

Abstract
Loss-of-function studies are valuable for elucidating kinase function and the validation of new drug targets. While genetic techniques, such as RNAi and genetic knockouts, are highly specific and easy to implement, in many cases post-translational perturbation of kinase activity, specifically pharmacological inhibition, is preferable. However, due to the high degree of structural similarity between kinase active sites and the large size of the kinome, identification of pharmacological agents that are sufficiently selective to probe the function of a specific kinase of interest is challenging, and there is currently no systematic method for accomplishing this goal. Here, we present a modular chemical genetic strategy that uses antibody mimetics as highly selective targeting components of bivalent kinase inhibitors. We demonstrate that it is possible to confer high kinase selectivity to a promiscuous ATP-competitive inhibitor by tethering it to an antibody mimetic fused to the self-labeling protein SNAPtag. With this approach, a potent bivalent inhibitor of the tyrosine kinase Abl was generated. Profiling in complex cell lysates, with competition-based quantitative chemical proteomics, revealed that this bivalent inhibitor possesses greatly enhanced selectivity for its target BCR-Abl, in K562 cells. Importantly, we show that both components of the bivalent inhibitor can be assembled in K562 cells to block the ability of BCR-Abl to phosphorylate a direct cellular substrate. Finally, we demonstrate the generality of using antibody mimetics as components of bivalent inhibitors by generating a reagent that is selective for the activated state of the serine/threonine kinase ERK2.
Keywords: bivalent, kinase, BCR-Abl, MAPK, antibody mimetic, inhibitor
Graphical Abstract
Introduction
Classical forward genetics has been a powerful technique for deconvoluting the cellular roles of proteins through loss-of-function studies, and is one of the principle means for discovering novel drug targets.1 Genetic methods that disrupt protein function prior to translation, like gene deletions and RNAi, are attractive for these studies due to their genome-wide scalability, high specificity, and ease of implementation.2 Such methods of disrupting protein function do so by significantly reducing expression of the entire protein from the cell, which for a multi-domain protein makes it difficult to attribute the observed phenotype to the inhibition of a specific function. For example, distinguishing between inhibition of catalytic activity or disruption of critical protein-protein interactions is not possible as both the catalytic and scaffolding functions have been removed simultaneously. Additionally, these methods introduce permanent changes, or slowly reversible in the case of RNAi, making it difficult to differentiate direct effects of inhibition of protein function from compensatory changes that occur due to chronic loss of protein function.
In some instances, small molecule inhibition of protein function, which occurs post-translationally, may be preferable for understanding the cellular function of proteins. This is especially true when validating putative drug targets because the mechanism of action is the same as that of a pharmacological agent. In many cases, pharmacological inhibition and RNAi do not lead to the same phenotype.2 The challenge in using small molecule inhibitors for studying protein function is in achieving a sufficiently high degree of selectivity and proteome-wide scalability. Therefore, technologies that elicit the pharmacological consequences of post-translational protein target inhibition without requiring the extensive effort needed for the development a mono-selective inhibitor are valuable.
Protein kinases are especially attractive targets for the development of more efficient post-translational inhibition strategies because the identification of selective inhibitors for specific members of this large enzyme family is challenging, and the inherent plasticity and compensatory changes observed in protein kinase signaling cascades complicates the use of genetic-based methods for probing function.3,4 In an attempt to identify selective and potent inhibitors of protein kinases, bivalent inhibition strategies have been employed wherein a small molecule targeting the highly conserved ATP-binding site is tethered to a specificity ligand that binds to a unique interaction motif of a kinase of interest. If the selected specificity ligand is capable of discriminating between closely related kinases, and the linkage between the two monovalent components is favorable, potent and selective bivalent inhibitors can be generated.5,6 We have developed a bivalent inhibitor strategy based on an engineered version of the self-labeling DNA repair enzyme O6-alkylguanine-DNA alkyltransferase (commonly referred to as SNAPtag).7–9 By selectively labeling SNAPtag fusion proteins, which display a specificity ligand from their N- or C-termini, with an ATP-competitive inhibitor, potent and selective bivalent inhibitors of several kinases have been generated.10–13 Our previously described SNAPtag-based bivalent inhibitors relied on the use of genetically-encoded peptide ligands that had previously been characterized to be selective for a targeted site of interest in biochemical screens (referred to here as consensus-binding motifs) as secondary specificity ligands. While this strategy led to impressive selectivity amongst closely related kinases, consensus-binding motifs often possess modest affinity and selectivity for their respective targets. Additionally, consensus-binding motifs for many kinases are unknown or poorly characterized, which limits the ability of this strategy to be generally applied throughout the kinome.
A truly general approach for generating bivalent kinase inhibitors would require no prior biochemical understanding of the kinase target in order to identify specificity ligands; especially given the sheer number of poorly characterized protein kinases. To this end, new classes of non-immunoglobulin scaffolds have been identified that, when coupled with in vitro display technologies, allow for the rapid identification of potent and selective affinity capture reagents suitable for intracellular studies. In fact, antibody mimetics, based on two different protein scaffolds (monobodies and DARPins), that selectively target several kinases have been identified.14–21 Unfortunately, the utility of antibody mimetics for studying kinase function is often limited because many of these reagents target binding sites that do not overlap, or only partially overlap, with active site features. On the other hand, this trait makes antibody mimetics potentially attractive candidates as second site specificity ligands of SNAPtag-based bivalent inhibitors.
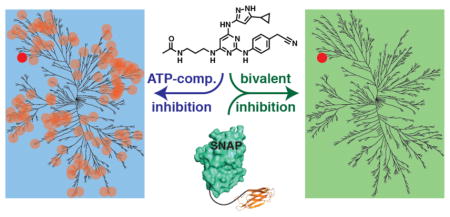
Here, we show that antibody mimetics can be used as highly effective secondary specificity ligands for SNAPtag-based bivalent kinase inhibitors (Figure 1). By linking a promiscuous pan-ATP-competitive inhibitor to a SNAPtag-monobody fusion, a potent bivalent inhibitor of Abl was obtained. Competition-based quantitative chemical proteomics was used to demonstrate that this Abl-directed bivalent inhibitor is selective for BCR-Abl over 205 other endogenously expressed kinases in K562 cell lysate. Importantly, we find that this Abl-selective bivalent inhibitor can be readily assembled in K562 cells, and inhibit the ability of BCR-Abl to phosphorylate a direct cellular substrate. Finally, the generality of using intracellular antibodies as specificity elements was demonstrated by using a SNAPtag-DARPin fusion to generate a bivalent inhibitor that is selective for the activation loop-phosphorylated form of ERK2. The observed selectivity over the non-phosphorylated form of ERK2 also demonstrates the feasibility of using bivalent inhibitors to differentially modulate target subpopulations that differ only in a specific post-translational modification (PTM), which is representative of a specific activation state.
Figure 1.

SNAPtag-based bivalent inhibitors of protein kinases containing a pan-kinase inhibitor tethered to an antibody mimetic. (Top panel) A promiscuous ATP-competitive inhibitor (blue star) blocks the activity of the majority of the kinome (cellular kinase targets are shown as bean-shaped objects and shading represents inhibition of kinase catalytic activity). Non-kinase targets are represented as black shapes. (Middle panel) An intracellular antibody-SNAPtag fusion (SNAPtag is shown in teal and the antibody mimetic is shown in orange) selectively interacts with its kinase target (shown in grey) but does not block catalytic activity. (Bottom panel) A bivalent inhibitor containing a non-selective ATP-competitive inhibitor and an antibody mimetic selectively interacts with its kinase target and blocks catalytic activity.
Results
To generate bivalent inhibitors based on SNAPtag, two components are necessary: (1) an ATP-competitive inhibitor linked to a chemoselective SNAPtag-labeling moiety and (2) a ligand that selectively interacts with unique regions of a kinase of interest. For the ATP-competitive inhibitor, we were particularly interested in a single ligand that could be used to target the largest subset of the kinome possible. This would allow the rapid assembly of potent bivalent inhibitors of diverse kinases without the need to identify a target-specific pharmacophore. Furthermore, using a promiscuous kinase inhibitor would provide a true metric of the degree of selectivity that can be gained with an antibody mimetic-directed bivalent inhibitor. For these reasons, a previously reported 5-cyclopropyl-3-aminopyrazolo-based inhibitor (1) was of particular interest (Figure 2A).22 This pharmacophore contains functional groups that are able to interact with active site features that are conserved in the ATP-binding sites of most kinases.23 Additionally, a co-crystal structure with a quinazoline analog bound to the tyrosine kinase Src indicated a straightforward site of linker attachment that would likely not interfere with interactions between the inhibitor and kinase ATP-binding sites (Figure 2A). Indeed, a derivative of 1 (2), which contains a linker that can be modified with diverse acylating agents, possesses a broad inhibitory profile as assessed with KINOMEscan™ (Supporting Information (SI), Figure S1).
Figure 2.
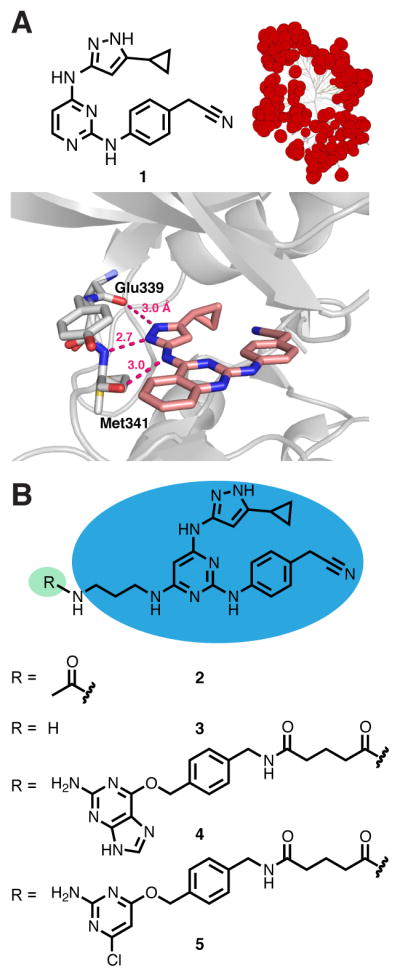
A derivatizable, promiscuous inhibitor of protein kinases. (A) (Top panel, left) A previously reported promiscuous ATP-competitive kinase inhibitor (1). (Top panel, right) The pan-kinase inhibitor scaffold used in the design of the bivalent inhibitors in this study retains a broad spectrum of kinase inhibition as assessed by KINOMEscan (Figure S1). (Bottom panel) A crystal structure of a quinazoline derivative of 1 bound to the kinase Src suggests a site for linker attachment. PDB ID 3F6X. (B) Reagents based on the pan-kinase inhibitor 1.
Due to the linkable and modular nature of 2, a single component kinase affinity matrix was readily generated by reacting an amine-containing version of this scaffold (3) to NHS-activated sepharose. With this tool, the ability of 2 to bind to a variety of full-length protein kinases in cellul lysate was determined (Figure 3A). K562 lysates were pre-incubated with varying concentrations of free 2 or DMSO control, after which, the lysates were incubated with the affinity matrix. The resin was washed, proteins were eluted under denaturing conditions, and the eluent was analyzed using quantitative mass spectrometry. Protein kinases enriched in the DMSO control sample represent those kinases capable of binding to 2 when attached to a solid support matrix. Those kinases exhibiting concentration-dependent competition upon the addition of free 2 are those that exhibit the highest affinity for 2 when in solution (Figure 3B).
Figure 3.

A single component affinity matrix based on pan-kinase inhibitor 2. (A) Schematic of quantitative chemical proteomics workflow. Lysates are pre-treated with free 2 or DMSO before enrichment with 3-conjugated resin. Quantitative mass spectrometry is then employed to determine the affinity of cellular protein kinases for 2. (B) The average half-maximal residual binding (RB50) values of 182 protein kinases from K562 lysate for inhibitor 2 that were determined in two independent quantitative chemical proteomics experiments. Each bar in the graph represents an individual kinase RB50 value and the number of kinases in each group is shown in parentheses below the group name. RB50 values are listed in SI, Table S1.
From this analysis, >260 protein kinases were identified between replicate DMSO control samples, establishing the utility of 2 as a single component kinase profiling tool compound. To our knowledge, this represents the largest swath of the kinome to be profiled by a single compound via chemical proteomics; narrowly edging CTx-0294885.24 For 182 protein kinases, a relative affinity for 2 could be estimated by calculating the concentration at which 50% of a given protein remained bound to the affinity matrix (residual binding (RB50) value). As depicted in Figure 3B, full-length protein kinases from the various branches of the kinome tree exhibit measurable affinity for 2. The number and diversity of kinase active sites targeted by 2 highlights the potential of this single compound to serve as both a versatile building block of bivalent inhibitors as well as a profiling tool for assessing bivalent inhibitor selectivity.
Analogs of 2 that are capable of labeling SNAPtag (4 and 5) were generated by derivatizing the free amine of 3 with either O6-benzylguanine (BG) or 4-(benzyloxy)-6-chloropyrimidin-2-amine (CLP) moieties (Figure 2B). BG-derivatized compound 4 is ideally suited for in vitro labeling because of its ability to provide highly reproducible and nearly quantitative labeling of purified SNAPtag constructs. While CLP derivatives provide incomplete SNAPtag labeling in vitro (SI, Figure S2), we have found that CLP-linked kinase inhibitors are cell permeable and efficiently label SNAPtag in cells,12 which is a requirement for in situ bivalent inhibitor assembly. Therefore, CLP-derivatized analog 5 was also generated. As expected, derivatizing 3 with CLP or BG does not alter the broad kinase profile of the 5-cyclopropyl-3-aminopyrazolo pharmacophore (data not shown).
The first kinase targeted with our bivalent reagents was the tyrosine kinase c-Abl. This multi-domain kinase interacts with cellular substrates through its Src homology 2 (SH2) and Src homology 3 (SH3) domains.25 A monobody-based antibody mimetic, called HA4, that potently binds to the SH2 domains of Abl1 and Abl2 has been identified.15 When HA4 was tested against a microarray containing 84 SH2 domains, only one (the SH2 domain of the tyrosine kinase SYK) demonstrated comparable affinity to that of Abl1 and Abl2, while two others (the tyrosine kinase BLK and non-kinase Grb7) were within 10-fold.15 To test the suitability of HA4 to serve as a component of a bivalent inhibitor, a SNAPtag fusion (SNAP(HA4)) displaying HA4 from the C-terminus through a four amino acid linker (SI, Figure S3) was expressed and purified (Figure 4A). 4-labeled constructs were then assembled, purified, and tested (SI, Figure S4).
Figure 4.

A bivalent inhibitor of Abl. (A) (Top panel) SNAPtag-HA4 fusion that was generated for this study. GT = glycine-threonine. The exact sequence for this construct is shown in SI, Figure S3. (Bottom panel) The binding sites for the two ligands of the bivalent inhibitor are superimposed on a crystal structure of Abl3D, PDB ID 2FO0. (B) In vitro activities of 4, SNAPtag proteins, and assembled SNAPtag-4 conjugates against two different Abl constructs. Abl3D contains the kinase, SH2, and SH3 domains, while AblKD is the kinase domain alone. Values shown are the average of assays performed in triplicate ± SEM.
We next determined the ability of the bivalent inhibitor SNAP(HA4)-4 and its constituent monovalent components to block the activity of a construct (Abl3D) that contains Abl’s catalytic, SH2 and SH3 domains (Figure 4B). Unconjugated 4 is a modest inhibitor of Abl3D (IC50 = 720 nM). This potency is diminished by conjugation to SNAPtag (SNAPtag-4, IC50 = >15,000 nM), presumably due to unfavorable steric interactions. However, the assembled bivalent inhibitor (SNAP(HA4)-4) exhibits an IC50 of 8.3 nM against Abl3D, which is an 87-fold increase in potency relative to unconjugated 4 and a >1,800-fold increase in potency compared to monovalent SNAPtag-4. SNAP(HA4) alone does not demonstrate any activity against Abl3D, suggesting that the observed increase in affinity is due to the conjugation of the pharmacophore to the HA4 monobody. Furthermore, size-exclusion chromatography supports a model wherein the SNAPtag-based bivalent inhibitor forms a 1:1 complex (heterodimer) with Abl3D (SI, Figure S5).
To further confirm that the observed increase in potency of bivalent SNAP(HA4)-4 for Abl is due to its engagement of Abl’s catalytic and SH2 domains, it was tested against an Abl construct (AblKD) that contains only a catalytic domain. While unconjugated 4 demonstrates an almost identical IC50 for AblKD and Abl3D, SNAP(HA4)-4 shows almost no detectable inhibition of AblKD (Figure 4B). Thus, in the absence of an SH2 domain to target, bivalent SNAP(HA4)-4 behaves similarly to monovalent ligands SNAPtag-4 and SNAP(HA4). These results demonstrate that 4’s interaction with Abl’s catalytic domain and HA4’s interaction with Abl’s SH2 domain are together necessary for potent inhibition of Abl.
Our biochemical experiments have demonstrated that conjugation of inhibitor 4 to SNAP(HA4) leads to a dramatic increase in Abl inhibitory potency. However, in order for bivalent inhibitors to be useful in studying kinase function they must be selective for their target over other potential kinase targets. To determine how conjugation of the HA4 monobody to non-selective ATP-competitive inhibitor 4 affects Abl selectivity, we profiled unconjugated inhibitor 2, SNAPtag-4, and bivalent SNAP(HA4)-4 against the endogenously expressed protein kinase targets in K562 cell lysate using the quantitative competition-based chemical proteomics method shown in Figure 3A. With this method, the potency (RB50 values) of this reagent for its target (BCR-Abl) can be assessed in parallel with any potential kinase off-targets. Furthermore, by comparing RB50 values for unconjugated inhibitor 2 and bivalent construct SNAP(HA4)-4, the overall contribution of the HA4 monobody to Abl selectivity can be quantitatively determined.
The binding profile of unconjugated inhibitor 2 demonstrates the challenge in obtaining selectivity for Abl (Abl and BCR-Abl in K562 cells) with a 5-cyclopropyl-3-aminopyrazolo-based pharmacophore (Figure 5A). In total, 61 of the 182 kinases profiled have RB50 values that are equal to or lower than BCR-Abl’s RB50 value for 2 (SI, Table S1). Ten of these kinases have RB50 values that are at least 10-fold lower, with AAK1 possessing a greater than 100-fold lower RB50 value. Conjugation of the 5-cyclopropyl-3-aminopyrazolo pharmacophore to SNAPtag has a generally detrimental effect on inhibitor potency, as most kinases have a higher RB50 value (an average of a 14-fold higher for all kinases in which an RB50 value could be calculated) for SNAPtag-4 relative to inhibitor 2 (SI, Figure S6), which is consistent with the observed loss in potency for SNAPtag-4 versus inhibitor 4 in the in vitro Abl inhibition assay (Figure 4B). However, the overall unfavorable selectivity profile of SNAPtag-4 is comparable to inhibitor 2 for BCR-Abl over other kinases and, thus, reflects a property of the shared ATP-competitive pharmacophore.
Figure 5.

Quantitative competition-based chemical proteomics. (A) Log10[RB50 values] (average of 2 independent experiments) of 182 kinases for unconjugated inhibitor 2. Each kinase is represented as a square. BCR and Abl are shown as red squares. Kinases with RB50 values ≤150 nM are labeled (green squares). Kinases with RB50 values >150 nM are unlabeled and shown in blue. (B) Log10[RB50 values] (average of 2 independent experiments) of 206 kinases for SNAP(HA4)-4. The color coding scheme for each kinase was retained from panel A. (C) Log10[Relative RB50 values] (average of 2 independent experiments) of kinases for SNAP(HA4)-4 compared to 2. The dotted line represents ≥3-fold increase in potency for the assembled bivalent inhibitor. Kinases with ≥3-fold increase in potency for the assembled bivalent inhibitor compared to 2 are labeled (cyan squares). All other kinases are unlabeled and shown in blue. (D) Bar graphs showing selectivity ratios (log10[RB50 for kinase listed/RB50 for BCR-Abl]) for 2 and SNAP(HA4)-4.
Conjugation of 4 to SNAP(HA4) markedly increases the potency and selectivity of BCR-Abl inhibition in K562 cell lysate (Figure 5B). For bivalent inhibitor SNAP(HA4)-4, BCR-Abl has the lowest RB50 value (RB50 = 0.021 μM) of any of the kinases profiled. This dramatic increase in selectivity is due to both reduced potency for most other kinases and an enhanced affinity for BCR-Abl; SNAP(HA4)-4 is 71-fold more potent than unconjugated inhibitor 2 (RB50 = 1.5 μM) and greater than 470-fold more potent than SNAPtag-4 (RB50 >10 μM) against BCR-Abl (Figure 5C). The next lowest RB50 values for SNAP(HA4)-4 (RB50 values = 0.1–0.3 μM) are for kinases that possess an inherently high affinity for ATP-competitive inhibitor 2 (AAK1, BMP2K, GSK3α, and GSK3β). Comparison of relative RB50 values for 2 and SNAP(HA4)-4 show the dramatic increase in BCR-Abl selectivity that the HA4 monobody confers (Figure 5D). For example, 2 is 179-fold selective for AAK1 over BCR-Abl, but SNAP(HA4)-4 is 4-fold selective for BCR-Abl over AAK1, which is a >700-fold increase in selectivity ratio for the desired target (BCR-Abl). Similar increases in selectivity for BCR-Abl over BMP2K, GSK3α, and GSK3β are observed (Figure 5D).
While comparison of absolute RB50 values (Figure 5B) is useful for determining potential cellular off-targets of our bivalent inhibitor, a global comparison of the ratio of RB50 values for SNAP(HA4)-4 versus 2 for individual kinases (Figure 5C) provides information on which kinases are recognized by the monobody component of SNAP(HA4). Beyond BCR-Abl, only three protein kinases have ≥3-fold lower RB50 values for SNAP(HA4)-4 relative to 2 (Figure 5C): Fyn (RB50 = 0.94 μM; 10-fold increase), MARK4 (RB50 = 0.54 μM; 5.0-fold increase), and MAST3 (RB50 = 0.93 μM; 3.5-fold increase). For SYK, which contains an SH2 domain that has an affinity for HA4,15 SNAP(HA4)-4 showed only a 2-fold increase in overall potency relative to 2. BLK, which also contains an SH2 domain recognized by HA4, was not detected in the K562 lysate.
In the original characterization of the HA4 monobody,15 this reagent was shown to enrich FYN from HEK293T and K562 lysates, but based on the lack of observed binding of HA4 to the isolated SH2 domain of FYN, the authors concluded that FYN enrichment was likely due to its interaction with BCR-Abl rather than direct binding to the monobody itself. While well reasoned, we did not want to exclude the possibility that HA4 exhibits appreciable affinity for FYN in the context of the full length kinase; similarly for MAST3 and MARK4, which do not contain SH2 domains. To determine if the enhanced potency observed in the chemical proteomics experiments was the result of direct binding by the bivalent inhibitor or as indirect consequence of binding to Abl, the IC50 values of 4 and SNAP(HA4)-4 for purified MARK4 and FYN were determined (SI, Figure S7). SNAP(HA4)-4 is 6-fold less potent against MARK4 than 4, which is consistent with MARK4 forming a complex with BCR-Abl in the chemical proteomic experiments. However, SNAP(HA4)-4 is an 8-fold more potent inhibitor of FYN than monovalent 4. Therefore, it appears that HA4 recognizes the SH2 domain of FYN in the context of the full-length protein. Finally, the IC50 values of 4 and SNAP(HA4)-4 for purified BLK were also determined and, despite the reported interaction of HA4 with the SH2 domain of BLK, SNAP(HA4)-4 is a >20-fold less potent inhibitor of BLK than 4 (SI, Figure S7).
Competition experiments in K562 lysates verify that our bivalent inhibitor, SNAP(HA4)-4, is selective for BCR-Abl over other kinases present in K562 cells. For this bivalent strategy to be truly useful, it is important that bivalent inhibitors can be readily assembled inside cells and capable of inhibiting the activities of their kinase targets. Prior to performing cellular experiments, we verified that probe 5 is cell permeable and able to label SNAPtag in K562 cells using a fluorescent cellular blocking experiment (SI, Figure S8).
Next, we determined whether the Abl-selective bivalent inhibitor SNAP(HA4)-5 is able to inhibit BCR-Abl activity in K562 cells. To do this, intracellular phospho-STAT5 levels were determined with fluorescence-activated cell sorting. In K562 cells, STAT5 is a direct substrate of constitutively active BCR-Abl, and its level of tyrosine phosphorylation is commonly used as read-out for monitoring BCR-Abl activity.26 K562 cells were transfected with various SNAPtag constructs and then treated with different concentrations of 5 for 1 hour. Flow cytometry was used to sort transfected from non-transfected cells, and phospho-STAT5 levels were determined with a phospho-specific antibody (SI, Figure S9). Gratifyingly, cells expressing SNAP(HA4) demonstrated a concentration-dependent decrease in phospho-STAT5 levels after incubation with 5 for 1 hour (Figure 6A). Importantly, 5 alone is not capable of inhibiting BCR-Abl, as control-transfected cells subjected to the same concentrations of 5 do not show a concentration-dependent decrease in phospho-STAT5 levels (Figure 6B), consistent with phospho-STAT5 inhibition occurring through bivalency. Furthermore, covalent conjugation between 5 and SNAP(HA4) is necessary for robust phospho-STAT5 inhibition as incubating 5 (5 μM) with a catalytically dead SNAPtag-HA4 fusion (cdSNAP(HA4)) fails to reduce phospho-STAT5 levels beyond that of cdSNAP(HA4) alone (Figure 6C). Therefore, assembly of both components into a bivalent inhibitor is necessary for robust BCR-Abl inhibition in K562 cells.
Figure 6.

BCR-Abl activity in K562 cells as assessed by phosphorylated STAT5 levels after 1 hour of treatment with varying concentrations of 5. (A) Flag-SNAP(HA4)-transfected cells were treated with varying concentrations of 5, sorted using a Flag-FITC antibody, and compared to Flag-control-transfected cells treated with DMSO. (B) Flag-control-transfected cells were treated with varying concentrations of 5, sorted using a Flag-FITC antibody, and compared to Flag-control-transfected cells treated with DMSO. (C) An overlay of phospho-STAT5 levels for cells transfected with a catalytically dead SNAPtag mutant, Flag-cdSNAP(HA4) treated with DMSO or 5 μM 5. *K562 cells treated with 5 for short periods of time do not show reduced viability (SI, Figure S10).
With Abl, we have demonstrated that antibody mimetics can impart appreciable degrees of potency and selectivity as secondary specificity ligands of bivalent inhibitors. However, their use as selectivity conferring agents can, in principle, go beyond selectively inhibiting one particular kinase to inhibiting a specific subpopulation of a given kinase. For example, members of the mitogen-activated protein kinase (MAPK) family of protein kinases primarily exist in two different phospho-forms in cells that differ by >50,000-fold in catalytic activity: activation loop unphosphorylated (inactive) or activation loop phosphorylated (active).27 In the presence of diverse stimuli, unphosphorylated MAPKs are activated by the phosphorylation of a threonine and a tyrosine residue on their activation loops, initiating signaling cascades that exert control over numerous signaling networks.28
Recently, DARPin-based antibody mimetics have been reported that selectively recognize activation loop-phosphorylated or unphosphorylated forms of the MAPK extracellular signal-regulated kinase 2 (ERK2).21 For this study, we explored the use of a DARPin (pE59) that has been shown previously to recognize the phosphorylated activation loop of ERK2, thus, potentially affording a bivalent inhibitor that selectively inhibits the activated form of this MAPK. With this in mind, a SNAPtag fusion (SNAP(pE59)) containing pE59 displayed from SNAPtag’s C-terminus through an eight amino acid linker was generated (Figure 7A). Monovalent and bivalent inhibitor complexes containing SNAP(pE59) were assembled, purified, and tested (SI, Figure S4).
Figure 7.

A bivalent inhibitor of ERK2. (A) (Top panel) SNAPtag-DARPin fusion that was generated for this study. The exact sequence for this construct is shown in SI, Figure S11. (Bottom panel) The binding sites for the ligands of the bivalent inhibitor are superimposed on a crystal structure of ERK2, PDB ID 3ZUV. (B) In vitro activities of 4, SNAPtag proteins and assembled SNAPtag-4 conjugates against doubly phosphorylated ERK2. Values shown are the average of assays performed in triplicate ± SEM. ‡ indicates that only partial inhibition of ERK2 was observed (see plot below). (C) In vitro activity of 4 and assembled SNAPtag-4 construct against activated ERK2, JNK2, and p38α. IC50 values are shown as ratios and plotted on a log10-based scale. A blue dashed line denotes an IC50 ratio of one, indicating equipotent inhibition of ERK2 and either JNK2 or p38α. (D) RB50 curves of the phosphorylated and nonphosphorylated ERK activation-loop peptide for SNAP(pE59)-4. Curves are shown in pink for the phosphorylated peptide and blue for the nonphosphorylated peptide.
The ability of SNAP(pE59)-4, and its constituent monovalent components, to block ERK2 function were assessed with an activity assay using an activation loop-phosphorylated form of this kinase (Figure 7B). Similar to Abl, unconjugated 4 is a moderately potent inhibitor (IC50 = 170 nM) of ERK2, with conjugation to SNAPtag leading to reduced potency against ERK2 (IC50 = 3,900 nM). Unlike monobody HA4’s lack of inhibition of in vitro Abl activity, monovalent SNAP(pE59) partially inhibits ERK2 with maximum inhibition plateauing at ~60%. Gratifyingly, the bivalent inhibitor SNAP(pE59)-4 completely suppresses ERK2’s catalytic activity, exhibiting significantly enhanced potency (IC50 = 0.80 nM) compared to either monovalent component.
As our ERK2-directed bivalent inhibitor relies on the use of an antibody mimetic that recognizes phosphorylated activation loops in order to obtain selectivity, we wished to assess its effects amongst kinases that possess a similar activation state-dependent phosphorylation-motif. The closely-related MAPKs c-Jun N-terminal kinase 2 (JNK2) and p38α contain the same diphosphorylation motif (TXY) as ERK2 and are likely candidates for off-target inhibition.29 Therefore, we tested 4, SNAPtag-4, and SNAP(pE59)-4 against phosphorylated JNK2 and p38α (Figure 7C). 4 is essentially an equipotent inhibitor of ERK2 and JNK2, whether as a free BG derivative or conjugated to SNAPtag. This is in striking contrast to the conjugation of 4 to SNAP(pE59), which increases selectivity ~10,000-fold for ERK2 over JNK2. A similar trend is observed for p38α, despite a slight preference of 4 for ERK2 over p38α. Thus, phosphorylation-specific antibody mimetics are able to confer exquisite selectivity in the context of bivalent inhibitors, even amongst closely-related MAPKs.
Assessing the kinome-wide specificity of SNAP(pE59)-4 proved less straightforward than for SNAP(HA4)-4 because we could not identify activated ERK2 in the K562 lysates used in this study; both mass spectrometry- and western blot-based methods failed to detect the presence of phosphorylated ERK2 (data not shown). As MAPKs are typically activated in response to extracellular stimulation, we attribute the lack of activated ERK2 to the basal growth conditions of the K562 cells prior to cell lysis, which did not include any such stimulation. To circumvent this issue, competition-based quantitative chemical proteomics experiments were performed with SNAP(pE59)-4 in K562 lysates spiked with exogenous activation loop-phosphorylated ERK2 (phospho-ERK2). The relative affinities of the bivalent inhibitor for unphosphorylated and phosphorylated ERK2 were monitored by quantification of an activation loop tryptic peptide that contains the TXY motif. SNAP(pE59)-4 shows potent and robust competition of phospho-ERK2 (RB50 = 50 nM) and little affinity for unphosphorylated ERK2 (RB50 = >10,000 nM), resulting in >200-fold selectivity (Figure 7D). While admittedly a controlled system, these results highlight the potential use of antibody-based mimetics as secondary specificity ligands towards obtaining selective inhibition of PTM-specific kinase states.
Discussion
Starting from a single pan-kinase inhibitor, bivalent inhibitors for either BCR-Abl or phospho-ERK2 were obtained with dramatically increased potencies for their targets (>80-fold and >200-fold, respectively) and overall enhanced selectivity. The enhanced selectivities and affinities observed for both bivalent inhibitors in vitro were confirmed in complex cell lysates with competition-based quantitative chemical proteomics. For the BCR-Abl-directed bivalent inhibitor, the most potently competed kinase in our quantitative proteomic experiment was the intended target despite the relatively unfavorable selectivity profile of the monovalent ATP-binding site ligand. In targeting ERK2, a bivalent inhibitor that is selective for activation loop-phosphorylated ERK2 over its non-phosphorylated form was obtained. The ability to develop bivalent reagents that selectively inhibit specific post-translationally modified forms of kinases holds promise for studying signal transduction, where different subpopulations of a kinase may play distinct roles in the cell (for ERK2, only the activation loop phosphorylated form is believed to mediate downstream signaling). For example, there are 22 validated sites of phosphorylation on AKT1 alone.30 Upon treatment with stimulatory conditions, such as insulin, the levels and patterns of phosphorylation change markedly. With the proper antibody mimetic, it may be possible to survey the molecular function of a precise subset of a protein population using the bivalent inhibitor strategy described here.
Similar to previous efforts,31–34 we have converted a highly promiscuous ATP-competitive inhibitor into a selective bivalent inhibitor. The key to obtaining selectivity with our methodology was the use of intracellular antibodies, specifically DARPins and monobodies, as second site specificity ligands. While there are only a limited number of selective DARPins and monobodies currently available for use as components of bivalent kinase inhibitors, continuous advances in high-throughput selection strategies should greatly expand the reserve of suitable reagents. Furthermore, the fact that two different non-immunoglobin scaffolds, with very different topologies, could successfully be used as SNAPtag fusions suggests that it should be straightforward to rapidly generate bivalent inhibitors with these reagents once they become available.
An important step in the evaluation of the SNAPtag-based bivalent inhibitor strategy was the discovery and characterization of the general kinase ligand 2. The enabling feature of 2 is its very broad kinome coverage, which allows it to serve as a potential starting point in the development of next-generation bivalent inhibitors. We envision that 4 and 5 could be thought of as off-the-shelf building blocks in the continued development of SNAPtag-based bivalent inhibitors; although, if one wanted to achieve the goal of mono-specific kinase inhibition, a more selective starting point would likely be needed. The ability to access a single compound that can be efficiently transformed into a selective inhibitor for kinases irrespective of their degree of homology enables a significantly simplified strategy. For example, BCR-Abl and ERK2 differ significantly in their respective ATP-binding sites, and compounds do not commonly engage both kinases.35 Because of the versatility of 2, we were able to use this single reagent to generate potent and selective bivalent inhibitors for either BCR-Abl or ERK2. Additionally, the promiscuity of 2 was leveraged to create a general selectivity-profiling tool for hundreds of endogenously expressed kinases. While many different scaffolds already exist for profiling the kinome via chemical proteomics,36–40 a single compound covering a large swath of the kinome offers some technical advantages. These advantages include: generally reduced synthetic burden/cost as well as the inherent simplicity of making one affinity matrix versus multiple affinity matrices and combining the various matrices into a cocktail for profiling. Furthermore, due to the conservation of general pharmacological characteristics of kinase ATP-binding sites across organisms, a 2-based affinity matrix should be of utility for profiling any lysate.41
Perhaps the most critical step towards the application of SNAPtag-based bivalent inhibitors is the demonstration that these reagents can be used to perturb an intra-cellular signaling pathway. Treating K562 cells expressing SNAP(HA4) with 5 led to a significant and concentration-dependent decrease in phospho-STAT5 levels. These results demonstrate that SNAPtag-based bivalent inhibitors can assemble in cells and engage the intended target protein to modulate signaling pathways. An inherent challenge in using inhibitors that target multiple sites of a protein of interest is that these reagents tend to be large and, therefore, lack cell permeability. While there has been some recent success in targeting intra-cellular kinases with bivalent inhibitors by appending cell penetrating peptides,42,43 our approach is fundamentally different in that it relies on the intracellular assembly of the bivalent reagent. Since one component is expressed in the cell line of interest, the size of the small molecule portion of the bivalent inhibitor can be greatly reduced, which increases the likelihood of cell permeability.
While the focus of this manuscript has been on kinases, the approach described can be applied to other enzyme and protein families. As mentioned previously, the availability of small molecules that broadly target specific enzyme families is advantageous, and high quality probes have already been developed for PARPs, HDACs, and Bromodomain-containing proteins.44–46 Additionally, the increasing number of available non-immunoglobulin protein scaffolds, including DARPins and monobodies, will greatly facilitate the tailoring of specific and selective bivalent inhibitors to dissect complex biological phenotypes.
The resurgence of phenotypic-based screening has brought with it an increased interest in targeted and de novo target deconvolution technologies. However, the stark reality is that it is not uncommon for such experiments to identify multiple proteins capable of binding a compound of interest; any one of which could be the efficacy target, assuming that only one protein is responsible for the observed phenotype. This can, in part, be rationalized by the fact that while compounds coming from phenotypic screens can be optimized for potency in cellular assays, it is not until a comprehensive binding profile is obtained that the full spectrum of potential on- and off-targets can be appreciated. This is particularly true for ATP-competitive kinase inhibitors. In some instances a firm understanding of the biological pathway can guide and (de)prioritize proteins for follow-up efforts. However, this can be a misleading approach, especially if the pharmacology of a compound is ahead of the current biological understanding of a pathway. In these instances, it would be ideal to have a modular strategy for enhancing the potency and selectivity of a compound for each of the possible hypothesized targets, allowing one to determine to what extent, if any, each protein contributes to the overall phenotype. To this end, the design and application of bivalent inhibitors is an attractive approach for enhancing the potency and selectivity for a specific target. We believe that the advances presented in this work will help enable the field to develop increasingly potent and selective tools for tackling the key challenge of target validation in drug discovery.
Materials and Methods
Quantitative chemical proteomic methods and data are described in detail in Supporting Information. Chemical synthesis, compound characterization, protein expression and purification, bivalent inhibitor assembly, in vitro activity assay conditions, and cellular assay methods are also described in Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health grants R01GM086858 (D.J.M.) and R21CA177402 (D.J.M.); and the Camille and Henry Dreyfus Foundation (D.J.M.).
Footnotes
Conflict of interest statement: Jason Thomas, Edmund Harrington, Jason Murphy, Ivan Cornella-Taracido, Rishi Jain and Markus Schirle are employees of Novartis Institutes for Biomedical Research.
Supporting Information: Protocols for chemical synthesis and characterization, protein production, kinase activity assays, cellular assay methods, and quantitative chemical proteomic methods, as well as supporting figures and tables
References
- 1.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 2.Weiss WA, Taylor SS, Shokat KM. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P. Protein kinases--the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 4.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 5.Profit AA, Lee TR, Lawrence DS. Bivalent Inhibitors of Protein Tyrosine Kinases. J Am Chem Soc. 1999;121:280–283. [Google Scholar]
- 6.Gower CM, Chang MEK, Maly DJ. Bivalent inhibitors of protein kinases. Critical Reviews in Biochemistry and Molecular Biology. 2014;49:102–115. doi: 10.3109/10409238.2013.875513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gronemeyer T, Chidley C, Juillerat A, Heinis C, Johnsson K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for applications in protein labeling. Protein engineering, design & selection: PEDS. 2006;19:309–316. doi: 10.1093/protein/gzl014. [DOI] [PubMed] [Google Scholar]
- 8.Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem Biol. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 9.Juillerat A, Heinis C, Sielaff I, Barnikow J, Jaccard H, Kunz B, Terskikh A, Johnsson K. Engineering substrate specificity of O6-alkylguanine-DNA alkyltransferase for specific protein labeling in living cells. ChemBioChem. 2005;6:1263–1269. doi: 10.1002/cbic.200400431. [DOI] [PubMed] [Google Scholar]
- 10.Hill ZB, Perera BGK, Maly DJ. A Chemical Genetic Method for Generating Bivalent Inhibitors of Protein Kinases. J Am Chem Soc. 2009;131:6686–6688. doi: 10.1021/ja900871y. [DOI] [PubMed] [Google Scholar]
- 11.Hill ZB, Perera BGK, Maly DJ. Bivalent inhibitors of the tyrosine kinases ABL and SRC: determinants of potency and selectivity. Mol BioSyst. 2011;7:447. doi: 10.1039/c0mb00108b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill ZB, Perera BGK, Andrews SS, Maly DJ. Targeting Diverse Signaling Interaction Sites Allows the Rapid Generation of Bivalent Kinase Inhibitors. ACS Chem Biol. 2012;7:487–495. doi: 10.1021/cb200387g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregersen KAD, Hill ZB, Gadd JC, Fujimoto BS, Maly DJ, Chiu DT. Intracellular Delivery of Bioactive Molecules using Light-Addressable Nanocapsules. ACS Nano. 2010;4:7603–7611. doi: 10.1021/nn102345f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karatan E, Merguerian M, Han Z, Scholle MD, Koide S, Kay BK. Molecular recognition properties of FN3 monobodies that bind the Src SH3 domain. Chem Biol. 2004;11:835–844. doi: 10.1016/j.chembiol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nature Structural & Molecular Biology. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gulyani A, Vitriol E, Allen R, Wu J, Gremyachinskiy D, Lewis S, Dewar B, Graves LM, Kay BK, Kuhlman B, Elston T, Hahn KM. A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nat Chem Biol. 2011;7:437–444. doi: 10.1038/nchembio.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang R, Fang P, Kay BK. Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase. New Biotechnology. 2012;29:526–533. doi: 10.1016/j.nbt.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amstutz P, Koch H, Binz HK, Deuber SA, Plückthun A. Rapid selection of specific MAP kinase-binders from designed ankyrin repeat protein libraries. Protein engineering, design & selection: PEDS. 2006;19:219–229. doi: 10.1093/protein/gzl004. [DOI] [PubMed] [Google Scholar]
- 19.Bandeiras TM, Hillig RC, Matias PM, Eberspaecher U, Fanghänel J, Thomaz M, Miranda S, Crusius K, Pütter V, Amstutz P, Gulotti-Georgieva M, Binz HK, Holz C, Schmitz AAP, Lang C, Donner P, Egner U, Carrondo MA, Müller-Tiemann B. Structure of wild-type Plk-1 kinase domain in complex with a selective DARPin. Acta crystallographica Section D, Biological crystallography. 2008;64:339–353. doi: 10.1107/S0907444907068217. [DOI] [PubMed] [Google Scholar]
- 20.Kummer L, Hsu C-W, Dagliyan O, MacNevin C, Kaufholz M, Zimmermann B, Dokholyan NV, Hahn KM, Plückthun A. Knowledge-based design of a biosensor to quantify localized ERK activation in living cells. Chem Biol. 2013;20:847–856. doi: 10.1016/j.chembiol.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PRE, Kaufholz M, Zimmermann B, Herberg FW, Plückthun A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proceedings of the National Academy of Sciences. 2012;109:E2248–57. doi: 10.1073/pnas.1205399109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Statsuk AV, Maly DJ, Seeliger MA, Fabian MA, Biggs WH, Lockhart DJ, Zarrinkar PP, Kuriyan J, Shokat KM. Tuning a Three-Component Reaction For Trapping Kinase Substrate Complexes. J Am Chem Soc. 2008;130:17568–17574. doi: 10.1021/ja807066f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aronov AM, Murcko MA. Toward a pharmacophore for kinase frequent hitters. J Med Chem. 2004;47:5616–5619. doi: 10.1021/jm049793g. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Holmes IP, Hochgräfe F, Walker SR, Ali NA, Humphrey ES, Wu J, de Silva M, Kersten WJA, Connor T, Falk H, Allan L, Street IP, Bentley JD, Pilling PA, Monahan BJ, Peat TS, Daly RJ. Characterization of the novel broad-spectrum kinase inhibitor CTx-0294885 as an affinity reagent for mass spectrometry-based kinome profiling. J Proteome Res. 2013;12:3104–3116. doi: 10.1021/pr3008495. [DOI] [PubMed] [Google Scholar]
- 25.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 26.Goss, et al. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood. 2006;107:4888. doi: 10.1182/blood-2005-08-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prowse C, Hagopian J, Cobb M, Ahn N, Lew J. Catalytic reaction pathway for the mitogen-activated protein kinase ERK2. Biochemistry. 2000;39:14002. doi: 10.1021/bi005116m. [DOI] [PubMed] [Google Scholar]
- 28.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions 1. Endocrine Reviews. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 30.Guo H, Gao M, Lu Y, Liang J, Lorenzi PL, Bai S, Hawke DH, Li J, Dogruluk T, Scott KL, Jonasch E, Mills GB, Ding Z. Coordinate phosphorylation of multiple residues on single AKT1 and AKT2 molecules. Oncogene. 2014;33:3463–3472. doi: 10.1038/onc.2013.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA. Mechanism-based design of a protein kinase inhibitor. Nat Struct Biol. 2001;8:37–41. doi: 10.1038/83028. [DOI] [PubMed] [Google Scholar]
- 32.Meyer SC, Shomin CD, Gaj T, Ghosh I. Tethering Small Molecules to a Phage Display Library: Discovery of a Selective Bivalent Inhibitor of Protein Kinase A. J Am Chem Soc. 2007;129:13812–13813. doi: 10.1021/ja076197d. [DOI] [PubMed] [Google Scholar]
- 33.Shomin CD, Meyer SC, Ghosh I. Staurosporine tethered peptide ligands that target cAMP-dependent protein kinase (PKA): Optimization and selectivity profiling. Bioorganic & Medicinal Chemistry. 2009;17:6196–6202. doi: 10.1016/j.bmc.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider TL, Mathew RS, Rice KP, Tamaki K, Wood JL, Schepartz A. Increasing the Kinase Specificity of K252a by Protein Surface Recognition. Org Lett. 2005;7:1695–1698. doi: 10.1021/ol050179o. [DOI] [PubMed] [Google Scholar]
- 35.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 36.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 37.Oppermann FS, Gnad F, Olsen JV, Hornberger R, Greff Z, Kéri G, Mann M, Daub H. Large-scale proteomics analysis of the human kinome. Mol Cell Proteomics. 2009;8:1751–1764. doi: 10.1074/mcp.M800588-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ku X, Heinzlmeir S, Helm D, Médard G, Kuster B. New affinity probe targeting VEGF receptors for kinase inhibitor selectivity profiling by chemical proteomics. J Proteome Res. 2014;13:2445–2452. doi: 10.1021/pr401247t. [DOI] [PubMed] [Google Scholar]
- 39.Schirle M, Petrella EC, Brittain SM, Schwalb D, Harrington E, Cornella-Taracido I, Tallarico JA. Kinase inhibitor profiling using chemoproteomics. Methods Mol Biol. 2012;795:161–177. doi: 10.1007/978-1-61779-337-0_11. [DOI] [PubMed] [Google Scholar]
- 40.Ku X, Heinzlmeir S, Liu X, Médard G, Kuster B. A new chemical probe for quantitative proteomic profiling of fibroblast growth factor receptor and its inhibitors. J Proteomics. 2014;96:44–55. doi: 10.1016/j.jprot.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 41.Urbaniak MD, Mathieson T, Bantscheff M, Eberhard D, Grimaldi R, Miranda-Saavedra D, Wyatt P, Ferguson MAJ, Frearson J, Drewes G. Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem Biol. 2012;7:1858–1865. doi: 10.1021/cb300326z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandvold KR, Santos SM, Breen ME, Lachacz EJ, Steffey ME, Soellner MB. Exquisitely specific bisubstrate inhibitors of c-Src kinase. ACS Chem Biol. 2015;10:1387–1391. doi: 10.1021/cb501048b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stebbins JL, De SK, Pavlickova P, Chen V, Machleidt T, Chen L-H, Kuntzen C, Kitada S, Karin M, Pellecchia M. Design and Characterization of a Potent and Selective Dual ATP- and Substrate-Competitive Subnanomolar Bidentate c-Jun N-Terminal Kinase (JNK) Inhibitor. J Med Chem. 2011;54:6206–6214. doi: 10.1021/jm200479c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell A-G, Pol E, Frostell A, Ekblad T, Oncu D, Kull B, Robertson GM, Pellicciari R, Schuler H, Weigelt J. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 30:283–288. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 45.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon A-M, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 46.http://www.thesgc.org/chemical-probes/bromosporine.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.