

Hypertension is the leading cause of stroke and cardiovascular diseases and a leading risk factor for global disease burden, affecting 30% of the adult population in Western cultures.1 Blood pressure (BP) can be elevated by vasoconstriction and/or by increasing the circulating volume. Evidence from studies of mutations in renal Na+ transporters, renal transplantation, and diuretic action all support Guyton’s hypothesis that long term regulation of effective circulating volume and blood pressure depend on fractional renal Na+ reabsorption.2–4 Ultimately, excess Na+ reabsorption (Fig 1, red arrows) raises effective circulating volume and BP which provoke counteracting natriuretic responses to match Na+ output to Na+ intake at the expense of elevated BP (Fig 1, blue arrow).2 We and others have determined that “pressure natriuresis” responses involve Na+ transporter inhibition at multiple levels of regulation.5–13 According to Guyton, kidneys possess the capacity to excrete enough Na+ and volume to normalize blood pressure in the face of expanded effective circulating volume.2 Thus, hypertension can be characterized as a failure of compensatory renal pressure natriuresis. Indeed, there is strong evidence that the pressure natriuresis response is impaired during experimental hypertension by inflammation, immune cell infiltration, and intrarenal AngII production, secondary to initiating stimuli such as AngII infusion, reduced nitric oxide (NO) production, high salt diet, or elevated renal sympathetic nerve activity (RSNA).14–17 This brief review focuses on the natriuretic effectors and addresses: 1) the renal tubular locations and transporters that participate in pressure natriuresis, and 2) the mechanisms that blunt the response in experimental models of hypertension.
Figure 1. Renal transporter profile reveals region specific transporter regulation during experimental hypertension.
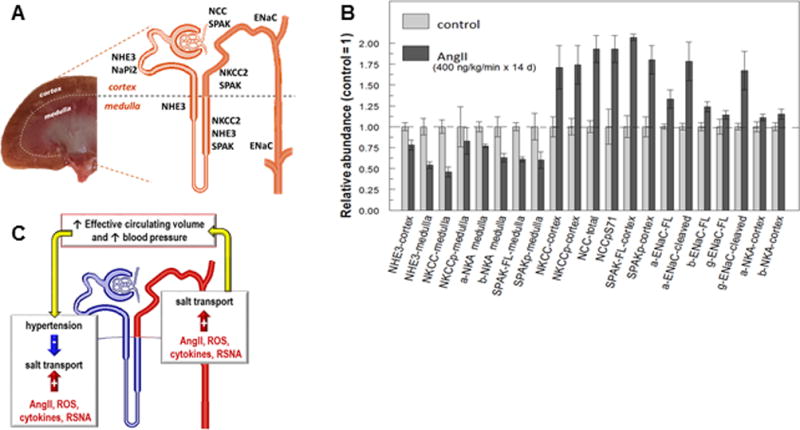
A. Anatomical arrangement of renal cortex and renal medulla in a kidney cross section with adjacent drawing of a single nephron indicating locations of sodium transporters, ENaC and SPAK. Cortical NHE3 and NaPi2 are primarily restricted to the proximal tubules where 2/3 of the filtered load is reabsorbed. Medullary NHE3 is expressed in the S3 portion of the proximal tubule that terminates in the medulla as well as in the thick ascending limb of the loop of Henle. NKCC2 (target of loop diuretics) is expressed in both medulla and cortex all along the thick ascending limb. NCC (target of thiazide diuretics) is localized to the cortical distal convoluted tubule and SPAK (kinase that activates NKCC2 and NCC) is expressed in both cortex and medulla from thick ascending limb through distal convoluted tubules. Epithelial sodium channel (ENaC, target of potassium sparing diuretics) alpha, beta and gamma subunits are expressed in the cortex from late distal convoluted tubule through to principal cells of the cortical collecting duct, as well as in the medullary collecting ducts. B. Effects of AngII infusion into rats (400 ng/kg/min for 14 days) replotted from Nguyen et. al.,28 expressed as protein abundance relative to mean abundance in control untreated rats, defined as 1.0. Definitions: FL = full length form, p = phosphorylated form, cleaved = ENaC subunits cleaved to smaller molecular weight forms associated with channel activation. Summary: Distal nephron NKCC, NCC, SPAK and their phosphorylated forms all increase significantly (NCCpT53 and NCCpS89 also significantly increase 5 and 3 fold, respectively); cortical alpha (a-ENaC) and beta (b-ENaC) as well as cleaved a-ENaC and g-ENaC subunits significantly increase. In contrast, cortical NHE3 and medullary thick limb NHE3, NKCC, NKCCp, sodium pump alpha and beta subunits (a-NKA, b-NKA), SPAK and SPAKp are all significantly depressed in abundance during AngII hypertension. C. Integration of responses to maintain fluid balance: antinatriuretic stimuli including AngII, reactive oxygen species (ROS), cytokines and renal sympathetic nervous system activity (RSNA) can stimulate salt reabsorption (red arrows) increasing effective circulating volume and blood pressure which can suppress salt transporters (blue arrow).
Effectors of Pressure Natriuresis
Acute Pressure Natriuresis describes the responses to acute or chronic increases in blood pressure. Chou and Marsh, using video microscopy, provided compelling evidence that the proximal tubule was a site of pressure natriuresis: raising BP by acutely constricting the vasculature18 rapidly increased the flow leaving the proximal tubule by 50%.19 They concluded that the depressed proximal tubule reabsorption increased fluid load signal at the macula densa, contributing to the GFR and RBF autoregulation evident during increased renal perfusion pressure, and that the measured increased distal delivery of fluid and salt could account for the magnitude of the pressure natriuresis and diuresis.19 Proximal tubule reabsorbs two-thirds of the filtered Na+ and volume at baseline and this fraction is decreased not only when BP is increased, but also during a high-salt diet (facilitated by local dopamine production), and when the renin angiotensin system (RAS) is inhibited. Working with the Marsh group, we discovered that these variables all regulate the distribution of the proximal tubule sodium-hydrogen exchanger isoform 3 (NHE3) and the sodium-phosphate cotransporter isoform 2 (NaPi2) between the top and the base of the apical microvilli of the proximal tubule.20–22 Natriuretic stimuli provoke the dynamic redistribution of these transporters, along with associated regulators, molecular motors (myosin VI, IIA), and cytoskeleton-associated proteins, to the base of the microvilli.7 During acute hypertension, the lipid raft-associated NHE3 remains at the base, and the non-raft-associated NaPi2 is endocytosed, culminating in decreased Na+ transport activity measured as increased proximal tubule flow rate. Further along the nephron, an analysis of the response of the distal convoluted tubule to acute hypertension revealed that the sodium chloride co-transporter (NCC) retracted from apical membranes to sub apical cytoplasmic vesicles, providing evidence for its participation of NCC in pressure natriuresis. Importantly, the NCC redistribution was driven by the fall in AngII that accompanies acute hypertension rather than hypertension per se.10
In contrast to pressure natriuresis, anti-natriuretic stimuli (AngII, RSNA, and low-salt diet) redistribute the same proximal tubule transporters into the body of the microvilli associated with an increase in transport activity measured as decreased proximal tubule flow rate.23, 24 Along the distal nephron, acute AngII stimulation provokes redistribution of NCC into the apical plasma membrane. While abundance of the phosphorylated NCC (NCC-P) was not increased by acute AngII treatment, this NCC-P clustered within the apical membrane, as multimeric complexes, creating regions with elevated NCC-P to NCC total ratio (assessed by subcellular fractionation and blue native gels).9, 25 As reviewed below, anti-natriuretic stimuli are increase and oppose pressure natriuresis during chronic hypertension, necessitating a further increase in blood pressure, the error signal driving the response, in order to recruit additional anti-natriuretic mechanisms to balance Na+ output to Na+ intake.
Chronic Angiotensin II hypertension, one of the well-studied model of experimental hypertension, involves the controlled infusion of a subpressor dose of AngII over a couple of weeks.4, 15, 26 Since transporter regulation in one region of the nephron can drive transporter regulation in other regions, we analyzed renal transporter regulation to the entire nephron, building on the “transporter profiling” approach developed by the Knepper group.27 This approach, implemented in homogenates of renal cortex and medulla, is facilitated by the anatomical arrangement of sodium transporters and channels along the nephron (Figure 1A) and the availability of specific antibodies: renal cortical NHE3 and NaPi2 are localized to the proximal tubule, medullary NHE3 and sodium-potassium-2 chloride cotransporter (NKCC2) to the medullary thick ascending limb, cortical NKCC2 to the cortical thick ascending limb, cortical NCC to the distal convoluted tubule, and cortical epithelial sodium channel (ENaC) subunits to the cortical collecting duct.
We tested, in male rats, the hypothesis that AngII infusion (400 ng/kg/min for 14 days) would activate Na+ transporters in the distal nephron, which would drives compensatory inhibition of proximal tubule transporters in order to maintain Na+ and volume homeostasis.28 Cardiac hypertrophy and increased urinary sodium excretion were consistent with a pressure-natriuresis response. The results, summarized in Figure 1B, demonstrate that AngII infusion increased the abundance and activating phosphorylation of cortical transporters including NKCC2, NCC and the regulatory kinase Ste20/SPS-1 related proline-alanine rich kinase (SPAK). Likewise, activation of the cortical collecting duct ENaC was evident increased cleavage29 of alpha and gamma subunits, as well as increased subunit abundance.
In the proximal nephron during chronic AngII hypertension, NHE3 localized to the body of the apical microvilli consistent with acute AngII stimulation,24 and NHE3 abundance decreased 25%, a substantial decrease considering the proximal tubule usually reabsorbs two-thirds of the glomerular filtrate. Apical NHE3 of the medullary thick ascending limb decreased 50%, suggesting the medullary thick ascending limb as another locus of pressure natriuresis. Consistent with this idea, medullary NKCC2 and SPAK abundance were also depressed 40–50%, in contrast to their activation in the cortex during AngII hypertension. Medullary sodium pump Na,K-ATPase (NKA) subunits, driving sodium reabsorption, were likewise decreased significantly during AngII infusion. The concerted decreases in abundance of NHE3, NKCC2 and NKA (Figure 1B) support the conclusion that the thick ascending limb participates in pressure natriuresis along with the proximal tubule (Fig 1C blue nephron region).28 This conclusion was already evident from the Cowley lab findings demonstrating a key role of medullary thick ascending limb in driving normal pressure natriuresis, specifically, the elevated medullary blood flow driven by renal interstitial hydrostatic pressure, as well as the medullary redox state influenced by NO production, two responses blunted in chronic hypertension.30
These results revealed that AngII hypertension increases transporters’ abundance and activation from the cortical thick limb to the medullary collecting duct (NKCC2, NCC, ENaC and regulatory kinase SPAK) and that this stimulation is balanced by a compensatory inhibition of transporters from proximal tubule through medullary thick limb (cortical NHE3 and medullary: NHE3, NKCC2, NKA, SPAK), presumably driven by elevated BP (Fig 1B,1C). Can proximal suppression be attributed to the inhibitory actions of AngII reported at concentrations above 10−7M?31 Direct measures of proximal tubule fluid [AngII] indicated concentrations 10 fold higher than in plasma at baseline, and further elevated by AngII infusion due to local AngII production, yet, concentrations were in the nanomolar range, 100 times less than the doses reported to inhibit proximal tubular reabsorption.32 The authors concluded that the elevated proximal tubule [AngII] impairs pressure natriuresis.32
This analysis of sodium transporter regulation along the nephron during AngII hypertension in rat defined how the effective circulating volume is maintained during the opposing forces of AngII and hypertension and revealed region and context specific regulation of NKCC2 and SPAK in medulla vs. cortex.
L-NAME hypertension
Nitric oxide (NO) is a vasodilator and natriuretic that inhibit NHE3, NKCC, and ENaC in vitro. L-NAME (Nω-L-arginine methyl ester) inhibits nitric oxide synthase (NOS), lowers NO levels and raises blood pressure.33 With Gonzalez-Villalobos and colleagues, we examined the transporter profile in kidneys from mice with L-NAME hypertension.16 In addition to low NO, this model exhibits low circulating AngII, elevated intrarenal AngII production and vasoconstriction. Thus, one might expect renal transporters stimulation in the absence of the NO, however, no distal transporter activation was evident. Rather, NHE3, NKCC2, ENaC subunits and the regulatory kinase SPAK were all suppressed; NCC was unaltered.34 L-NAME was previously reported to depress these sodium transporters in a model incorporating aldosterone plus high salt diet.35 These decreases theoretically facilitate sodium and volume excretion during persistent vasoconstriction and illustrate that the pressure natriuresis mediators can extend to the collecting duct if needed to match sodium excretion to sodium intake.
Genetic Models with Resistance to AngII Hypertension
Many genetic mouse models exhibit blunted hypertensive responses to AngII infusion compared to the response in wild type mice. We reasoned that a transporter profile approach could be applied to determine whether the blunting was due to less transporter activation by AngII or due to more effective pressure natriuresis. The results of three genetic models will be discussed. One caveat is that these studies were carried out in whole kidneys before we characterized differential regulation along the thick limb of the loop of Henle in the rat model.
Proximal tubule specific knockout of the angiotensin type 1 receptor (AT1R)
Gurley and Coffman created a mouse that did not express the AT1R in the proximal tubule (AT1R PTKO) and found that, in response to 2 weeks AngII infusion, blood pressure increased 10 mmHg less than wild type controls.36 While NHE3 and NKCC2 were depressed 25 and 30%, respectively, in AngII infused wild type mice, NHE3 was reduced further, to 50% of baseline, and proximal NaPi2 was reduced 40% in AngII infused AT1R PTKO. Supporting improved pressure-natriuresis in the AT1R PTKO, Schnermann demonstrated by micropuncture that proximal tubule fractional absorption was reduced from 44% to 36%.36 AngII activation of proximal tubule AT1R is also implicated in the stimulation of local synthesis and accumulation of AngII,37 a pathway that is likely suppressed in this model.
Mice with no kidney ACE
Gonzalez-Villalobos and Bernstein analyzed the responses to AngII hypertension in mice that were engineered to express angiotensin converting enzyme (ACE) only in myelomonocytic cells.38 These mice (termed ACE 10/10) have normal BP and negligible amounts of intrarenal ACE, thus, cannot produce intrarenal ACE during experimental hypertension. During chronic AngII infusion, BP rose 20 mmHg less in ACE 10/10 than wild type mice indicating that intrarenal ACE contributes to the hypertension.39 Transporter profiling of wild type and ACE 10/10 mice infused with AngII, revealed that the activation of distal transporters and regulatory kinases (including NKCC2, NCC, pendrin and SPAK) evident in the wild type, was effectively prevented in the ACE 10/10 mice supporting the idea that this activation was dependent on intrarenal AngII production. The physiologic significance of the transporter changes during AngII were demonstrated using diuretic tests that showed increased thiazide and furosemide sensitive Na+ excretion in wild type but not in ACE 10/10.39
Interestingly, ENaC subunits’ activation by cleavage was similar in both wild type and ACE 10/10 during AngII infusion, suggesting that the remaining BP elevation is due to ENaC persistently activated by the infused AngII or aldosterone stimulation.39 Unlike the results in the rat and PTAT1R KO studies,28, 36 NHE3 abundance did not significantly decrease in wild type or ACE10/10 mice in response to AngII in this study. Perhaps this is due to the lower amount of AngII infused and lower BP attained in the Gonzalez et al study (400 ng/kg/min and 150 mmHg)39 compared to the Gurley et al study (1000 ng/kg/min and 170 mmHg).36 Regardless, the NHE3 abundance was not increased by AngII infusion. The dependence of NHE3 abundance regulation on the degree of hypertension versus the amount of AngII remains to be explored. The effects of chronic L-NAME treatment were also examined in the ACE 10/10 mice.34 While BP increased to near 140 mmHg in the wild type mice treated with L-NAME, ACE 10/10 mice exhibited a strong natriuretic response to the NOS inhibitor and were completely protected from L-NAME hypertension.34 L-NAME provoked larger decreases in transporters’ abundance and phosphorylation all along the nephron in the ACE 10/10 (≥50% decreases in NHE3, phosphorylated NKCC, phosphorylated NCC and SPAK) compared to those observed in the wild types. Thus, the full potential of the pressure natriuresis response in L-NAME hypertension becomes evident in the absence of local AngII production. In both AngII and L-NAME hypertension, local production of AngII activates sodium transporters which “puts the brakes” on pressure natriuretic adjustments, thus, blood pressure must increase further to activate natriuretic mediators that suppress transporter activity.16, 34
Mice lacking cytokine production
Harrison and colleagues demonstrated that mice lacking T-lymphocytes present blunted hypertensive responses to experimental hypertension, restored by adoptive transfer of T-cells.40, 41 The same group recently provided evidence that a population of CD8+ T cells infiltrate the kidneys during AngII infusion and produce the cytokines interferon gamma (IFN-γ) and interleukin 17 (IL-17).41 In collaboration with the Harrison group, we investigated the roles of these specific cytokines by profiling transporters in interferon gamma knockout (IFN-γ−/−) and interleukin 17A knockout mice after 2 week Ang II (490 ng/kg/min).42 Systolic pressure increased more than 40 mmHg in wild type mice and less than 20 mmHg in IFN-γ−/− and IL-17A−/− mice. Additionally, natriuretic responses to a saline volume expansion were suppressed in wild types, but not in IL-17A−/− and IFN-γ−/− during AngII infusion. Despite similar blunting of hypertension in both IL-17A−/− and IFN-γ−/−, the transporter profiles during AngII infusion were quite distinct. Activation (increased phosphorylation) of distal NKCC2, NCC and SPAK, evident in the wild type mice, was preserved in IL-17A−/− but prevented in the IFN-γ−/− genotype; ENaC activation persisted in all three genotypes. In both IL-17A−/− and IFN-γ−/−, proximal NHE3, NaPi2, and the motor myosin VI were significantly depressed during Ang II infusion (25 – 50%). Taken together, the similar blunting of hypertension in the two distinct cytokine knockout genotypes can be attributed to the suppression of proximal NHE3, NaPi2 and myosin VI evident in both genotypes, and the role of the distal activation of NKCC, NCC and SPAK during AngII infusion, evident in IL-17A−/− but not IFN-γ−/− remains an open issue. As in the ACE 10/10 genotype, the remaining hypertension in the IL-17A−/− and IFN-γ−/− may be attributed to the persistent ENaC activation.
In conclusion, transporter profiling can pinpoint where transporters and channels are activated and suppressed along the nephron during hypertension. While not the focus of this review, distal anion transporters and potassium channels can also impact the blood pressure set point and warrant inclusion in future comprehensive profiling.43, 44 The profiles of the four genotypes illustrate improved pressure natriuresis potential during experimental hypertension, and shed some light on how this potential is blunted in wild type mice, i.e., infused and locally produced AngII and cytokines “put the brakes” on natriuresis in response to elevated effective circulating volume and hypertension (Fig.1C). Eliminating proximal tubule AT1R, intrarenal ACE, or whole body IFN-γ or IL-17A releases the brakes, at least in part, and improves natriuretic potential. Understanding the molecular mechanisms connecting these anti-natriuretic mediators to the transporter activation has the potential to lead to strategies to amplifying the pressure natriuretic responses of the proximal tubule and thick ascending limb and, theoretically, reduce the blood pressure set point needed to maintain effective circulating volume homeostasis.
Mediators of Pressure Natriuresis
This exploration of transporter regulation during hypertension brings us to the question, “What are the signals driving the transporter changes that cause pressure natriuresis?” Multiple signals have the potential to suppress sodium reabsorption and we assume the pressure natriuresis response is the sum of the prevailing natriuretic and anti-natriuretic influences along the nephron. Previous studies established that acutely raising BP stimulates non-autoregulating preglomerular vascular elements to release mediator(s) that can inhibit sodium transport in nearby tubules.45 Mediators may include cytochrome P-450 metabolites, nitric oxide, and factors suppressing the RAS. The cytochrome P-450 metabolite 20-hydroxyeicosatetraenoic acid (20-HETE), was one of the first candidates tested because it is a natriuretic and diuretic,46 potentiates tubuloglomerular feedback and autoregulates renal vascular tone.47 Inhibiting cytochrome P-450 metabolism blunts the diuretic response to acute hypertension and prevents redistribution of NHE3 and inhibition of Na,K-ATPase.13, 48–50 The Carey lab,51 provided evidence that an increase in renal interstitial cGMP is important to drive the natriuresis, supporting a role for intrarenal NO.45 Interestingly, this response would be lacking in L-NAME hypertension, where transporters’ abundance is nevertheless suppressed, presumably via a different path.34
Clamping AngII at a subpressor levels during acute hypertension blunts pressure natriuresis about 50%, and prevents retraction of both NHE3 from the proximal tubule microvilli8, 52 and NCC from apical membranes of the distal convoluted tubule.10 These findings are relevant to the blunting of pressure natriuresis in chronic AngII or L-NAME hypertension in which intrarenal AngII levels are elevated.16, 39 Activation of the local production of dopamine in the proximal tubule warrants consideration as a pressure-natriuretic mediator as it inhibits NHE3 and NKA activity, dopamine receptor inactivation raises BP, and reductions in activity of either the local RAS or the dopamine system increases the activity of the other.53, 54 Thus, suppression of natriuretic dopamine is predicted if local production of AngII is amplified. Another important factor to consider is renal sympathetic nervous system activation secondary to renal injury which prevents retraction of NHE3 out of the microvilli in the face of hypertension and blunts natriuresis.5, 55, 56
We propose, for further investigation, that the initial natriuretic response to acute hypertension is driven by rapid generation of mediators (20-HETE, NO, perhaps dopamine) released locally and that subsequent RAS and RSNA inhibition are important to sustain the natriuresis during chronic hypertension by reducing sodium transport along the nephron (as illustrated in L-NAME hypertension).16, 34 The pressure natriuresis response can be impaired during chronic hypertension by inflammation, immune cell infiltration, intrarenal AngII production, reduced NO production, high salt diet, and/or elevated RSNA provoking chronic hypertension (Fig 1C).14–17
Perspectives and Future Directions
This critical blood-pressure-setting response is understudied relative to its therapeutic potential. The studies summarized in this brief review focused on sodium transport regulation in the face of simultaneous AngII stimulation and the opposing anti-natriuretic signal of hypertension and demonstrate the rationale and necessity for addressing transporter regulation along the entire nephron utilizing whole animal models. While we can assess transporters’ abundance, covalent modifications, and subcellular distribution, fewer tools are available to investigate the impact of transporter changes besides lithium clearance, saline challenges and diuretic tests. An exception is the study, discussed above, visualizing hypertension stimulated redistribution of NHE3 to the base of the microvilli using in vivo microscopy.57 Further innovative efforts to examine in vivo responses to stimuli such as hypertension and renal injury, especially over a longer time courses, would undoubtedly answer open questions about how pressure natriuresis interfaces with anti-natriuretic stimuli and intrarenal controls, including autoregulation, to produce a natriuresis that equals sodium intake. At the level of transporter regulation, we have focused on abundance and phosphorylation in this brief review, but we and others have also provided evidence for importance of membrane lipid domain localization, ubiquitination and protein-protein interactions. Other potential transporter modifications warrant investigation in in vivo models. Regarding the integration of activating and inhibitory signaling pathways during hypertension, genetic models have proven invaluable, and have generated even more questions for study, e.g. the key role of immune cell infiltration. Future studies will likely benefit from tubule specific and inducible genetic modifications. In conclusion, while the field (my lab included) has focused on how and why sodium transport is aberrantly elevated in specific tubular regions during hypertension, we provide a rationale for broadening investigations to the entire nephron to obtain an integrated understanding of how the kidney generates a natriuresis to maintain fluid and electrolyte balance in the face of effective circulating volume and BP elevation.
Acknowledgments
I am honored to be chosen by the Kidney in Cardiovascular Disease to deliver the 2014 Donald Seldin lecture. American Heart Association support, to myself and my trainees during the past 30 years, provided us the opportunity to explore novel ideas and directions.
Sources of Funding
National Institutes of Health DK083785 and American Heart Association Grant in Aid 15GRNT23160003.
Footnotes
Disclosures - none
References
- 1.Bromfield S, Muntner P. High blood pressure: The leading global burden of disease risk factor and the need for worldwide prevention programs. Current hypertension reports. 2013;15:134–136. doi: 10.1007/s11906-013-0340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guyton AC. Blood pressure control–special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 3.Rossier BC, Staub O, Hummler E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: Importance in the control of blood pressure and hypertension. FEBS Lett. 2013;587:1929–1941. doi: 10.1016/j.febslet.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Crowley SD, Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest. 2014;124:2341–2347. doi: 10.1172/JCI72274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang LE, Leong PK, Ye S, Campese VM, McDonough AA. Responses of proximal tubule sodium transporters to acute injury-induced hypertension. Am J Physiol Renal Physiol. 2003;284:F313–322. doi: 10.1152/ajprenal.00134.2002. [DOI] [PubMed] [Google Scholar]
- 6.Yang LE, Maunsbach AB, Leong PK, McDonough AA. Differential traffic of proximal tubule na+ transporters during hypertension or pth: Nhe3 to base of microvilli vs. Napi2 to endosomes. Am J Physiol Renal Physiol. 2004;287:F896–906. doi: 10.1152/ajprenal.00160.2004. [DOI] [PubMed] [Google Scholar]
- 7.Yang LE, Maunsbach AB, Leong PK, McDonough AA. Redistribution of myosin vi from top to base of proximal tubule microvilli during acute hypertension. J Am Soc Nephrol. 2005;16:2890–2896. doi: 10.1681/ASN.2005040366. [DOI] [PubMed] [Google Scholar]
- 8.Leong PK, Yang LE, Holstein-Rathlou NH, McDonough AA. Angiotensin ii clamp prevents the second step in renal apical nhe3 internalization during acute hypertension. Am J Physiol Renal Physiol. 2002;283:F1142–1150. doi: 10.1152/ajprenal.00178.2002. [DOI] [PubMed] [Google Scholar]
- 9.Lee DH, Maunsbach AB, Riquier-Brison AD, Nguyen MT, Fenton RA, Bachmann S, Yu AS, McDonough AA. Effects of ace inhibition and ang ii stimulation on renal na-cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol. 2013;304:C147–163. doi: 10.1152/ajpcell.00287.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DH, Riquier AD, Yang LE, Leong PK, Maunsbach AB, McDonough AA. Acute hypertension provokes acute trafficking of distal tubule na-cl cotransporter (ncc) to subapical cytoplasmic vesicles. Am J Physiol Renal Physiol. 2009;296:F810–818. doi: 10.1152/ajprenal.90606.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2010;298:R851–861. doi: 10.1152/ajpregu.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris RC, Zhang MZ. Dopamine, the kidney, and hypertension. Current hypertension reports. 2012;14:138–143. doi: 10.1007/s11906-012-0253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dos Santos EA, Dahly-Vernon AJ, Hoagland KM, Roman RJ. Inhibition of the formation of eets and 20-hete with 1-aminobenzotriazole attenuates pressure natriuresis. Am J Physiol Regul Integr Comp Physiol. 2004;287:R58–68. doi: 10.1152/ajpregu.00713.2003. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Iturbe B, Franco M, Johnson RJ. Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens. 2013;22:37–44. doi: 10.1097/MNH.0b013e32835b3d54. [DOI] [PubMed] [Google Scholar]
- 15.Trott DW, Harrison DG. The immune system in hypertension. Advances in physiology education. 2014;38:20–24. doi: 10.1152/advan.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giani JF, Janjulia T, Taylor B, Bernstein EA, Shah K, Shen XZ, McDonough AA, Bernstein KE, Gonzalez-Villalobos RA. Renal generation of angiotensin ii and the pathogenesis of hypertension. Current hypertension reports. 2014;16:477. doi: 10.1007/s11906-014-0477-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol. 2014;307:F499–508. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roman RJ, Cowley AW., Jr Characterization of a new model for the study of pressure-natriuresis in the rat. Am J Physiol. 1985;248:F190–198. doi: 10.1152/ajprenal.1985.248.2.F190. [DOI] [PubMed] [Google Scholar]
- 19.Chou CL, Marsh DJ. Time course of proximal tubule response to acute arterial hypertension in the rat. Am J Physiol. 1988;254:F601–607. doi: 10.1152/ajprenal.1988.254.4.F601. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Magyar CE, Norian JM, Holstein-Rathlou NH, Mircheff AK, McDonough AA. Reversible effects of acute hypertension on proximal tubule sodium transporters. Am J Physiol. 1998;274:C1090–1100. doi: 10.1152/ajpcell.1998.274.4.C1090. [DOI] [PubMed] [Google Scholar]
- 21.Yang LE, Sandberg MB, Can AD, Pihakaski-Maunsbach K, McDonough AA. Effects of dietary salt on renal na+ transporters’ subcellular distribution, abundance, and phosphorylation status. Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajprenal.90235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leong PK, Devillez A, Sandberg MB, Yang LE, Yip DK, Klein JB, McDonough AA. Effects of ace inhibition on proximal tubule sodium transport. Am J Physiol Renal Physiol. 2006;290:F854–863. doi: 10.1152/ajprenal.00353.2005. [DOI] [PubMed] [Google Scholar]
- 23.Leong PK, Yang LE, Landon CS, McDonough AA, Yip KP. Phenol injury-induced hypertension stimulates proximal tubule na+/h+ exchanger activity. Am J Physiol Renal Physiol. 2006;290:F1543–1550. doi: 10.1152/ajprenal.00392.2005. [DOI] [PubMed] [Google Scholar]
- 24.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin ii stimulates trafficking of nhe3, napi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol. 2010;298:F177–186. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. Ang ii provokes acute trafficking of distal tubule na+-cl(−) cotransporter to apical membrane. Am J Physiol Renal Physiol. 2007;293:F662–669. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 26.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knepper MA, Masilamani S. Targeted proteomics in the kidney using ensembles of antibodies. Acta Physiol Scand. 2001;173:11–21. doi: 10.1046/j.1365-201X.2001.00880.x. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of na+ transporters along nephron during ang ii-dependent hypertension: Distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol. 2013;305:F510–519. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleyman TR, Carattino MD, Hughey RP. Enac at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284:20447–20451. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cowley AW., Jr Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension. 2008;52:777–786. doi: 10.1161/HYPERTENSIONAHA.107.092858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris PJ, Navar LG. Tubular transport responses to angiotensin. Am J Physiol. 1985;248:F621–630. doi: 10.1152/ajprenal.1985.248.5.F621. [DOI] [PubMed] [Google Scholar]
- 32.Wang CT, Navar LG, Mitchell KD. Proximal tubular fluid angiotensin ii levels in angiotensin ii-induced hypertensive rats. J Hypertens. 2003;21:353–360. doi: 10.1097/00004872-200302000-00027. [DOI] [PubMed] [Google Scholar]
- 33.Zatz R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension. 1998;32:958–964. doi: 10.1161/01.hyp.32.6.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giani JF, Janjulia T, Kamat N, Seth DM, Blackwell WL, Shah KH, Shen XZ, Fuchs S, Delpire E, Toblli JE, Bernstein KE, McDonough AA, Gonzalez-Villalobos RA. Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol. 2014;25:2752–2763. doi: 10.1681/ASN.2013091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turban S, Wang XY, Knepper MA. Regulation of nhe3, nkcc2, and ncc abundance in kidney during aldosterone escape phenomenon: Role of no. Am J Physiol Renal Physiol. 2003;285:F843–851. doi: 10.1152/ajprenal.00110.2003. [DOI] [PubMed] [Google Scholar]
- 36.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. At1a angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navar LG, Satou R, Gonzalez-Villalobos RA. The increasing complexity of the intratubular renin-angiotensin system. J Am Soc Nephrol. 2012;23:1130–1132. doi: 10.1681/ASN.2012050493. [DOI] [PubMed] [Google Scholar]
- 38.Giani JF, Shah KH, Khan Z, Bernstein EA, Shen XZ, McDonough AA, Gonzalez-Villalobos RA, Bernstein KE. The intrarenal generation of angiotensin ii is required for experimental hypertension. Curr Opin Pharmacol. 2015;21C:73–81. doi: 10.1016/j.coph.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ace protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. The Journal of experimental medicine. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wall SM, Pech V. Pendrin and sodium channels: Relevance to hypertension. Journal of nephrology. 2010;23(Suppl 16):S118–123. [PubMed] [Google Scholar]
- 44.Holtzclaw JD, Grimm PR, Sansom SC. Role of bk channels in hypertension and potassium secretion. Curr Opin Nephrol Hypertens. 2011;20:512–517. doi: 10.1097/MNH.0b013e3283488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans RG, Majid DS, Eppel GA. Mechanisms mediating pressure natriuresis: What we know and what we need to find out. Clin Exp Pharmacol Physiol. 2005;32:400–409. doi: 10.1111/j.1440-1681.2005.04202.x. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi K, Capdevila J, Karara A, Falck JR, Jacobson HR, Badr KF. Cytochrome p-450 arachidonate metabolites in rat kidney: Characterization and hemodynamic responses. Am J Physiol. 1990;258:F781–789. doi: 10.1152/ajprenal.1990.258.4.F781. [DOI] [PubMed] [Google Scholar]
- 47.Zou AP, Imig JD, Kaldunski M, Ortiz de Montellano PR, Sui Z, Roman RJ. Inhibition of renal vascular 20-hete production impairs autoregulation of renal blood flow. Am J Physiol. 1994;266:F275–282. doi: 10.1152/ajprenal.1994.266.2.F275. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YB, Magyar CE, Holstein-Rathlou NH, McDonough AA. The cytochrome p-450 inhibitor cobalt chloride prevents inhibition of renal na,k-atpase and redistribution of apical nhe-3 during acute hypertension. J Am Soc Nephrol. 1998;9:531–537. doi: 10.1681/ASN.V94531. [DOI] [PubMed] [Google Scholar]
- 49.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: A new target for the treatment of hypertension. J Cardiovasc Pharmacol. 2010;56:336–344. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension. 2007;49:687–694. doi: 10.1161/01.HYP.0000255753.89363.47. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed F, Kemp BA, Howell NL, Siragy HM, Carey RM. Extracellular renal guanosine cyclic 3′5′-monophosphate modulates nitric oxide and pressure-induced natriuresis. Hypertension. 2007;50:958–963. doi: 10.1161/HYPERTENSIONAHA.107.092973. [DOI] [PubMed] [Google Scholar]
- 52.Leong PK, Zhang Y, Yang LE, Holstein-Rathlou NH, McDonough AA. Diuretic response to acute hypertension is blunted during angiotensin ii clamp. Am J Physiol Regul Integr Comp Physiol. 2002;283:R837–842. doi: 10.1152/ajpregu.00089.2002. [DOI] [PubMed] [Google Scholar]
- 53.Li D, Scott L, Crambert S, Zelenin S, Eklof AC, Di Ciano L, Ibarra F, Aperia A. Binding of losartan to angiotensin at1 receptors increases dopamine d1 receptor activation. J Am Soc Nephrol. 2012;23:421–428. doi: 10.1681/ASN.2011040344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carey RM. The intrarenal renin-angiotensin and dopaminergic systems: Control of renal sodium excretion and blood pressure. Hypertension. 2013;61:673–680. doi: 10.1161/HYPERTENSIONAHA.111.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang LE, Zhong H, Leong PK, Perianayagam A, Campese VM, McDonough AA. Chronic renal injury-induced hypertension alters renal nhe3 distribution and abundance. Am J Physiol Renal Physiol. 2003;284:F1056–1065. doi: 10.1152/ajprenal.00317.2002. [DOI] [PubMed] [Google Scholar]
- 56.Pontes RB, Crajoinas RO, Nishi EE, Oliveira-Sales EB, Girardi AC, Campos RR, Bergamaschi CT. Renal nerve stimulation leads to the activation of the na+/h+ exchanger isoform 3 via angiotensin ii type i receptor. Am J Physiol Renal Physiol. 2015;308:F848–856. doi: 10.1152/ajprenal.00515.2014. [DOI] [PubMed] [Google Scholar]
- 57.Brasen JC, Burford JL, McDonough AA, Holstein-Rathlou NH, Peti-Peterdi J. Local ph domains regulate nhe3 mediated na+ reabsorption in the renal proximal tubule. Am J Physiol Renal Physiol. 2014 doi: 10.1152/ajprenal.00174.2014. ajprenal 00174 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]