

Abstract
RATIONALE
Apolipoprotein(a) [apo(a)] is the defining protein component of lipoprotein(a) [Lp(a)], an independent risk factor for cardiovascular disease. The regulation of Lp(a) levels in blood is poorly understood in part due to technical challenges in measuring Lp(a) kinetics. Improvements in the ability to readily and reliably measure the kinetics of apo (a) using a stable isotope labeled tracer is expected to facilitate studies of the role of Lp(a) in cardiovascular disease. Since investigators typically determine the isotopic labeling of protein-bound amino acids following acid-catalyzed hydrolysis of a protein of interest [e.g., apo(a)], studies of protein synthesis require extensive protein purification which limits throughput and often requires large sample volumes. We aimed to develop a rapid and efficient method for studying apo(a) kinetics that is suitable for use in studies involving human subjects.
METHODS
Microfluidic device and tandem mass spectrometry were used to quantify the incorporation of [2H3]-leucine tracer into protein-derived peptides.
RESULTS
We demonstrated that it is feasible to quantify the incorporation of [2H3]-leucine tracer into a proteolytic peptide from the non-kringle repeat region of apo(a) in human subjects. Specific attention was directed toward optimizing the multiple reaction monitoring (MRM) transitions, mass spectrometer settings, and chromatography (i.e., critical parameters that affect the sensitivity and reproducibility of isotopic enrichment measurements). The results demonstrated significant advantages with the use of a microfluidic device technology for studying apo(a) kinetics, including enhanced sensitivity relative to conventional micro-flow chromatography, a virtually drift-free elution profile, and a stable and robust electrospray.
CONCLUSIONS
The technological advances described herein enabled the implementation of a novel method for studying the kinetics of apo(a) in human subjects infused with [2H3]-leucine.
Lipoprotein(a) [Lp(a)] has received considerable attention recently as it is known to be an important biomarker for dyslipidemia and is associated with cardiovascular disease.[1] Lp(a) is similar in size and composition to low density lipoprotein (LDL) having a relatively high cholesterol content and apolipoprotein (apo) B100 as a major structural protein. In addition, Lp(a) contains a highly glycosylated protein, apo(a), which is covalently attached to apoB100. Not only do the plasma concentrations of Lp(a) mass vary widely but the mechanisms of assembly, secretion, and catabolism are poorly understood. The ability to quantify the rate of apo(a) synthesis, the defining protein component of Lp(a), in human subjects might help shed light on the mechanism of Lp(a) production and thereby yield a better understanding of how to lower its levels in blood.
Protein kinetic studies frequently involve the administration of a stable isotope labeled amino acid for a given amount of time with subsequent measurements of the incorporation of that precursor into a given protein.[2] The synthesis rate is estimated by comparing the rate of change in isotopic labeling of the target protein over time with that of the precursor amino acid labeling or by determining the amount of time required for the target protein to reach steady-state labeling.[3,4] Although this logic has been used for many years, studies have often been limited to cases in which purified proteins can be obtained in large quantities. For example, the traditional approach generally entails isolating the protein of interest, degrading the isolated protein into its component amino acids (e.g., via hydrolysis in a strong acid), and then measuring the amount of amino acid labeling [e.g., via gas chromatography/mass spectrometry (GC/MS)].[2,5]
Recent advances in proteomic techniques enable the rapid, high-throughput analysis of complex protein mixtures. Therefore, it is possible to envision highly targeted studies of protein kinetics by coupling stable isotope based protocols with novel analytical algorithms, with the potential ultimate goal of global proteome kinetics.[6–9] However, a major challenge associated with the development of such a technique is the ability to adequately measure the incorporation of a tracer molecule into a given protein, or, more specifically, measuring the isotopic enrichment of a selected proteolytic peptide.[9] While this idea may appear straightforward, there are several points that one needs to consider when developing a novel proteome kinetic methodology. First, one can optimize the amount of raw material (e.g., via maximizing plasma volume) used for extraction and minimized dilution factors [e.g., via employing micro-flow liquid chromatography/mass spectrometry (LC/MS) methods]. Second, one can examine other factors, including (i) the identification of proteolytic peptides that yield the highest mass spectrometric response, and (ii) carefully selecting multiple reaction monitoring (MRM) transitions for the purpose of measuring the incorporation of [2H3]-leucine (tracer) into apo(a). Although prior studies have described methods for studying apo(a) kinetics,[10–13] several days are typically required to prepare a limited set of samples.
EXPERIMENTAL
Subjects and infusion protocol
Individuals were enrolled in an ongoing Merck-sponsored phase II study examining the effects of a cholesterol ester transfer protein (CETP) inhibitor, anacetrapib, on lipoprotein kinetics (unpublished data on file; Merck Sharp & Dohme Corp.). To establish proof of concept for the methodology, apo(a) kinetics were examined in three subjects who did not receive anacetrapib treatment. All subjects were recruited at Columbia University Medical Center (New York City, NY, USA) wherein each subject received a primed infusion of [5,5,5-2H3]-L-leucine as previously described.[14] Briefly, while consuming a low-fat liquid diet every 2 h, subjects were given a bolus injection of [5,5,5-2H3]-L-leucine (10 μmol/kg body weight) immediately followed by a constant infusion of [5,5,5-2H3]-L-leucine (10 μmol/kg body weight/h) for 15 h. Blood samples were collected at 0, 2, 4, 6, 8, 10, 12, and 15 h and stored at −80 °C until analysis. The Columbia University Investigational Review Committee approved the study protocol. Informed consent was obtained from each of the study participants before the commencement of study procedures.
Sample preparation
The majority of Lp(a) particles (~95%) are located in the density range between 1.019 and 1.21 g/mL following ultracentrifugation of plasma samples.[15] To enrich plasma samples in apo(a) relative to other abundant plasma proteins, such as albumin, equal volumes (62.5 μL) of ultracentrifuged fractions in the low-density lipoprotein (LDL) (1.019–1.063 g/mL) and high-density lipoprotein (HDL) density ranges (1.063–1.21 g/mL) were combined. The combined ultracentrifuged fractions were desalted and the buffer was exchanged using 3 mL of 50 mM ammonium bicarbonate (pH ~8) and a 5-kDa molecular weight cut-off (MWCO) filter. The retentate (final volume ~100 μL) was mixed with 88 μL of 50 mM ammonium biocarbonate, 10 μL of 10% sodium deoxycholate (SDC), and 2 μL of 500 mM dithiothreitrol (DTT), and subsequently incubated at 60 °C for 30 min. Following incubation, 6.7 μL of 300 mM iodoacetamide was added to the samples followed by incubation at room temperature in the dark for 60 min. Finally, 30 μL of 0.5 mg/mL trypsin Gold (Promega Corp., Madison, WI, USA) was added to each sample and incubated overnight at 37 °C. Protein digestion was terminated by the addition of 10 μL of 20% formic acid. Samples were mixed well to precipitate SDC, which was subsequently removed via centrifugation (16 100 relative centrifugal force for 5 min). The samples were filtered (0.22-μm spin filter; EMD Millipore Corp., Bellerica, MA, USA) prior to LC/MS/MS analysis. The final filtration step was critical for prolonging the lifetime of the microfluidic device.
Measurement of apo(a): LC/MS/MS for determining isotopic enrichment
A MRM method was used to monitor the precursor product ion transitions for the unlabeled and labeled forms: 786.5 → 1069.6 (unlabeled → unlabeled), 788 1069.6 (labeled → unlabeled) and 788 1072.6 → (labeled → labeled). The digested samples (10 μL) → were first tested on a triple quadrupole mass spectrometer (TSQ Vantage, Thermo Fisher) coupled with a nanoAcquity ultra-performance liquid chromatography (UPLC) system (Waters) and a CaptiveSpray ionization source (Michrom Bioresources). The separation was achieved using a Magic C18 column (3 μm, 500 μm × 50 mm), which was maintained at 50 °C. The gradient was 97% A (0.1% formic acid in water)/3% B (0.1% formic acid in acetonitrile) ramped linearly to 10% A at 8 min, then re-equilibrated at initial condition (total run time: 10 min; flow rate: 15 μL/min). The settings on the quadrupoles were 0.3 full width at half maximum (FWHM) on Q1, 0.5 FWHM on Q3. The collision energy was 19 eV and S-lens was 120.
In addition, digested samples (3 μL) were also partial-loop injected onto a microfluidic device (150 μm × 100 mm packed with BEH C18, 1.7-μm particle, prototype obtained from Waters) using a nanoAcquity UPLC system interfaced with a triple quadrupole mass spectrometer (Xevo TQS, Waters). The gradient was 97% A (0.1% formic acid in water)/3% B (0.1% formic acid in acetonitrile) ramped linearly to 10% A at 5 min, then re-equilibrated at initial condition (total run time: 8 min; flow rate: 3 μL/min). The microfluidic device was maintained at 60 °C throughout the chromatographic gradient elution. An infusion microfluidic device (no packing material) was used to optimize emitter position, signal intensity, and transitions. The precursor ion (m/z 786.5, 2+) and product ion (m/z 1069.6 and 1072.6, 1+) were monitored with a Q1 low-mass resolution of 2.8 and a high-mass resolution of 14.7 (equivalent of 0.7 FWHM), collision energy of 25 eV, span set 0.2, and scan time of 17 ms.
For LC/MS method evaluation, both natural and 2H3-labeled peptide LFLEPTQADIALLK (2H3 labeled at N-terminal second leucine) were synthesized (New England Peptide). 100 nM of the natural peptide was serial diluted with pooled rhesus plasma digests, and M3/M0 ratio of the peptide was measured by LC/MS/MS with n = 6 replicate injections. Rhesus plasma digest was used as sample matrix because it resembled the complexity of human plasma digest and rhesus apo(a) did not produce the same LFLEPTQADIALLK peptide sequence. To evaluate the accuracy for measuring changes in enrichment, equimolar amounts of labeled and natural peptides were diluted with rhesus plasma digests to a final concentration of 10 nM. Then, the mixture was 2-fold serially diluted with 10 nM of the natural peptide solution to achieve expected enrichment ranging between 0 to 6.25%. These samples were analyzed with n = 4 replicate injections.
Measurement of VLDL apoB100 leucine asymptotic enrichment
ApoB100 enrichment was analyzed by the IDOM Metabolic Tracer Resource at the University of Pennsylvania as previously described.[16] Briefly, apoB100-containing lipoprotein particles in the density range associated with very low density lipoprotein (VLDL) (density range <1.006 g/mL) were isolated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and hydrolyzed to constituent amino acids with HCl. Amino acids were purified using cation-exchange chromatography, derivatized to heptafluorobutyryl isobutyl esters and subsequently analyzed by GC/MS to determine [5,5,5-2H3]-L-leucine enrichment. The asymptotic enrichment of VLDL apoB [5,5,5-2H3]-L-leucine was estimated using compartmental modeling as previously described.[2]
Measurement of Lp(a) concentrations
Lp(a) protein levels were measured using an enzyme-linked immunosorbent assay (ELISA)[17] technique at Northwest Lipid Research Laboratories, University of Washington. The assay measures the non-repeating region of Lp(a).
Kinetics calculations
Studies examining protein synthesis require calculation of the following critical values: (i) an estimate of the pool size of the protein of interest (which is often inferred by multiplying its concentration by its volume of distribution) and (ii) an estimate of the fractional synthesis rate (FSR) of the protein of interest.[3,18] The FSR is typically estimated using one of two general approaches. First, if the protein turnover is rapid enough during the time course of the tracer study, one can model the rate of change in protein labeling as it approaches steady-state labeling (e.g., using a single exponential curve) and therein obtain the FSR:
| (1) |
The equation noted above describes the general scenario for a single well-mixed compartment. Foster et al.[3] discussed specific considerations for studies of lipoprotein kinetics, including delays to account for secretion. A second approach for estimating FSR, which is often used in cases when the turnover of the protein is slow and, therefore, the labeling does not approach steady state (i.e., when there is a pseudo-linear change in the labeling), is to compare the protein labeling with a measure of the precursor labeling:
| (2) |
In cases where labeled leucine is infused one can substitute various values for the term ‘precursor labeling’. The most obvious endpoint to consider is the labeling of plasma leucine (i.e., plasma 2H3-leucine enrichment). However, this will likely always lead to an underestimate of the true FSR. For example, one expects a gradient (or dilution) between the plasma labeling and that of the true intracellular labeling of many amino acids due to dilution by intracellular free amino acids generated by protein turnover. Thus, some investigators have proposed to use different surrogates to represent the intracellular labeling (e.g., in certain instance the α-keto acid of leucine, α-keto-isocaproate, is assumed to be a more accurate surrogate of the true intracellular leucine labeling).[19] Another approach is to measure the isotopic enrichment of a protein that was made in the same cells as the product of interest but has a much faster turnover time. The rationale is that its labeling will approximate a steady state and therein reflect the true labeling of the intracellular amino acid.[20] In our study we used the latter approach, the steady-state enrichment of leucine in VLDL-apoB was used as a measure of ‘precursor labeling’ in Eqn. (2).[2] The ‘change in product labeling’ was determined using a linear regression analyses between 6 and 15 h. Because there were two leucines in the product ion sequence, the measured M3/M0 labeling ratio was divided by 2 prior to linear regression analysis.[21] Finally, it should be noted that under metabolic steady-state conditions, in which the pool size of a given product is constant over the course of the tracer study, the fractional synthetic rate (FSR) is equivalent to the fractional catabolic rate (FCR).[5]
Using the logic outlined above, the production rate (PR) of apo(a) is the product of the FSR and the apo(a) pool size.[11]
RESULTS
Selection of peptides and MRM transitions
The ability to measure the synthetic rate of a protein depends on several factors. First, the degree of precursor labeling determines the potential maximum increase in product labeling. From an analytical perspective, it becomes easier to measure protein synthesis when greater quantities of tracer are administered during an experiment. Since there are often limits regarding the quantity of tracer that can be administered, the identification of peptides containing multiple copies of the precursor amino acid may help by amplifying the expected amount of product labeling.[21,22] Second, the faster the synthesis rate the greater the change in product labeling at any given sampling time. Third, a good signal-to-noise (S/N) ratio associated with the analytical measurement will improve the measurement precision. Finally, minimizing the natural background will enhance the technique’s ability to detect small changes in product labeling. In the present case, where the infusion protocol and sampling intervals were predetermined in the study protocol, we aimed to optimize the analytics by improving the S/N ratio of the measurement and minimizing the contribution from natural background noise.
Since apo(a) has been shown to possess a relatively slow FSR,[13] the methodology employed to measure apo(a) kinetics must have an adequate S/N ratio to allow for a reliable measurement of labeled product. To this end, we selected a protein-specific apo(a) peptide sequence that contains four leucines, i.e., LFLEPTQADIALLK (Fig. 1(A)). As noted above, the presence of multiple leucines in this peptide sequence will enhance total 2H3-leucine enrichment compared with a peptide containing a single leucine, thereby allowing for a more reliable quantification of product labeling.[21,22] In addition, the peptide LFLEPTQADIALLK yields a high MRM signal due to a proline in the peptide sequence at the y10+ product ion (Fig. 1(B)). Figure 1(C) illustrates that both the y10+ and the b4+ product ions can be used to quantify 2H3-leucine enrichment; however, the y10+ product ion provided a superior signal response and less background noise compared with the b4+ ion. Adequate sensitivity of the measurement is needed given the wide concentration range of apo(a) in human plasma.[1]
Figure 1.
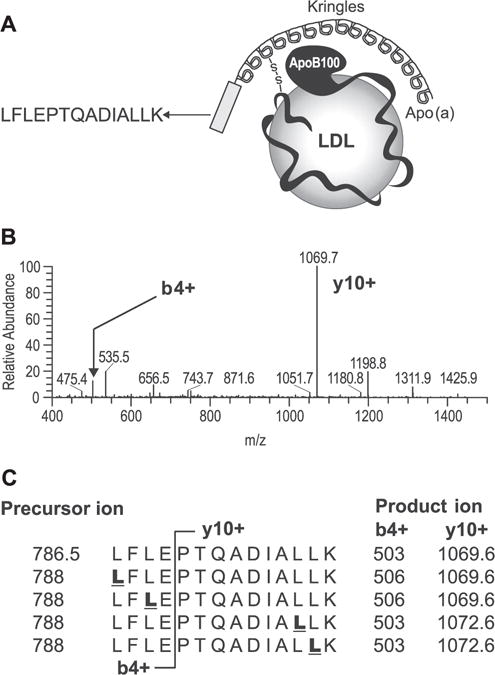
Selection of peptides and MRM transitions for quantifying 2H3-leucine enrichment in plasma apo(a). (A) The kringles are identical repeats, and the peptide LFLEPTQADIALLK was selected as the best candidate because it was in the non-repeating region and unique to apo(a). The peptide also contained multiple leucines and produced high MRM intensity. (B) MS/MS spectra of peptide LFLEPTQADIALLK (m/z 786.5, 2+) show that the most intense product ion was y10+ (PTQADIALLK). (C) Both y10+ and b4+ will allow quantification of 2H3-leucine enrichment. However, product ion y10+ gave better signal-to-noise than b4+. Underlined bold letters in the sequence indicate potential 2H3-leucine-labeled sites.
The effects of the first quadrupole (Q1) resolution and different MRM transitions were tested in an effort to minimize the natural background noise mainly contributed by the 13C isotope. For example, for the peptide LFLEPTQADIALLK, the most intense precursor → product ion transition was doubly charged (m/z 786.5) to singly charged y10+ (m/z 1069.6). Since the peptide contained four leucine residues, one could set up MRM transitions to quantify the incorporation of 2H3-leucine into one of the two leucine residues, closer to the N-terminus (788 → 1069.6), or one of the two leucine residues closer to the C-terminus of the peptide (788 → 1072.6). Since one assumes that the peptide is synthesized de novo (and that fragments are not recycled during degradation and synthesis), we expected that the enrichment kinetics of these two settings should be identical. However, when the Q1 resolution was set at 0.2 or 0.4 FWHM, the transition 788 → 1069.6 resulted in ~6 times less natural background than the transition 788 → 1072.6 (Figs. 2(A) and 2 (B), respectively). Under these conditions, one might expect that it would be better to use the transition with the lower natural background (i.e., 788 → 1069.6) versus the other transition (i.e. 788 → 1072.6). When the Q1 resolution was set at 0.7 FWHM, both transitions (i.e., 788 → 1069.6 and 788 → 1072.6) were associated with similar natural background (Fig. 2(C)). Our observations are in strong agreement with those previously reported by MacCoss and colleagues.[23]
Figure 2.
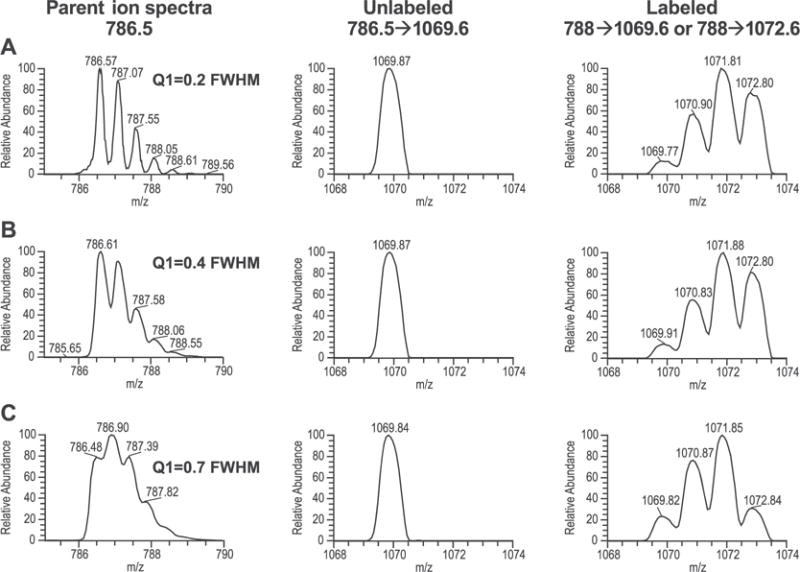
Effect of the first quadrupole resolution on the background of M3/M0 ratio. Peptide LFLEPTQADIALLK was infused into mass spectrometer under conditions in which the resolution on the third quadrupole (Q3) was fixed at 0.7 FWHM and the resolution for the first quadrupole (Q1) was varied. Precursor ion and MRM spectra are shown for cases in which Q1 was varied from 0.2, 0.4 and 0.7 FWHM (A, B and C, respectively). Setting Q1 resolution to 0.2 or 0.4 resulted in the background for transition 788 → 1072.6 approximately 6 times higher than transition 788 → 1069.6. In contrast, when the Q1 resolution was set to 0.7 (unit resolution), comparable background was observed for both transitions, but higher than that obtained with higher Q1 resolution.
From the exercise described above, it seemed reasonable to set the Q1 resolution at 0.2 or 0.4 FWHM. However, a sizeable decrease in the signal was observed when 0.2 or 0.4 FWHM was used compared with 0.7 FWHM when tested on both mass spectrometers (i.e., TSQ Vantage and Xevo TQS). Furthermore, changes in measured isotope ratios for the same sample were observed over 24 h. This finding is probably due to the fact that the mass accuracy for these instruments (triple quadrupole) does not remain constant for a long period, and the degree of deviation will be highly dependent on the laboratory environment when Q1 resolution is set at 0.2 or 0.4 FWHM. As a result, the analyses would have to be interrupted to perform daily mass calibrations. Such methodological limitations are not practical when analyzing hundreds to thousands of samples in a clinical environment. To test whether similar data could be obtained using a unit resolution on Q1 for apo(a) kinetics measurements, the analysis was performed using two transitions and two different resolution settings on samples from a subject who had been infused with [2H3]-leucine. As illustrated in Fig. 3, regardless of the Q1 settings and transitions used, all analyses yielded similar slopes (i.e., rates of change in the product labeling) when the data points were fitted using a linear regression line. This finding indicated that we could use unit resolution setting for the measurement. The p value determined from the analysis of covariance testing of the slopes was non-significant at 0.9867.
Figure 3.
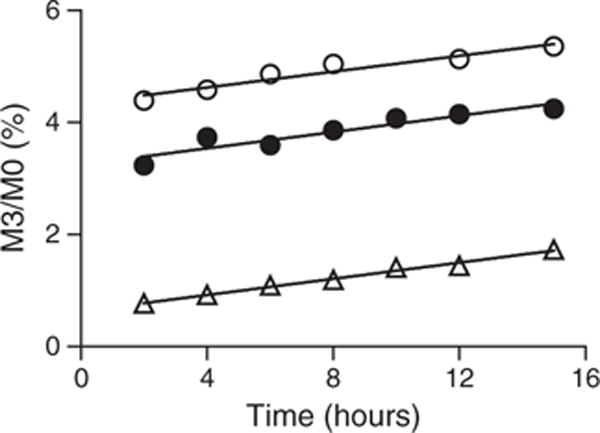
Effect of Q1 resolution on the measurement of 2H3-leucine enrichment. Samples from a subject were digested and analyzed using microfluidic device with the Xevo TQS mass spectrometer. Open triangles: transition 788 → 1069.6, and Q1 was set at 0.4 FWHM; closed circles: transition 788 → 1069.6, and Q1 was 0.7 FWHM; open circles: transition 788 → 1072.6, and Q1 was 0.7 FWHM. The data were analyzed using linear regression fit. The slopes were 0.07199 ± 0.003533 (open triangles), 0.07267 ± 0.01209 (closed circles), and 0.0706 ± 0.009510 (open circles), respectively. Analysis of covariance test yielded a p value of 0.9867, indicating that the three plots had similar slopes (i.e., rates of change in the product labeling).
Sensitivity and accuracy of 2H3-leucine enrichment measurement
We employed an MRM method to take advantage of the S/N gain from precursor to product selection. To estimate the lower limit of quantification (LLOQ) for the natural M3/M0 ratio measurement, known amounts of natural LFLEPTQADIALLK peptide were serially diluted with pooled rhesus plasma digests to yield a concentration range of ~0.4 to 200 nM. The samples were analyzed using the method described above (n = 6 replicate injections per standard). The LLOQ for the natural M3/M0 ratio measurement was determined as ~1.6 nM, where the precision and accuracy was below 10% (Fig. 4(A)).
Figure 4.
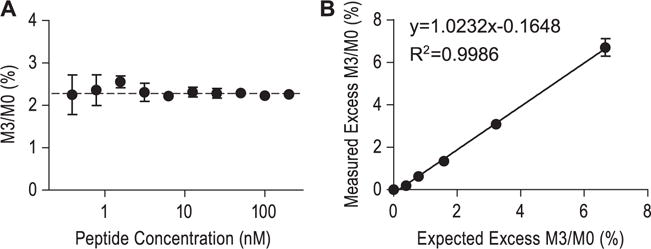
Performance of leucine enrichment measurement using microfluidic device with tandem mass spectrometry. (A) To estimate the lower limit of quantification, natural peptide LFLEPTQADIALLK was serially diluted using pooled rhesus plasma digests and then analyzed by LC/MS (n = 6 LC/MS injections per sample). The dashed line represents the expected natural abundance M3/M0 ratio in the sample. The lower limit of quantification was 1.6 nM where both the precision and the accuracy were below 10% coefficient of variation. (B) To examine the possibility of quantifying [2H3]-leucine enrichment, synthetic peptide standards of known enrichment were analyzed. The measured enrichments were plotted against expected enrichments. The correlation coefficient r2 was 0.9986, and the slope of the linear regression curve was 1.0232, demonstrating that this LC/MS method is capable of quantifying low levels of leucine enrichment in peptides present in a complex sample matrix.
To gain confidence in these ratio measurements, we constructed a standard curve by adding 10 nM of the natural peptide and variable amounts of labeled peptide into rhesus plasma digests. An excellent agreement was seen between the measured and expected enrichments (n = 4 replicate LC/MS injections; the correlation coefficient r2 and the slope of the linear regression analysis was 0.9986 and 1.0232, respectively), confirming that the method is able to reliably quantify low levels of labeling in a complex plasma matrix (Fig. 4(B)).
Reproducibility and robustness
In general, it is necessary to collect multiple samples from an individual subject over a defined time interval in order to obtain adequate kinetic information. The methodologies employed by kinetic studies need be both sensitive (i.e., capable of detecting small amounts of label in low abundance proteins) and robust. To this end, 10 nM labeled peptide (spiked in pooled rhesus plasma digests) was used as quality controls and analyzed alongside subject samples. Given the total number of samples analyzed in this study (~800), the quality control samples added 13 additional injections over the entire sequence. The variability of peptide peak area (M0: 786.5 → 1069.6) during the course of 800 LC/MS/MS injections (i.e., spanning 6 days of uninterrupted analyses) was approximately 3.5% (Fig. 5(A)). Presumably this high degree of robustness reflects the stable backpressure of the microfluidic device (Fig. 5(B)) coupled with virtually no retention time shift (Fig. 5(C)). In addition, the shapes of the peak profiles were nearly identical between the first and last injections (data not shown).
Figure 5.
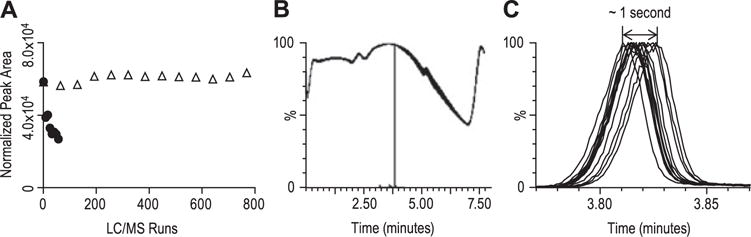
Reproducibility and robustness of the microfluidic device with tandem mass spectrometry. The peptide LFLEPTQADIALLK was synthesized with a 2H3-label on the second leucine from the N-terminus. Labeled peptide (10 nM) was added to rhesus plasma digests and used as quality controls, which was analyzed along with plasma samples from human subjects (800 LC/MS injections). (A) The variability of peptide peak area (M0: 786.5 → 1069.6): the plot with closed circles was from CaptiveSpray-TSQ Vantage, and open triangles was from the microfluidic device and the Xevo TQS. Using the CaptiveSpray source, the peak area decreased 50% within 50 LC/MS injections, and eventually the ion transfer tube became clogged. Using microfluidic device ionization source, the CV of the peak area was approximately 3.5% in the course of 800 LC/MS injections. (B) Overlay of backpressure traces of all QC samples, which was stable. (C) Overlay of selected ion chromatograms of all QC samples. The retention time shift was within 1 s.
It is important to note that although the entire LC/MS analysis reported herein was performed on a single prototype microfluidic device, the overall device-to-device performance was found to be fairly reproducible.[24] In comparison, signal intensities dropped ~50% after the first 50 LC/MS/MS injections when the CaptiveSpray-TSQ Vantage system was used as compared with the microfluidic device (Fig. 5(A)). Continued analysis using the CaptiveSpray-TSQ Vantage system quickly resulted in a complete loss of signal due to clogging of the ion transfer tube (unpublished observations).
Apo(a) kinetics measurement in individual subjects
To demonstrate proof-of-concept for this novel methodology, the LC/MS method was used to measure apo(a) kinetics in three subjects. As shown in Fig. 6, over the course of the tracer infusion protocol, the total change in 2H3-leucine labeling in these three subjects was below 1.5%. As shown in Table 1, the calculated FSRs were 0.128, 0.164, 0.368 pools/day, respectively, and the PRs were 1.22, 1.47, 1.14 nmol/kg/day, respectively. These values are consistent with reports in the literature in which other investigators have used more traditional GC/MS methods.[10–13]
Figure 6.
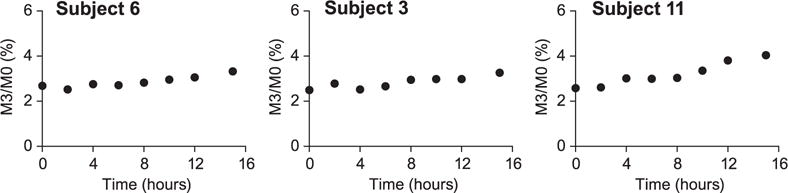
Measurement of 2H3-leucine labeling in apo(a) in three subjects. Samples were analyzed from humans displaying a range of plasma apo(a) levels (211.9, 199.7, 68.8 nM, respectively). The LDL and HDL lipoprotein fractions were prepared by ultracentrifugation and then combined for a given subject and trypsinized into peptides, and analyzed using the microfluidic device and the Xevo TQS using unit resolution on both Q1 and Q3 quadrupoles.
Table 1.
Plasma apo(a) concentrations, fractional synthesis rate (FSR), and production rate (PR) from three human subjects. Note that the change in isotope labeling of apo(a) reflects the incorporation of two leucines; therefore, calculation of FSR requires dividing by 2 (see Experimental section). The literature values reported here were expressed as FCR, which we have assumed a metabolic steady state (i.e., FSR = FCR) for purposes of comparison
| apo(a) | Subject 6 | Subject 3 | Subject 11 | Literature values |
|---|---|---|---|---|
| Plasma concentration (nM) | 211.9 | 199.7 | 68.8 | |
| Change in isotope labeling (slope/h) | 0.062 | 0.056 | 0.129 | |
| VLDL-apoB leucine enrichment (%) | 5.25 | 4.57 | 4.20 | |
| FSR (pools/day) | 0.128 | 0.164 | 0.368 | 0.22; 0.24; 0.25a |
| production rate (nmol/kg/day) | 1.22 | 1.47 | 1.14 |
DISCUSSION
It is possible to rigorously examine human biology by coupling the administration of stable isotope tracer protocols with advances in mass spectrometry and closely related areas of analytical chemistry (e.g., chromatography methods). Several reports in the literature have demonstrated the ability to readily and reliably quantify isotopic labeling of multiple analytes present in complex mixtures.[8,9,25,26] In some cases, such novel approaches have used either unbiased or untargeted designs. For example, we recently demonstrated that one can obtain information regarding lipidomic flux by utilizing an MSE logic.[27,28] Others have demonstrated the ability to quantify proteome kinetics using high-resolution mass spectrometry.[8] Although reports of untargeted analyses are effective in certain scenarios, the ability to quantify targeted kinetic profiles is of equal importance.[9]
In the current study, we tested certain fundamental issues involved in conducting studies of targeted protein kinetics which rely on LC/MS/MS-based analyses of proteolytic peptides. A critical consideration for most analyses involves the specificity of the analyte under measurement. One way to build confidence regarding the ability of a method to identify the analyte of interest is to alter the MS resolution settings. For example, even quadrupole-based mass spectrometers typically allow some options for mass resolution settings (sometimes referred to simply as ‘low’ and ‘high’ resolution acquisition). As the resolution increases, the specificity increases, which results in less interference and lower background yet the sensitivity decreases because of reduced ion transmission. We explored this approach by using high Q1 resolution settings (FWHM of 0.2 or 0.4 Da). However, these settings required frequent instrument calibrations and intensive quality control monitoring to maintain the mass accuracy of the MRM transitions of interest. Although this logic is not inherently flawed,[23] the practical implementation of such an approach is less than ideal for studies involving large patient cohorts. The results of our study show that the resolution setting can have a substantial impact on the apparent isotope labeling, and thus is a critical parameter that deserves attention for some analyses. Even with Q1 resolution setting at 0.7 FWHM, the measured isotope ratios can vary over months. For example, the measured natural abundance M3/M0 ratio at t0 in Fig. 3 (closed circles) was 3.2%, but the t0 samples measured in Fig. 6 were close to 2.6%. As shown in Fig. 3, although the measured natural abundance M3/M0 ratio ranged from 0.7 to 4.4% (depending on the instrument settings), we were able to reliably quantify the change in labeling over time. Presumably, since all time points from the same subjects were analyzed in a sequence, and as long as the ratio measurement was stable over those analysis periods, the data acquired would still be reliable.
An alternative approach for influencing the specificity of the analysis is the chromatographic peak capacity. In general, good chromatographic fidelity will ensure adequate separation of co-eluting biomolecules and minimize interference in the absence of high resolution mass spectral separation. How to best achieve good chromatography is expected to vary on a case-by-case scenario. In the present study, we demonstrated that a prototype microfluidic device with sub-2-μm particles provided excellent stability and reproducibility for apo(a) measurements. The major disadvantages of traditional micro-flow and nano-flow separations are the high complexity of use and limited robustness of these techniques. The microfluidic device technique enables the chromatographic column and ion-spray emitter to be built under the same design casing, thus removing the need for manual connections and finger-tight fittings and thereby reducing dead volumes and flow inconsistencies that have had a direct impact on chromatographic fidelity.
A final consideration regarding studies of targeted protein kinetics involves determining which transition(s) to monitor. Although it is obvious that a candidate peptide must contain at least one copy of the infused precursor amino acid, the impact of utilizing fragments that contain multiple copies of the infused precursor may be underappreciated.[21] Proteins are effectively biopolymers of amino acids and, at a very basic level, one can ascribe the labeling profiles to a binomial distribution in cases where a single-labeled amino acid is administered (e.g., [2H3]-leucine).[22] Thus, the ability to identify proteolytic peptides and/or fragment ions that contain multiple copies of the infused amino acid increases the experimental window when measuring change in enrichment over time. Although a noticeable shift in the abundance of different mass isotopomers will be observed with increased enrichment of the precursor labeling,[22] a detectable shift in the M3 isotopomer is almost exclusively expected for cases such as the one described in this report. For example, since the intracellular tRNA leucine (i.e., the true precursor pool) reached ~5% 2H3 (Table 1, leucine labeling of VLDL-apoB is used as a surrogate), the maximum shift in the M3 and M6 isotopomers should be ~20 and ~2%, respectively, whereas the shift in M9 and M12 is expected to be <0.1% for each and therefore virtually nondetectable.[21] The calculated data above assume the shift in labeling of the precursor ion; however, the fragment ion that we monitored contained only two of the four leucine residues so the expected maximal increases in labeling would be further reduced. The fact that we only observed a small increase in the isotopic labeling (i.e., M3 ~1.0 to 1.5% over the natural background, Fig. 6) is consistent with the fact that apo(a) has a relatively slow turnover rate.[13]
In summary, the results reported herein describe a novel approach for investigating protein kinetics in vivo in human subjects. The findings outlined in this report can be utilized by other researchers to further expand knowledge in the field. We expect that the methodology and data described in this report can offer a specific advantage in studies aimed at elucidating the potential role of apo(a) in dyslipidemia and cardiovascular disease.[1] As we have shown, it is possible to readily and reliably determine the incorporation of a labeled amino acid into a distinct apo(a)-derived peptide using a fairly high-throughput method.
Acknowledgments
The authors thank Professor Santica M. Marcovina (University of Washington) for performing apo(a) ELISA measurements, as well as Jay Johnson and James Murphy from Waters Corporation for their technical support. In addition, the authors wish to thank Dr. Amy O. Johnson-Levonas and Kathleen Newcomb of Merck Sharp & Dohme, Corp. for their assistance with the editing and preparation of this manuscript for publication. Supported, in part, by the National Center for Research Resources, Grant UL1RR024134 (now at the National Center for Advancing Translational Sciences, Grant UL1TR000003).
References
- 1.Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichtenstein AH, Cohn JS, Hachey DL, Millar JS, Ordovas JM, Schaefer EJ. Comparison of deuterated leucine, valine, and lysine in the measurement of human apolipoprotein A-I and B-100 kinetics. J Lipid Res. 1990;31:1693. [PubMed] [Google Scholar]
- 3.Foster DM, Barrett PH, Toffolo G, Beltz WF, Cobelli C. Estimating the fractional synthetic rate of plasma apolipoproteins and lipids from stable isotope data. J Lipid Res. 1993;34:2193. [PubMed] [Google Scholar]
- 4.Ramakrishnan R. Studying apolipoprotein turnover with stable isotope tracers: correct analysis is by modeling enrichments. J Lipid Res. 2006;47:2738. doi: 10.1194/jlr.M600302-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parhofer KG, Hugh P, Barrett R, Bier DM, Schonfeld G. Determination of kinetic parameters of apolipoprotein B metabolism using amino acids labeled with stable isotopes. J Lipid Res. 1991;32:1311. [PubMed] [Google Scholar]
- 6.Price JC, Holmes WE, Li KW, Floreani NA, Neese RA, Turner SM, Hellerstein MK. Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Anal Biochem. 2012;420:73. doi: 10.1016/j.ab.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Reckow S, Webhofer C, Boehme M, Gormanns P, Egge-Jacobsen WM, Turck CW. Proteome scale turnover analysis in live animals using stable isotope metabolic labeling. Anal Chem. 2011;83:1665. doi: 10.1021/ac102755n. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H, Li W, Wang SP, Mendoza V, Rosa R, Hubert J, Herath K, McLaughlin T, Rohm RJ, Lassman ME, Wong KK, Johns DG, Previs SF, Hubbard BK, Roddy TP. Quantifying apoprotein synthesis in rodents: coupling LC-MS/MS analyses with the administration of labeled water. J Lipid Res. 2012;53:1223. doi: 10.1194/jlr.D021295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rader DJ, Cain W, Zech LA, Usher D, Brewer HB., Jr Variation in lipoprotein(a) concentrations among individuals with the same apolipoprotein (a) isoform is determined by the rate of lipoprotein(a) production. J Clin Invest. 1993;91:443. doi: 10.1172/JCI116221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenner JL, Seman LJ, Millar JS, Lamon-Fava S, Welty FK, Dolnikowski GG, Marcovina SM, Lichtenstein AH, Barrett PH, deLuca C, Schaefer EJ. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005;54:361. doi: 10.1016/j.metabol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Frischmann ME, Kronenberg F, Trenkwalder E, Schaefer JR, Schweer H, Dieplinger B, Koenig P, Ikewaki K, Dieplinger H. In vivo turnover study demonstrates diminished clearance of lipoprotein(a) in hemodialysis patients. Kidney Int. 2007;71:1036. doi: 10.1038/sj.ki.5002131. [DOI] [PubMed] [Google Scholar]
- 13.Demant T, Seeberg K, Bedynek A, Seidel D. The metabolism of lipoprotein(a) and other apolipoprotein B-containing lipoproteins: a kinetic study in humans. Atherosclerosis. 2001;157:325. doi: 10.1016/s0021-9150(00)00732-2. [DOI] [PubMed] [Google Scholar]
- 14.Millar JS, Brousseau ME, Diffenderfer MR, Barrett PH, Welty FK, Cohn JS, Wilson A, Wolfe ML, Nartsupha C, Schaefer PM, Digenio AG, Mancuso JP, Dolnikowski GG, Schaefer EJ, Rader DJ. Effects of the cholesteryl ester transfer protein inhibitor torcetrapib on VLDL apolipoprotein E metabolism. J Lipid Res. 2008;49:543. doi: 10.1194/jlr.M700268-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Fless GM, Rolih CA, Scanu AM. Heterogeneity of human plasma lipoprotein (a). Isolation and characterization of the lipoprotein subspecies and their apoproteins. J Biol Chem. 1984;259:11470. [PubMed] [Google Scholar]
- 16.Millar JS, Maugeais C, Ikewaki K, Kolansky DM, Barrett PH, Budreck EC, Boston RC, Tada N, Mochizuki S, Defesche JC, Wilson JM, Rader DJ. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. 2005;25:560. doi: 10.1161/01.ATV.0000155323.18856.a2. [DOI] [PubMed] [Google Scholar]
- 17.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a) Clin Chem. 1995;41:246. [PubMed] [Google Scholar]
- 18.Wolfe RR, Chinkes DL. Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analyses. Wiley-Liss; Hoboken, NJ: 2005. [Google Scholar]
- 19.Belloto E, Diraison F, Basset A, Allain G, Abdallah P, Beylot M. Determination of protein replacement rates by deuterated water: validation of underlying assumptions. Am J Physiol Endocrinol Metab. 2007;292:E1340. doi: 10.1152/ajpendo.00488.2006. [DOI] [PubMed] [Google Scholar]
- 20.Jahoor F, Burrin DG, Reeds PJ, Frazer M. Measurement of plasma protein synthesis rate in infant pig: an investigation of alternative tracer approaches. Am J Physiol. 1994;267:R221–R227. doi: 10.1152/ajpregu.1994.267.1.R221. [DOI] [PubMed] [Google Scholar]
- 21.Brunengraber H, Kelleher JK, DesRosiers C. Applications of mass isotopomer analysis to nutrition research. Annu Rev Nutr. 1997;17:559. doi: 10.1146/annurev.nutr.17.1.559. [DOI] [PubMed] [Google Scholar]
- 22.Papageorgopoulos C, Caldwell K, Shackleton C, Schweingrubber H, Hellerstein MK. Measuring protein synthesis by mass isotopomer distribution analysis (MIDA) Anal Biochem. 1999;267:1. doi: 10.1006/abio.1998.2958. [DOI] [PubMed] [Google Scholar]
- 23.Tomazela DM, Patterson BW, Hanson E, Spence KL, Kanion TB, Salinger DH, Vicini P, Barret H, Heins HB, Cole FS, Hamvas A, MacCoss MJ. Measurement of human surfactant protein-B turnover in vivo from tracheal aspirates using targeted proteomics. Anal Chem. 2010;82:2561. doi: 10.1021/ac1001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waters Corporation. Chromatographic reproducibility of TRIZAIC nanoTile technology [serial online] 2012 Available: http://www.waters.com/waters/library.htm?cid=511436&lid=134652984 (accessed January 7, 2013)
- 25.Bereman MS, Tomazela DM, Heins HS, Simonato M, Cogo PE, Hamvas A, Patterson BW, Cole FS, MacCoss MJ. A method to determine the kinetics of multiple proteins in human infants with respiratory distress syndrome. Anal Bioanal Chem. 2012;403:2397. doi: 10.1007/s00216-012-5953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacCoss MJ, Wu CC, Matthews DE, Yates JR., III Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal Chem. 2005;77:7646. doi: 10.1021/ac0508393. [DOI] [PubMed] [Google Scholar]
- 27.Castro-Perez J, Previs SF, McLaren DG, Shah V, Herath K, Bhat G, Johns DG, Wang SP, Mitnaul L, Jensen K, Vreeken R, Hankemeier T, Roddy TP, Hubbard BK. In vivo D2O labeling to quantify static and dynamic changes in cholesterol and cholesterol esters by high resolution LC/MS. J Lipid Res. 2011;52:159. doi: 10.1194/jlr.D009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castro-Perez JM, Roddy TP, Shah V, McLaren DG, Wang SP, Jensen K, Vreeken RJ, Hankemeier T, Johns DG, Previs SF, Hubbard BK. Identifying static and kinetic lipid phenotypes by high resolution UPLC-MS: unraveling diet-induced changes in lipid homeostasis by coupling metabolomics and fluxomics. J Proteome Res. 2011;10:4281. doi: 10.1021/pr200480g. [DOI] [PubMed] [Google Scholar]