed] [Google Scholar]
- 11.Greene RF, Jr, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease,lysozyme, α -chymotrypsin and β-lactoglobulin. J Biol Chem. 1974;249:5388–93. [PubMed] [Google Scholar]
- 12.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W242–52. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40:W294–297. doi: 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berendsen HJ, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 15.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 16.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, MacKerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257–73. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–90. [Google Scholar]
- 18.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52:7182–90. [Google Scholar]
- 19.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson EG, Thornton JM. The Greek key motif: extraction, classification and analysis. Protein Eng. 1993;6:233–45. doi: 10.1093/protein/6.3.233. [DOI] [PubMed] [Google Scholar]
- 21.Augusteyn RC, Chandrasekher G, Ghiggiano KP, Vassett P. Probing the microenvironments of tryptophan residues in the monomeric crystallins of the bovine lens. Biochim Biophys Acta. 1994;1205:89–96. doi: 10.1016/0167-4838(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 22.Vendra VP, Chandani S, Balasubramanian D. The mutation V42M distorts the compact packing of the human gamma-S-crystallin molecule, resulting in congenital cataract. PLoS One. 2012;7:e51401. doi: 10.1371/journal.pone.0051401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Z, Piszczek G, Wingfield PT, Sergeev YV, Hejtmancik JF. The G18V CRYGS mutation associated with human cataracts increases gammaS-crystallin sensitivity to thermal and chemical stress. Biochemistry. 2009;48:7334–41. doi: 10.1021/bi900467a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brubaker WD, Martin RW. 1H, 13C, and 15N assignments of wild-type human γS-crystallin and its cataract-related variant γS-G18V. Biomol NMR Assign. 2011;6:63–7. doi: 10.1007/s12104-011-9326-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brubaker WD, Freitas JA, Golchert KJ, Shapiro RA, Morikis V, Tobias DJ, Martin RW. Separating instability from aggregation propensity in γS-crystallin variants. Biophys J. 2011;100:498–506. doi: 10.1016/j.bpj.2010.12.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills IA, Flaugh SL, Kosinski-Collins MS, King JA. Folding and stability of the isolated Greek key domains of the long-lived human lens proteins gammaD-crystallin and gammaS-crystallin. Protein Sci. 2007;16:2427–44. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallivan JP, Dougherty DA. Cation- pi interactions in structural biology. Proc Natl Acad Sci USA. 1999;96:9459–64. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar S, Nussinov R. Salt bridge stability in monomeric proteins. J Mol Biol. 1999;293:1241–55. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]