

Abstract
Neurogranin is a calmodulin binding protein that has been implicated in learning and memory, long-term potentiation and synaptic plasticity. Neurons expressing neurogranin in the cortex degenerate in late stages of Parkinson’s disease with widespread α-synuclein pathology. While analyzing neurogranin gene expression levels through rtPCR in brains of mouse models overexpressing human α-synuclein, we found levels were elevated 2.5 times when compared to nontransgenic animals. Immunohistochemistry in the cortex revealed colocalization between α-synuclein and neurogranin in mouse transgenics when compared to control mice. Coimmunoprecipitation studies in the superior temporal cortex in humans confirmed interaction between α-synuclein and neurogranin, and decreased interaction between α-synuclein and neurogranin was noticed in patients diagnosed with Parkinson’s disease when compared to normal control brains. Additionally, phosphorylated neurogranin levels were also decreased in the human superior temporal cortex in patients diagnosed with Parkinson’s disease and patients diagnosed with dementia with Lewy bodies. Here, we show for the first time that neurogranin binds to α-synuclein in the human cortex, and this interaction decreases in Parkinson’s disease along with the phosphorylation of neurogranin, a molecular process thought to be involved in learning and memory.
Keywords: Parkinson’s disease, Dementia with Lewy bodies, Neurogranin, α-Synuclein, Calmodulin, LTP
1. Introduction
In Parkinson’s disease (PD), the traditional emphasis on research has been the degeneration of the substantia nigra and the motor deficits which arise as a result of the depletion of dopaminergic neurons in this region. However, as PD progresses, the onset of Parkinson’s disease dementia (PDD) and cell loss in the cortex and hippocampus is likely (McKeith et al., 2005). Dementia with Lewy bodies (DLB) is diagnosed when dementia occurs concurrently or before parkinsonism while PDD is described as when dementia occurs more than a year after a diagnosis of PD (McKeith et al., 2005). PD, PDD and DLB are all characterized by α-synuclein (α-syn) accumulation extracellularly in cortical and subcortical regions. It is believed that α-syn protein functions normally in cell body and presynaptic terminal (Maroteaux et al., 1988), but is capable from moving from cell to cell as it begins to aggregate (Lee et al., 2010).
In cortical regions in PD and DLB, it is not understood what makes specific cells pregnable to disease. Typical corticalbasal atrophy occurs in parasagittal posterior frontal and parietal regions with more generalized degeneration of the inferior frontal and temporal lobes after onset of dementia (McKeith et al., 2005). Swollen neurons are found throughout affected cortical areas, but are most frequent in layers III, IV and VI (Rebeiz et al., 1967). Neurogranin (Ng) expression is prominent in neurons in layers IV and VI of the cortex: Areas which project to and receive projections from the thalamus (Gerendasy and Sutcliffe, 1997; Neuner-Jehle et al., 1996; Represa et al., 1990; Watson et al., 1992, 1994). Ng is also found in the pyramidal neurons of the hippocampus. These areas of the cortex and hippocampus are damaged in later stages of Parkinson’s disease during the onset of dementia (McKeith et al., 2005).
Ng is a postnatally expressed protein and a member of the calpactin family involved in calcium signaling (Represa et al., 1990). It is located in soma and dendritic spines and traditionally thought to bind calmodulin (CaM) in the absence of calcium (Baudier et al., 1991; Hoffman et al., 2014b; Prichard et al., 1999). CaM has been shown in vitro to bind to the N-terminus of α-syn (Gruschus et al., 2013; Martinez et al., 2003). Ng is also a specific substrate for the γ-isoform of protein kinase C (PKC), and is phosphorylated at Ser36 location in the IQ domain when calcium is available to CaM, and is closely associated with cell membranes (Baudier et al., 1991; Dominguez-Gonzalez et al., 2007; Liu et al., 1990). Because of site proximity, Ng-CaM binding and Ng phosphorylation by protein kinase C appear to be mutually exclusive (Baudier et al., 1991). Ng-CaM binding can accelerate CaM dissociation from calcium in postsynaptic neurons (Kubota et al., 2007; Kumar et al., 2013). Additionally, recent studies have shown that Ng bound to CaM can lower the threshold for long-term potentiation (LTP) (Zhong and Gerges, 2012).
Ng knockout mice look similar to nontransgenic mice phenotypically but exhibit difficulties in learning and memory tasks while also developing emotional disruptions (Miyakawa et al., 2001). Decreases in Ng expression levels was asseverated in the hippocampus of amyloid precursor protein (APP) transgenic mice and blocking phosphorylation of Ng can prevent learning indicating a role for Ng in dementia (Alexander et al., 1987; Liu et al., 1990). Additionally, Ng expression has been affected differentially in Alzheimer’s and Frontal Temporal dementia (Chang et al., 1997). Ng levels are increased in the cerebral spinal fluid in Alzheimer’s disease (Thorsell et al., 2010). A genetic variant upstream of the Ng gene (NRGN) has also been shown in patients diagnosed with schizophrenia, further revealing a role for Ng in cognition (Ohi et al., 2013; Stefansson et al., 2009; Steinberg et al., 2011; Walton et al., 2013).
The degeneration of cells in areas that express Ng in Parkinson’s disease dementia indicate that Ng may be involved in pathways which lead to dementia in the brain. Additionally, Ng and α-syn both appear to interact with CaM in calcium signalling pathways, suggesting Ng likely contributes a role to α-syn molecular functionality.
2. Results
2.1. Ng expression in Tg mice and colocalization with α-syn
A 2.5-fold increase of Ng gene expression in Tg mice over-expressing human α-syn versus nonTg mice was shown by quantitative real time PCR (Fig. 2; 2.58±1.21 vs. 0.98±0.45 relative expression, respectively; p=0.008; 2-sided t-test) (Fig. 1).
Fig. 2.

Neurogranin interaction with α-synuclein in Tg mouse brain. Immunohistochemical neurogranin expression in nonTg mouse brains (green, (A)) when compared with α-syn (red, (B)) revealed no colocolization among cell bodies ((C) and (D)) but possible colocalization in the synapse. In Tg mouse brains, neurogranin (E) and α-syn (F) showed obvious yellow colocalization in cell bodies and synapes ((G) and (H)). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 1.
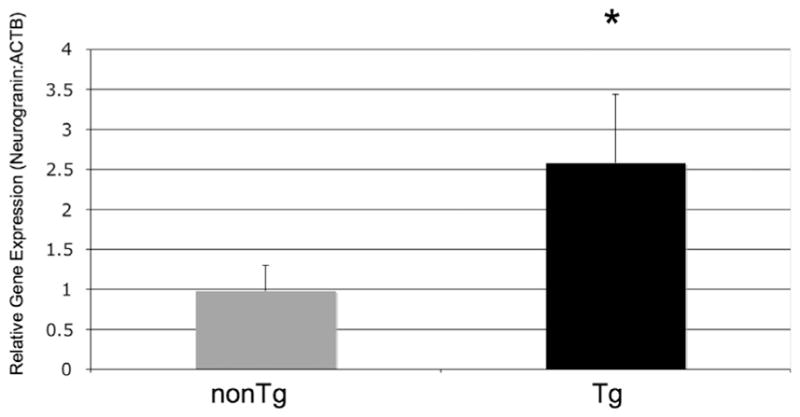
Neurogranin expression levels. Neurongranin RNA levels in the brains of Tg mice over expressing human α-syn. Expression increased 2.5 times in Tg mice when compared to normal controls (Fig. 2; 2.58±1.21 vs. 0.98±0.45 relative expression, respectively; p=0.008; 2-sided t-test).
In nonTg mice, in areas of the cortex dorsal to the hippocampus, Ng immunohistochemistry was observed through confocal microscopy most closely associated with the membrane in the cell body and dendrite (Fig. 2A). α-syn immunohistochemistry in nonTg mice was relegated to pixelated synaptic expression (Fig. 2B). When merged, no colocalization was seen in cell bodies (Fig. 2C). A higher level magnification of Ng expressing cells with α-syn expression can be seen in Fig. 2D.
Ng immunohistochemistry in Tg mice was also widespread in the dendrites and the cytosol of cell bodies in areas dorsal to the hippocampus in Tg mice (Fig. 2E). However, in these mice, immunohistochemistry for α-syn revealed large cell body inclusions (Fig. 2F). In Tg mice, colocalization of Ng and α-syn was widespread throughout the cortex (Fig. 2G and Fig. 3H).
Fig. 3.

Neurogranin and α-synuclein Western blot in superior temporal cortex in human brains. α-Syn protein levels as analyzed by Western blot and compared to actin were increased between PD and DLB brains when compared to normal controls (Fig. 4A and B) (*=<.05; **=<.01). Neurogranin levels remained the same between normal controls, PD and DLB brains (Fig. 4A and C).
2.2. Ng and α-synuclein levels in human superior temporal cortex
In Fig. 3A and B, Western blot analysis of α-syn in the superior temporal cortex revealed increases when compared to actin (*=<.05; **=<.01). The increase was larger in patients with DLB than with PD, but were both significantly higher than controls. Ng protein expression did not differ between normal controls, PD patients and DLB patients (Fig. 3A and C).
2.3. Ng interaction with α-synuclein in human superior temporal cortex
Immunoprecipitation with Ng in human superior temporal cortex also revealed interactions when probing on Western blot for α-synculein, and reverse coimmunoprecipitation confirmed these results (Fig. 4A). As a control, when immunoprecipitating with mouse and rabbit IgG, no expression of α-syn or Ng was observed through Western blot analysis (Fig. 4B). In Fig. 4C, levels from Ng immunoprecipitation and α-synuclein probing through Western blot showed a significant decrease was noticed in patients with PD (**=p<.01). When comparing DLB and PD patients Ng levels in Fig. 3B with Ng-α-syn interaction in Fig. 4A, a dramatic reduction in Ng-α-syn interaction compared to Ng protein levels was observed. In DLB brains, Ng-α-syn interaction was not different between normal control brains and DLB patients. However, DLB patients had a 320% increase in α-syn levels as shown in Fig. 3B. This large amount of α-syn present in DLB brains might explain why the levels of interaction remain similar between Ng and α-syn when comparing normal control brains and DLB brains.
Fig. 4.

Neurogranin interaction with α-synuclein in human superior temporal cortex. In human superior temporal cortex, when immunoprecipitated for neurogranin followed by Western blot of precepitate for α-syn, an interaction was observed in normal control, PD and DLB patients; additionally, reverse coIP, immunoprecipitation for α-syn and Western for neurogranin revealed interaction as well (A) (N=no antibody included in immunoprecipitation with DLB sample, B=blank, no sample included in immunoprecipitation). In (B) when immunoprecipitating with non specific IgG, no interaction was revealed, indicating that the interaction was specific for neurogranin and α-syn. In (C) pixel density analysis indicates a statistical reduction in PD brains in the interaction between neurogranin and α-syn (**=p<.01). In (D) when comparing the levels of neurogranin from (C) with the levels of neurogranin in the coimmunoprecipitation assay as percent of normal control, normal neurogranin levels were slightly diminished in PD brains, whereas α-syn bound neurogranin levels were dramatically reduced by half in PD brains indicating a pathological difference in neurogranin-α-syn in this disease state. No difference was seen in DLB brains in normal neurogranin or neurogranin bound with α-syn.
2.4. p-Ng levels in the membrane and cytosolic fraction in human superior temporal cortex
In order to determine if phosphorylated Ng (p-Ng) levels had changed due to decreased binding of Ng with α-syn in patients diagnosed with Parkinson’s disease, Western blot analysis of p-Ng was completed. Ng is bound to CaM in the absence of calcium but is phosphorylated by protein kinase C when it is unbound, and similar mechanisms may be involved in Ng interactions with α-syn. Interestingly, p-Ng levels were diminished in the membrane fraction of patients diagnosed with PD and in the brains of patients diagnosed with DLB (Fig. 5A and B) *=p<.05. However, no difference was seen in the cytosolic fraction in p-Ng levels in the superior temporal cortex, as shown in Fig. 5C.
Fig. 5.

Phosphorylated neurogranin Western blot in the membrane and cytosolic fraction in superior temporal cortex in human brains. p-Ng protein levels as analyzed by Western blot and compared to actin were decreased in the membrane faction of superior temporal cortex in PD and DLB brains when compared to normal controls ((A) and (B)) (*=<.05; **=<.01). p-Ng levels in the cytosolic fraction remained the same between normal controls, PD and DLB brains ((A) and (C)).
3. Discussion
Ng, a CaM binding protein implicated in intracellular calcium modulation in neurons (Guadano-Ferraz et al., 2005; Zhabotinsky et al., 2006), interacts with α-synuclein in the human superior temporal cortex.
The binding of these two proteins is pathologically decreased in the cases of patients with Parkinson’s disease. We demonstrate in mice expressing human α-syn that Ng gene expression increases. However, Ng protein expression is steady when comparing levels in the superior temporal cortex of normal human controls, PD and DLB patients. Subsequently, we postulate that cells manufacture more Ng in order to compensate for increases in α-synuclein in transgenic mice.
Although Ng protein expression is not increased in the superior temporal cortex in human brains with PD, Ng-α-syn interactions decrease in patients with PD. These studies suggest that Ng interaction with α-syn is a naturally occurring process that is disrupted during onset of dementia.
As it is known that α-syn is likely a CaM substrate in the presence of calcium, in order to determine whether a decrease in Ng-α-syn interaction in patients with Parkinson’s disease coincided with an increase in calcium dependent phosphorylation of Ng, we studied the membrane and cytosolic fractions of superior temporal cortex for p-Ng. Surprisingly, p-Ng levels were also diminished along with the interaction of Ng and α-syn, indicating that phosphorylation of Ng and Ng-α-syn interaction processes were both disrupted, while total Ng levels remain constant.
In humans, as PD progresses, abated Ng-α-syn interactions may contribute to the onset of Parkinson’s disease dementia (PDD). However, it is possible that α-syn misfolding (Chiti and Dobson, 2006; Dauer and Przedborski, 2003) or disruption of α-syn protein renewal resulting in depleted functional α-syn might also result in the drop in Ng-α-syn binding (Jaeger and Wyss-Coray, 2009; Lee et al., 2008). Interestingly, since decreased Ng phosphylation is also noticed, Ng functionality may be disrupted in disease states, which may account for decreased Ng-α-syn interaction. It’s possible that the interaction between α-syn and Ng is essential to human synaptic function and integrity.
Ng interaction with CaM has been indicated in synaptic plasticity (Roberson and Sweatt, 1999; Xia et al., 1993). Ng has also been involved in Nitric Oxide Synthase activity through its CaM binding properties (Martzen and Slemmon, 1995; Slemmon and Martzen, 1994). It is known that after protein kinase C (PKC) phosphorylates Ng, it becomes unbound to CaM because they compete for the same binding site (Kubota et al., 2007; Liu et al., 1990). Blocking phosphorylation of Ng seems to be disruptive to learning (Liu et al., 1990; Zhong et al., 2011). Ng has also been established to play a role in schizophrenia, further demonstrating known roles for the protein in cognition (Ohi et al., 2013); additionally, decreased expression of Ng has recently been shown after cocaine abuse (Kovalevich et al., 2012), while increased expression of Ng was observed after morphine administration (Shukla et al., 2006). Furthermore, Ng bound to CaM in postsynaptic neurons causes CaM dissociation with calcium, and Ng binds PKC in the presence of calcium, a process that seems essential to the mechanistic processes in LTP (Liu et al., 1990).
Interestingly, it has been recently been reported that although Ng-CaM binding is 2–3 fold stronger in the absence of calcium, and that this binding facilitates LTP, Ng-CaM binding can also occur in the presence of low levels of calcium (Hoffman et al., 2014a). Conversely, it appears that CaM binds the N-terminus of α-syn in the presence of calcium, and that this binding is associated with the membrane (Gruschus et al., 2013). Ng is also known to be localized to the membrane when it is unbound to CaM (Dominguez-Gonzalez et al., 2007). It is possible Ng-α-syn binding occurs in the presence of calcium, as the studies presented here demonstrate decreased binding in PD correlates with decreased p-Ng as seen in the membrane fraction, indicating the result could be a by-product of calcium disturbances in degeneration (Branch et al., 2014). However, in DLB there is also decreased p-Ng in the membrane, but not decreased Ng-α-syn interaction compared to age-matched controls. It is also possible, that in disease states, less Ng is phosphorylated and more is bound to CaM to prime the cell for LTP to compensate for degeneration (Zhong and Gerges, 2012), and that Ng-α-syn binding occurs independently of the process. The discrepancy in Ng-α-syn binding and p-Ng levels between the diseases could warrant future studies that may illuminate the molecular cause of symptomatic differences in the diagnoses.
Additionally, it is also possible that Ng and α-syn can be bound to CaM simultaneously in the presence of calcium, or that CaM and Ng could instead be complexed to α-syn simultaneously, and associated with the membrane. Ng is currently believed to bind PKC and CaM through its IQ motif (Diez-Guerra, 2010), but binding of α-syn could occur through a different region, as α-syn-CaM binding also occurs at a different region on CaM than Ng bound to CaM (Gruschus et al., 2013).
Ng-α-syn binding could have an effect on LTP as evidenced by the decreased interaction in Parkinson’s disease patients compared with normal brains. Although where the interaction occurs between these proteins will largely give clues to the function of their interaction in human brains. α-Syn is traditionally thought to be pre-synaptic (Maroteaux et al., 1988), and Ng as post-synaptic (Prichard et al., 1999). It could be that Ng-α-syn interactions may be occurring extracellularly at the synapse (Lee et al., 2010), in non-neuronal cells, or in the cell body (Chang et al., 1997; Maroteaux et al., 1988; Prichard et al., 1999). For example, recent research in Alzheimer’s disease has shown that Ng in the cerebrospinal fluid can be a useful marker for synaptic degeneration (Thorsell et al., 2010).
Ng appears to play a vital function in cognition. Future studies need to determine further the role Ng interaction with α-syn has in neurodegenerative disease, where the cellular location of bound Ng-α-syn occurs, and how this interaction is related to: calcium signalling, Ng bound to CaM, CaM-α-syn interactions, Ng phosphorylation, and ultimately the cognitive deficits observed in disease.
4. Experimental procedures
4.1. Animals
For this study, heterozygous transgenic (Tg) mice (Line D) expressing hα-syn under the regulatory control of the platelet-derived growth factor-β (PDGFβ) promoter (Masliah et al., 2000b) were used. These animals were selected because they develop hα-syn-immunoreactive inclusion-like structures in the brain. Although some nuclear staining has been observed in this model, distinct cytoplasmic inclusion-like structures have been consistently identified by confocal and electron microscopy (Masliah et al., 2000b, 2005; Masliah, 2001; Rockenstein et al., 2002). Furthermore, these animals also display neurodegenerative and motor deficits that mimic certain aspects of PD and DLB. A total of 16 animals were used: 3 nontransgenic (nonTg) and 3 transgenic (Tg) for Western blot and immunohistochemical analysis; and separately, 5 nonTg and 5 Tg for gene expression studies. Animals were sacrificed at 1 year of age and the brains removed. All procedures were completed under the specifications set forth by the Institutional Animal Care and Use Committee.
4.2. Human samples
A total of 18 subjects were used for coimmunoprecipitation and Western blot analysis: 6 normal controls, 6 subjects diagnosed with disease with Lewy bodies, 6 subjects diagnosed with Parkinson’s disease. All subjects were between 67–85 years of age (Table 1). The brains were dissected and a 0.1 g portion of the superior temporal cortex removed. Samples were generously provided by the Alzheimer’s disease Research Center Brain Bank at the University of California, San Diego. All experiments on human subjects were conducted in accordance with the Declaration of Helsinki.
Table 1.
Diagnostic information of human samples.
For each Western blot using human samples, the samples were loaded from left to right, with sample number 1 on the left, and sample number 18 on the right in the figures. All samples were taken from superior temporal cortex in the brains indicated.
| Control | Diagnosis |
|---|---|
| 1 | Normal |
| 2 | Normal |
| 3 | Normal |
| 4 | Normal Lacunar infarct, chronic (old) |
| Infarct, other; Ischemic changes | |
| 5 | Normal |
| 6 | Normal |
| PD | |
| 7 | Parkinson’s disease, idiopathic |
| 8 | Parkinson’s disease, idiopathic |
| Alzheimer’s disease | |
| 9 | Parkinson’s disease, idiopathic |
| Alzheimer’s changes, mild | |
| 10 | Parkinson’s disease, idiopathic |
| 11 | Parkinson’s disease, idiopathic |
| 12 | Parkinson’s disease, idiopathic |
| Alzheimer’s changes | |
| DLBD | |
| 13 | Diffuse Lewy body disease |
| Dementia with Lewy bodies | |
| 14 | Diffuse Lewy body disease |
| 15 | Diffuse Lewy body disease |
| 16 | Diffuse Lewy body disease |
| Micro infarcts | |
| 17 | Diffuse Lewy body disease |
| Hippocampal sclerosis | |
| 18 | Diffuse Lewy body disease |
| Alzheimer’s changes | |
4.3. Gene expression study
For gene expression studies, 10 transgenic mice overexpressing human wildtype α-synuclein and 6 non-transgenic age-matched littermates as controls were used.
RNA was extracted from mouse hemi-brain homogenates using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Reverse transcription was performed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with RNase inhibitor following the standard protocol provided by the manufacturer. cDNA quantity and quality were analyzed by spectrophotometry (at 260 and 280 nm) and the samples were normalized to 25 ng/μl. For Real-Time PCR, 50 ng of cDNA were used. Real-time PCR was performed using pre-designed TaqMan Gene Expression Assays for Ng (Applied Biosystems; Reference sequence: NM_022029.2) and for β-actin as endogenous control (TaqMan Endogenous Control; Reference sequence NM_007393.1). Both probes were labelled with FAM/MGB. Mastermixes consisted of the 2 × TaqMan Universal PCR Mastermix (Applied Biosystems), double distilled water and the 20 × primer/probe pre-designed assay in a 25 μl reaction setup. Amplification conditions consisted of 2 min at 50 °C, 10 min at 95 °C then 40 cycles of 15 s at 95 °C and 1 min at 60 °C. PCR reactions were performed and threshold cycles (Ct) were calculated using the Applied Biosystems 7500 Real-Time PCR system. Relative gene expression quantities were calculated according to the ΔΔCt-method (2−ΔΔCt) with β-actin as the endogenous control and the average ΔCt (CtNg−CTβ-actin) of non-transgenic controls as the reference for normalization.
4.4. Immunohistochemical analysis of α-Syn and Ng
Mouse sections were first washed 3 times for 5 min each in PBS. They were then pretreated with 1% triton-X, 3% hydrogen peroxide in PBS for 15 min, washed again three times for 5 min in PBS and blocked in 10% serum matching the animal the secondary antibody was raised in for 1 h and then washed again three times for 5 min each in PBS. Sections were then placed in primary antibody rabbit anti-(h) α-syn (Chemicon, Temecula, CA (1:2500) overnight in 4 °C. Sections were washed three times for 5 min in PBS and then placed in goat anti-rabbit biotynilated secondary antibody (1:100) (Vector Laboratories, Burlingame, CA) for 1 h. Secondary antibody was detected with the Tyramide Signal Amplification-Direct (Red) System (NEN Life Sciences) (Masliah et al., 2000a), followed by an overnight incubation with the mouse monoclonal anti-microtubule-associated protein-2 (MAP2, 1:500; Abcam, Cambridge, MA) primary antibody. This antibody was then detected with FITC-conjugated anti-mouse secondary antibody (1:75) (Vector Laboratories, Burlingame, CA) for 1 h. The double fluorescent immunolabeled sections were imaged by laser scanning confocal microscope LSCM (MRC1024, Bio-Rad) to determine whether or not (h) α-syn and Ng colabel.
4.5. Preparation of brain hemogenates for Western blot and immunoprecipitation
Mouse brains were removed and divided sagittally. One hemibrain was snap frozen and stored at −70 °C for RNA and protein analysis, whereas the other hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4 °C for 48 h, sectioned at 40 μm with a Vibratome 2000 (Leica, Nussloch, Germany) and placed in cryosolution for immunohistochemical analysis. Preparation of brain hemogenates, immunoprecipitation and immunoblot analysis was performed identically for mouse hemisectioned brain and superior temporal cortex from human brains. Preparation of whole brain homogenates was performed as previously described with slight changes (Ho et al., 2005; Petrucelli et al., 2004). Briefly, brain samples were lysed in TNE buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% tween) containing 1X protease and phosphotase inhibitors (Sigma-Aldrich) and centrifuged for 10 min at 5000 ×g at 4 °C. The supernatant was collected and total protein concentration of each sample was determined using BCA protein assay reagents (Pierce, Rockford, IL). For immunoblot assays analyzing soluble (cytosolic) and insoluble (membrane) fractions, for p-Ng, which is more abundant in insoluble fractions, brain homogenates were lysed in PDGF buffer (HEPES 1.0 mM, Benzamidine 5 mM, 2-mercaptoethanol 2 mM, EDTA 3 mM, Magnesium sulfate 0.5, Sodium Azide 0.05%) containing 1X protease and phosphatase inhibitors (Sigma-Aldrich) and centrifuged for 10 min at 5000 ×g. The resulting supernatent was ultracentrifuged (274,000 ×g, 1 h, 4 °C), and the detergent soluble proteins were collected in the supernatant fraction. The pelleted detergent insoluble proteins were dissolved in PDGF buffer containing 1 × protease and phosphotase inhibitors. Total protein concentration of each sample was also determined using BCA protein assay reagents (Pierce, Rockford, IL).
4.6. Western blot analysis
Samples were separated on 4–12% SDS-PAGE gels (NuPAGE, Invitrogen) and transferred onto 0.22 μM PDVF membranes (Schleicher & Schunell, Keene, NH) using 1 × 3-[cyclohexylamino]-1-propaneosulfonic acid (CAPS) transfer buffer containing 20% methanol. Membranes were blocked with 3% milk in PBS containing 0.1% Tween-20 (Sigma-Aldrich) (PBS-T), followed by incubation in primary antibody (1:1000) in PBS-T and 3% Bovine Serum Albumin (BSA) overnight at 4 °C. The primary antibodies used were as follows: anti-actin and anti-(h) α-syn from Chemicon International (Temecula, CA); anti-Ng (Abcam, Cambridge, MA) and anti-phospho-Ng (Chemicon, Temecula, CA). Membranes were further incubated with goat anti-mouse or anti-rabbit IgG secondary antibodies conjugated to horseradish peroxidase (1:5000, American Qualex, San Clemente, CA) and visualized by enhanced chemiluminescence (ECL, NEN Life Sciences, Boston, MA). For determinations of levels of immunoreactivity, ECL treated membranes were analyzed in the VersaDoc imaging system (Bio-Rad, Hercules, CA) using the Quantity One software (Bio-Rad), with blot pixel density adjusted for background.
4.7. Immunoprecipitation of brain samples
Brain homogenates (300 μg) were harvested as described above for Western blot. Supernatant fractions from cell lysates were immunoprecipitated in TNE buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Tween) containing 1 × protease and phosphotase inhibitors (Sigma-Aldrich) by using anti-Ng (1:500) (Abcam, Cambridge, MA) and added to 25 μl of a 1:1 slurry of protein A/G beads (1:20) (Santa Cruz Biotechnology). The beads were washed 3 times with additional TNE buffer identical to the incubation buffer after an overnight rotating incubation at 4 °C. Proteins were then eluted by boiling in 25 μl of loading buffer and resolved by SDS-PAGE on a 4–12% Bis–Tris gel as described above for Western blot. Following electrotransfer to PDVF 0.22 membrane, α-synuclein was recovered by an anti-(h)α-syn antibody, LB509 (Chemicon, Temecula, CA; 1:1000). Secondary antibody and immunoblot analysis was performed as described for Western blot. Reverse coimmunoprecipitation was performed with Zymed anti (h)α-syn 211 (Invitrogen) at same concentrations, with same buffer and Ng was recovered using Abcam Ng antibody.
4.8. Statistical analysis
All values in the figures are expressed as the means±SEM. To determine the statistical significance, the values were compared by two group t-tests using the Statview II statistical package for the Macintosh computer. The differences were considered to be significant if p values were less than 0.05.
Acknowledgments
The authors would like to acknowledge the Stein Institute for Research on Aging, Don and Marilyn Short Fellowship for Research in Parkinson’s disease at the University of California, San Diego for their support. Special thanks to Pearl Josephine Koob for help with manuscript preparation.
Contributor Information
Andrew O. Koob, Email: andrew.koob@uwrf.edu.
Eliezer Masliah, Email: emasliah@ucsd.edu.
References
- Alexander JA, et al. The diagnosis of dementia. J Arkansas Med Soc. 1987;83:365–368. [PubMed] [Google Scholar]
- Baudier J, et al. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J Biol Chem. 1991;266:229–237. [PubMed] [Google Scholar]
- Branch SY, Sharma R, Beckstead MJ. Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J Neurosci. 2014;34:9310–9318. doi: 10.1523/JNEUROSCI.4228-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, et al. Dendritic translocation of RC3/ neurogranin mRNA in normal aging, Alzheimer disease and frontotemporal dementia. J Neuropathol Exp Neurol. 1997;56:1105–1118. doi: 10.1097/00005072-199710000-00004. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Diez-Guerra FJ. Neurogranin, a link between calcium/ calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life. 2010;62:597–606. doi: 10.1002/iub.357. [DOI] [PubMed] [Google Scholar]
- Dominguez-Gonzalez I, et al. Neurogranin binds to phosphatidic acid and associates to cellular membranes. Biochem J. 2007;404:31–43. doi: 10.1042/BJ20061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerendasy DD, Sutcliffe JG. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol. 1997;15:131–163. doi: 10.1007/BF02740632. [DOI] [PubMed] [Google Scholar]
- Gruschus JM, et al. NMR structure of calmodulin complexed to an N-terminally acetylated alpha-synuclein peptide. Biochemistry. 2013;52:3436–3445. doi: 10.1021/bi400199p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadano-Ferraz A, et al. RC3/neurogranin is expressed in pyramidal neurons of motor and somatosensory cortex in normal and denervated monkeys. J Comp Neurol. 2005;493:554–570. doi: 10.1002/cne.20774. [DOI] [PubMed] [Google Scholar]
- Ho GJ, et al. Altered p59Fyn kinase expression accompanies disease progression in Alzheimer’s disease: implications for its functional role. Neurobiol Aging. 2005;26:625–635. doi: 10.1016/j.neurobiolaging.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Hoffman L, et al. Neurogranin alters the structure and calcium binding properties of calmodulin. J Biol Chem. 2014a;289:14644–14655. doi: 10.1074/jbc.M114.560656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman L, et al. Neurogranin alters the structure and calcium-binding properties of calmodulin. J Biol Chem. 2014b doi: 10.1074/jbc.M114.560656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalevich J, et al. Cocaine decreases expression of neurogranin via alterations in thyroid receptor/retinoid X receptor signaling. J Neurochem. 2012;121:302–313. doi: 10.1111/j.1471-4159.2012.07678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Putkey JA, Waxham MN. Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/ CaM signaling. Biophys J. 2007;93:3848–3859. doi: 10.1529/biophysj.107.106849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, et al. Structural basis for the interaction of unstructured neuron specific substrates neuromodulin and neurogranin with calmodulin. Sci Rep. 2013;3:1392. doi: 10.1038/srep01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, et al. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Lee SJ, et al. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hirono S, Moriguchi I. Quantitative structure–activity relationships for calmodulin inhibitors. Chem Pharm Bull (Tokyo) 1990;38:2184–2189. doi: 10.1248/cpb.38.2184. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, et al. Parkinson’s disease-associated alpha-synuclein is a calmodulin substrate. J Biol Chem. 2003;278:17379–17387. doi: 10.1074/jbc.M209020200. [DOI] [PubMed] [Google Scholar]
- Martzen MR, Slemmon JR. The dendritic peptide neurogranin can regulate a calmodulin-dependent target. J Neurochem. 1995;64:92–100. doi: 10.1046/j.1471-4159.1995.64010092.x. [DOI] [PubMed] [Google Scholar]
- Masliah E, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000a;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masliah E, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000b;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masliah E. Recent advances in the understanding of the role of synaptic proteins in Alzheimer’s disease and other neurodegenerative disorders. J Alzheimer’s Dis. 2001;3:1–9. doi: 10.3233/jad-2001-3117. [DOI] [PubMed] [Google Scholar]
- Masliah E, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- McKeith IG, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, et al. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11:763–775. doi: 10.1002/hipo.1092. [DOI] [PubMed] [Google Scholar]
- Neuner-Jehle M, Denizot JP, Mallet J. Neurogranin is locally concentrated in rat cortical and hippocampal neurons. Brain Res. 1996;733:149–154. [PubMed] [Google Scholar]
- Ohi K, et al. Influence of the NRGN gene on intellectual ability in schizophrenia. J Hum Genet. 2013;58:700–705. doi: 10.1038/jhg.2013.82. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Prichard L, Deloulme JC, Storm DR. Interactions between neurogranin and calmodulin in vivo. J Biol Chem. 1999;274:7689–7694. doi: 10.1074/jbc.274.12.7689. [DOI] [PubMed] [Google Scholar]
- Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc. 1967;92:23–26. [PubMed] [Google Scholar]
- Represa A, et al. Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci. 1990;10:3782–3792. doi: 10.1523/JNEUROSCI.10-12-03782.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Sweatt JD. A biochemical blueprint for long-term memory. Learn Mem. 1999;6:381–388. [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Shukla PK, Tang L, Wang ZJ. Phosphorylation of neurogranin, protein kinase C, and Ca2+/calmodulin dependent protein kinase II in opioid tolerance and dependence. Neurosci Lett. 2006;404:266–269. doi: 10.1016/j.neulet.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Slemmon JR, Martzen MR. Neuromodulin (GAP-43) can regulate a calmodulin-dependent target in vitro. Biochemistry. 1994;33:5653–5660. doi: 10.1021/bi00184a039. [DOI] [PubMed] [Google Scholar]
- Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, et al. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum Mol Genet. 2011;20:4076–4081. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010;1362:13–22. doi: 10.1016/j.brainres.2010.09.073. [DOI] [PubMed] [Google Scholar]
- Walton E, et al. The impact of genome-wide supported schizophrenia risk variants in the neurogranin gene on brain structure and function. PLoS One. 2013;8:e76815. doi: 10.1371/journal.pone.0076815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JB, Sutcliffe JG, Fisher RS. Localization of the protein kinase C phosphorylation/calmodulin-binding substrate RC3 in dendritic spines of neostriatal neurons. Proc Natl Acad Sci USA. 1992;89:8581–8585. doi: 10.1073/pnas.89.18.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JB, Szijan I, Coulter PM., 2nd Localization of RC3 (neurogranin) in rat brain subcellular fractions. Brain Res Mol Brain Res. 1994;27:323–328. doi: 10.1016/0169-328x(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Xia Z, et al. Type I calmodulin-sensitive adenylyl cyclase is neural specific. J Neurochem. 1993;60:305–311. doi: 10.1111/j.1471-4159.1993.tb05852.x. [DOI] [PubMed] [Google Scholar]
- Zhabotinsky AM, et al. Role of the neurogranin concentrated in spines in the induction of long-term potentiation. J Neurosci. 2006;26:7337–7347. doi: 10.1523/JNEUROSCI.0729-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Kaleka KS, Gerges NZ. Neurogranin phosphorylation fine-tunes long-term potentiation. Eur J Neurosci. 2011;33:244–250. doi: 10.1111/j.1460-9568.2010.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Gerges NZ. Neurogranin targets calmodulin and lowers the threshold for the induction of long-term potentiation. PLoS One. 2012;7:e41275. doi: 10.1371/journal.pone.0041275. [DOI] [PMC free article] [PubMed] [Google Scholar]