

Abstract
The L-iduronic acid (IdoA) residue is a critically important structural component in heparan sulphate polysaccharide for the biological functions. The pyranose ring of IdoA is present in 1C4-chair, 2SO-skew boat, and less frequently, in 4C1-chair conformations. Here, we analyzed the conformation of IdoA residue in eight hexasaccharides by NMR. The data demonstrate a correlation between the conformation of IdoA and sulphations in the surrounding saccharide residues. For the 2-O-sulpho IdoA residue, a high degree of sulphation on neighboring residues drives ring dynamics towards the 2SO-skew boat conformer. In contrast, the nonsulphated IdoA residue is pushed towards the 1C4-chair conformer when the neighboring residues are highly sulphated. Our data suggest that the conformation of IdoA is regulated by the sulphation pattern of nearby saccharides that is genetically controlled by the heparan sulphate biosynthetic pathway.
Heparan sulphate (HS) is a polysaccharide that consists of disaccharide repeating units of a glucuronic acid (GlcA) or iduronic acid (IdoA) residue linked to a glucosamine (GlcN) residue. Capable of carrying sulpho groups by IdoA, GlcA and GlcN residues, HS is a highly negatively charged macromolecule under physiological conditions. HS participates in numerous physiological and pathophysiological processes, such as embryonic development, inflammatory responses, blood coagulation and viral/bacterial infections1,2,3. Most notably, heparin, a special form of HS containing higher sulphation and IdoA levels, is a commonly used anticoagulant drug in clinics for treatment of patients with thrombotic disorders4.
The molecular mechanism that allows HS to interact with specific protein targets remains elusive. The existing evidence suggests that the binding selectivity of HS to different proteins is dominated by sulphation patterns5. The types of sulphation found in HS include the 2-OH of IdoA and GlcA residue, as well as the N-, 3-OH and 6-OH position of GlcN residues. The negatively charges from the sulpho groups interact with the positively charged amino acid residues from proteins. Moreover, the pyranose rings of 2-O-sulpho iduronic acid (IdoA2S) and IdoA residues adopt both chair (1C4) and skew boat (2SO) conformations. The pyranose rings of GlcA and GlcN are, however, predominantly present in 4C1 conformation6,7. The conformational flexibility of IdoA2S residues is known to be essential for the binding of HS to antithrombin to display anticoagulant activity8 and fibroblast growth factors to regulate cell growth9.
Whether sulphations of neighboring residues affects the ring conformation of IdoA has not been systematically examined. The previous conformational analysis of IdoA or IdoA2S residues were carried out using sporadic structures of heparin or HS oligosaccharides either isolated from depolymerized heparin or through chemical synthesis10,11,12,13,14,15,16. The only available systematic study was to limited to the combined effect of N-sulphation and 6-O-sulphation in synthetic trisaccharides17,18. The authors found that 6-O-sulphation drives the conformation equilibrium to 2SO for IdoA2S, but the study did not offer the information on the effects of 2-O- and 3-O-sulphations. In the present study, we demonstrate that the conformation of IdoA is influenced by combined effects from three sulphation types, including 2-O-, 3-O- and 6-O-sulphation using structurally defined model hexasaccharides. Our findings suggest a cellular mechanism to control the conformation of IdoA through the biosynthetic pathway, offering a new hypothesis for the genetic regulation of conformation of carbohydrates.
Results
Eight different hexasaccharides (Fig. 1) were synthesized using the chemoenzymatic method19,20. Hexasaccharides (1–4) contained an IdoA2S residue that is flanked by two saccharides on the reducing end and three residues at the nonreducing end with different degrees of sulphations. The sulphation types cover N-unsubstituted GlcN (1), N-sulphation (2), 6-O-sulphation (3) and 3-O-sulphation (4) (Fig. 1). Hexasaccharides (5–8) are nearly carbon copies of 1–4 with the exception that an IdoA residue was introduced to substitute the IdoA2S residue (Fig. 1). It should be noted that the syntheses of IdoA-containing hexasaccharides (5 to 8) were not previously reported using the chemoenzymatic method. The purity analysis and molecular mass determination was completed by anion exchange HPLC and electrospray ionization mass spectrometry (Supplementary Figs 1 and 2). The structures of hexasaccharide were also confirmed by 1H-NMR and 13C-NMR (Supplementary Figs 3–58). The NMR signals were fully assigned (Supplementary Table 1 and 2).
Figure 1. Chemical structures of hexasaccharides used in this study.

Structures of hexasaccharides synthesized in this study. Both IdoA2S (in 1 to 4) and IdoA (5 to 8) residues are colored in blue.
Both IdoA and IdoA2S residues interconvert between different conformations, including 1C4, 2SO21 and 4C1 (Fig. 2)22 Each conformer has a unique set of three bond proton-proton coupling constants (3JH-H)23,24, which were obtained for the IdoA2S and IdoA residues in the eight hexasaccharides (Table 1). The relative populations of IdoA/IdoA2S ring conformations for the eight compounds were generated by minimizing the residual sum of squares (RSS) between the observed NMR J-values and the total contribution back-calculated for at least 350,000 snapshots of each structure from the molecular dynamics (MD). Due to the slow rate of ring flipping for IdoA (μs timescale)25, three independent simulations were performed for 1–8 (1 μs each), in which the IdoA/IdoA2S ring was restrained in each of the three conformations. As shown in Table 1, the J-value RSS for each oligosaccharide was less than ~1.0 Hz, which compares favorably with the deviations in J-values reported recently in a study that employed single energy-minimized conformations of a related HS fragment26.
Figure 2. 3-D Images of the NOE cross-peaks of H2-H5 and H4-H5 signals from representative hexasaccharides.

The images of NOE cross-peak signals of H2-H5 and H4-H5 from compound 1, 4, 5 and 8 are presented. Cpd 1 and 4 are IdoA2S-containing hexasaccharides, and cpd 5 and cpd 8 are IdoA-containing hexasaccharides. The structures of three different conformers for IdoA or IdoA2S are presented on top of the figure, where R represents –H or –SO3H. The calculated distance between H2 and H5, as well as H4 and H5 and displayed in lines with doubled arrows.
Table 1. Measurement of the population of conformers for IdoA2S and IdoA residues in hexasaccharides.
| Hexasaccharides | Measured 3JH-H couplings (Hz) (Calculated 3JH-H couplings) |
Population of conformersd |
Sum of square differencee | NOE signals for measuring 2SO conformer population | |||||
|---|---|---|---|---|---|---|---|---|---|
| 3JH1-H2 | 3JH2-H3 | 3JH3-H4 | 3JH4-H5 | 1C4 a | 2SOb | 4C1 c | H2-H5/H4-H5f | ||
| 1-IdoA2S | 2.2 (2.7) | 4.3 (4.1) | 3.2 (3.0) | 2.2 (2.1) | 68% | 32% | – | 0.34 | 0.19 |
| 2-IdoA2S | 2.2 (2.7) | 4.3 (4.1) | 3.2 (3.0) | 2.3 (2.2) | 68% | 32% | – | 0.34 | 0.21 |
| 3-IdoA2S | 3.0 (3.6) | 5.8 (5.5) | 3.6 (3.6) | 2.7 (2.5) | 47% | 53% | – | 0.49 | 0.26 |
| 4-IdoA2S | 3.7 (4.5) | 7.4 (6.9) | 4.0 (4.2) | 3.1 (2.8) | 25% | 75% | – | 1.02 | 0.34 |
| 5-IdoA | 3.0 (3.3) | 5.2 (5.1) | 3.5 (3.4) | 2.7 (2.4) | 53% | 47% | – | 0.20 | 0.34 |
| 6-IdoA | 3.1 (3.4) | 5.3 (5.2) | 3.5 (3.5) | 2.7 (2.4) | 51% | 49% | – | 0.19 | 0.35 |
| 7-IdoA | 2.3 (2.8) | 4.3 (4.1) | 3.4 (3.2) | 2.4 (2.2) | 68% | 29% | 3% | 0.37 | 0.22 |
| 8-IdoA | 2.3 (2.7) | 3.9 (3.8) | 3.5 (3.2) | 2.2 (2.2) | 73% | 19% | 8% | 0.26 | 0.19 |
aCalculated 3JH-H values using Amber 14 with GLYCAM06 parameter39 for 1C4 conformer are 1.35 (3JH1-H2), 2.04 (3JH2-H3), 2.19 (3JH3-H4) and 1.69 Hz (3JH4-H5), respectively.
bCalculated 3JH-H values using Amber 14 with GLYCAM06 parameter39 for 2SO conformer are 5.55 (3JH1-H2), 8.48 (3JH2-H3), 4.82 (3JH3-H4) and 3.14 Hz (3JH4-H5), respectively.
cCalculated 3JH-H values using Amber 14 with GLYCAM06 parameter39 for 4C1 conformer are 7.85 (3JH1-H2), 8.03 (3JH2-H3), 8.5 (3JH3-H4) and 5.08 Hz (3JH4-H5), respectively.
dObtained by least sum of square difference analysis of the calculated and experimental values.
eThe residual sum of squares (RSS) was used to determine how well the calculated population ratios fit the experimental data42.
fThe calculated ratio of NOE H2-H5 signal and H4-H5 signal for pure 2SO, 1C4 and 4C1 conformer is 0.53, 0.05, and 0.03 respectively39.
A correlation between the sulphation degree and the population of 2SO conformer was observed. For IdoA2S, the ring with lower sulphation levels (1 and 2) predominantly populated the 1C4 conformation (~68%). As the level of sulphation increased by adding 6-O-sulphation (hexasaccharide 3) and 3-O-sulphation (hexasaccharide 4), the dominant conformation for IdoA2S was shifted to the 2SO conformer. In comparing the percentage of 2SO in 3 and 2, it appears that 6-O-sulphation drives the IdoA2S residue towards 2SO conformation, confirming the results by Munoz-Garcia and colleagues using trisaccharide model compounds17. In addition, the 2SO conformer was populated as high as 75% in hexasaccharide 4, the most highly sulphated hexasaccharide among the tested compounds (Table 1). The 2SO conformer population for 4 is similar to that recently reported for a closely-related pentasaccharide of HS26. Together, the data suggests that neighboring sulphation upregulates the 2SO conformer for an IdoA2S residue.
For the IdoA-containing hexasaccharides (5–8), the sulphation impact on the 2SO conformer for IdoA residues exhibited a different trend from that was observed for an IdoA2S residue. Here, the IdoA residue displayed 47% and 49% of 2SO conformer in compounds 5 and 6, the low sulphated hexasaccharides (Table 1). As the sulphation level increased, the population of 2SO diminished to 29% in hexasaccharide 7 and 19% in hexasaccharide 8, respectively (Table 1). In addition, a small percentage of 4C1 conformer was detected in 7 and 8. This low abundance of the 4C1 conformer is in agreement with previous studies11,21. Sulphation of the N-unsubstituted GlcN residue at the nonreducing end of 1 and 5 had no effects on the conformer distribution for IdoA2S and IdoA residue. This observation is not surprising considering that the GlcN residue is three saccharide residues away from the IdoA2S and IdoA residue.
The distributions of conformers were further confirmed by 1H-1H NOE analysis. This analysis is uniquely sensitive to detect the presence of the 2SO conformer due to the difference in the atomic distance of H2 and H5 in the 2SO conformer (2.6 Å), compared with the 1C4 and 4C1 conformer (4.0 Å and 4.2 Å, respectively) (Fig. 2)27 The shorter atomic distance of H2 and H5 in the 2SO conformer offers a stronger intensity for NOE signal. Since heparin oligosaccharides may exhibit anisotropic motion in solution28, inter-proton distances cannot be calculated using the two-spin approximation, and a more extended experimental procedure, such as quantifying the anisotropy by relaxation or by off-resonance ROESY measurements27, would be necessary. However, together with the analysis of 3JH-H couplings, the analysis of H2-H5/H4-H5 NOE ratio can be used to confirm the conformer population of IdoA2S and IdoA residues. To compare the relative intensities of the H2-H5 NOE among hexasaccharides, a ratio of the volume of H2-H5 cross peak and that of H4-H5 was obtained (Table 1 and Fig. 2). The relative intensity of H2-H5 NOE signal from the IdoA2S residue in 4 was 0.34, while this signal in 1 was only 0.19, suggesting that the IdoA2S residue in 4 displayed higher population of 2SO than that in 1 (Fig. 2). For IdoA residue in hexasaccharide 5 and 8, the relative intensity of H2-H5 NOE signal from IdoA residue in 8 was 0.19, and this signal in 5 was 0.34, suggesting that the IdoA residue in 8 displayed lower population of 2SO than that in 5 (Fig. 2). The intensity of H4-H5 cross-peak of NOE signals should remain unchanged as the atomic distance of H4-H5 is similar amongst the different conformers (Fig. 2). The ratio of H2-H5/H4-H5 NOE signals among 1–4 exhibited an increasing trend as the level of sulphation elevated (Table 1), consistent with the conclusion from the J analysis. In the IdoA-containing series of hexasaccharides (5–8), the NOESY analysis indicated a decreasing trend in the ratio of H2-H5/H4-H5 NOE signals, suggesting that the 2SO conformer becomes less abundant as the surrounding sulphation level increased.
The observed conformational preferences can be explained in part by an examination of the hydrogen bonds detected during molecular dynamics (MD) simulations of the oligosaccharides (Supplementary Table 3). For example, an intra-residue hydrogen bond between the 2-O-sulpho and the 3-OH group in IdoA2S stabilizes the 2So conformation; however, the distance between these two groups is prohibitive for hydrogen bond formation in the 1C4 conformation (Fig. 3A and Supplementary Video 1), potentially leading to the preference for the former conformation. Apparently the presence of this hydrogen bond in the 4C1 conformation provides insufficient stabilization to result in appreciable population of this higher energy form23. The absence of sulphation at the 2-position in compounds 5–8 prevents the described intra-residue hydrogen bond from forming. However, in IdoA-containing hexasaccharides, the 2-OH group can form a hydrogen bond with the N-sulpho moiety in the adjacent residue on the nonreducing end, but only when the IdoA is in the 1C4 conformation (Fig. 3B), potentially leading to the preference for this conformation. While the overall conformation of the oligosaccharides is relatively insensitive to the conformation of the IdoA ring under the conditions of MD simulations29, the position of the sulpho group in IdoA2S is markedly impacted by ring conformation, differing by 4.9 Å between the 2So and 1C4 structures (Fig. 3C).
Figure 3. Images of IdoA2S or IdoA and hexasaccharides from the MD simulations.

Atomic distances are indicated by dashed lines. The 1C4, 2SO, and 4C1 conformations of IdoA2S are colored orange, green, and purple, respectively. Panel A shows the IdoA2S residue in the 1C4 and 2SO conformations (from a representative snapshot of the simulations of 4). Panel B displays the GlcNS(3S,6S) and IdoA residues from a representative frame of the simulation of 8 in the 2SO conformation. Panel C contains a superimposition of the ring atoms in oligosaccharides from simulations of 4 in each of the three IdoA ring conformations. The sulfur atom of the IdoA2S residue is also displayed in yellow for each conformation. The RMSD of the ring atoms of the ligands with IdoA in the 1C4 and 4C1 conformation are 0.9 Å and 0.7 Å, respectively, relative to the molecule containing IdoA in the 2SO conformation. Each model is the snapshot most similar to the average structure during the MD simulations.
The biological outcome was analyzed using the antithrombin-HS binding model. Antithrombin deactivates coagulating factor Xa upon the binding to HS. Among the eight synthesized hexasaccharides, only 4 and 8 produce anticoagulant activity due to the presence of 3-O-sulphation30,31. The binding affinity (Kd) of 4 and 8 to antithrombin was determined to be 15.5 ± 3.4 nM, and was 32.6 ± 5.3 nM, respectively. The IC50 value for the inhibition of the activity of Xa, a measurement for the anticoagulant activity, was determined to be 19 nM and 29 nM for 4 and 8, respectively (Fig. 4). A small, but notable, difference in the binding affinity to antithrombin and potency for the anti-Xa activity between 4 and 8 was observed. The co-crystal structure analysis of antithrombin and a pentasaccharide, structurally very similar to 4, revealed no amino acid residues from antithrombin directly interacted with the 2-O-sulpho group from the IdoA2S residue in the pentasaccharide32,33, suggesting that a decrease in binding affinity from 8 was not due to the lack of 2-O-sulpho group. The IdoA2S residue is known to display the 2SO conformer when it interacts with antithrombin in the co-crystal structure32, the solution complex34, as well as from study of the interactions of antithrombin with a conformationally-fixed oligosaccharide8. Gurerrini et al. demonstrated that the presence of unnatural saccharide residues at the reducing end of the IdoA2S residue domain reduces the binding affinity to antithrombin and decreases the anti-Xa activity. Authors attributed to the decreased effects to lower population of the 2SO conformer of the IdoA2S residue35. Although the difference is small in our study, it is tempting to attribute the higher binding affinity to antithrombin and stronger anti-Xa activity of 4 to the increased 2SO conformer population.
Figure 4. Inhibition curve of Xa activity by compound 4 and 8.
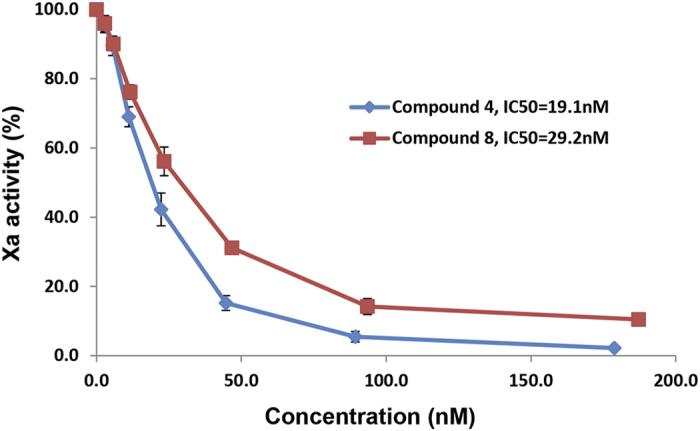
Different concentrations of compound 4 and 8 were incubated with antithrombin and Xa. The activity of Xa was then measured using a chromogenic substrate.
Discussion
Sulphation plays a critical role in determining the biological functions of HS. The interactions between HS and proteins are primarily mediated by the negative charges from the sulpho groups and the positive charges from lysine and arginine residues from the proteins as well as some hydrogen bonding interactions36,37. Here, we demonstrate that sulphation also affects the dynamics of the ring conformation of IdoA and IdoA2S. Interestingly, the sulphation of neighboring residues impacts the conformer population differently depending on whether the IdoA carries a 2-O-sulpho group. Our study relies on two crucial pieces of innovation that were unavailable to previous studies: the ability to design and synthesize HS oligosaccharides and a thorough computational analysis of these molecules. A uniquely designed group of eight hexasaccharides allowed us to investigate the effects of 2-O-sulphation, 6-O-sulphation and 3-O-sulphation on the conformation of IdoA. The oligosaccharides were synthesized by the chemoenzymatic method that offers an efficient synthesis of HS oligosaccharides19,20. It would be very difficult to obtain a similar set of oligosaccharides by isolation or the chemical synthetic approach. Improvements in molecular dynamics allowed us to extend the simulation from the traditional nanosecond timescale to a full microsecond for each compound. Considering the slow rate of conversion between conformations for IdoA29, simulations of the compounds were performed for each conformation (24 in total). As a result, the proton-proton coupling constants (3JH-H) obtained from NMR are close to the simulated values (Table 1). Furthermore, extending the length of the simulations allowed us to identify key intra-molecular interactions that stabilize the ring conformations.
This investigation offers insight into the control of the ring conformation of IdoA through the HS biosynthetic pathway. The pathway involves a series of sulphotransferases and a specialized C5-epimerase (Fig. 5). After the predominantly 4C1 GlcA residue is epimerized to IdoA, the HS sequence diverges into two separate pathways to form both IdoA2S-containing and IdoA-containing domains. Both paths are then subjected to 6-O-sulphotransferase and 3-O-sulphotransferase modifications to form matured HS polysaccharides. For the IdoA2S-containing domains, additional sulphation modifications, i.e. 6-O-sulphation and 3-O-sulphation, increase the 2SO conformer population, whereas for the IdoA-containing domains, additional sulphation modification decreases the 2SO conformer population. Given that IdoA and IdoA2S residues both exist in HS38, balancing the distribution of different conformers may represent an important structural feature for shaping the selectivity of HS towards potential binding partners. It has been well established that the amino acid sequences determine the secondary and tertiary structures of proteins. The availability of structurally defined oligosaccharides coupled with sophisticated NMR and computational techniques will open the door to unveil the structure of HS chain beyond the primary saccharide sequence.
Figure 5. Biosynthetic pathway of HS and relevant to the hexasaccharide model compounds.

HS biosynthesis is initiated from the polysaccharide containing -GlcA-GlcNAc- repeating units, followed by the modifications of N-deacetylase/N-sulphotransferase, C5-epimerase, 2-O-sulphotransferase, 6-O-sulphotransferase and 3-O-sulphotransferase. The 2-O-sulphotransferase modification can be incomplete, resulting the polysaccharide product consists of IdoA2S-containing domain and IdoA-containing domain. Hexasaccharide 2 and 6 represent these two domain structures. Hexasaccharide 3 and 7 represent the domain structures after 6-O-sulphotransferase modification, and hexasaccharide 4 and 8 represent the structures after 3-O-sulphotransferase modification. The wavy lines represent the linker between domains in HS polysaccharide with unknown structures.
Methods
Synthesis of compound 1 to 8
The synthesis of 1 to 8 was completed according to the chemoenzymatic method published previously6,19. Briefly, heparosan synthase-2 (PmHS2) from Pasteurella multocida to elongate the monosaccharide, p-nitrophenyl glucuronide (GlcA-pnp), to a hexasaccharide backbone. The hexasaccharide was then subjected to the modification of N-sulphotransferase, C5-epimerase, 6-O-sulphotransferase, and 3-O-sulphotransferase to generate IdoA residues and introduce the sulpho group at the desired position to produce the final products. For the synthesis of IdoA2S-containing hexasaccharides (1 to 4), 2-O-sulphotransferase was employed to introduce the 2-O-sulpho group to the IdoA residue. The 2-O-sulphotransferase modification step was omitted during the synthesis of 5 to 8. About 5–10 mg of each hexasaccharide was prepared for the current study. For the preparation of 5 to 8, a HPLC purification step was necessary after the hexasaccharide backbone modified by C5-epimerase to separate the hexasaccharide containing an IdoA residue.
MS analysis of oligosaccharides
MS analyses were performed at a Thermo LCQ-Deca. A syringe pump (Harvard Apparatus) was used to introduce the sample by direct infusion (35 μl min−1). Hexasaccharide (1 mg ml−1, 1 μl) was diluted by 200-fold with 10 mM ammonium bicarbonate. Experiments were carried out in negative ionization mode with the electrospray source set to 2 KV and 200 °C. The automatic gain control was set to 1 × 107 for full scan MS. The MS data were acquired and processed using Xcalibur 1.3.
Structural analysis of compound 1 to 8 by NMR
NMR experiments were performed at 298 K on Bruker Avance 700 MHz and 850 MHz spectrometer with Topsin 3.2 software. Samples (1.0 to 5.0 mg) were each dissolved in 0.5 ml D2O (99.996%, Sigma-Aldrich) and lyophilized three times to remove the exchangeable protons. The samples were re-dissolved in 0.4 ml D2O and transferred to NMR microtubes (O.D. 5 mm, Norrell). Chemical shifts are referenced to external 2,2-dimethyl-2-silapentane-5-sulphonate sodium salt (DSS, Sigma, Co.). Deuterated EDTA (Sigma, Co.) was added to remove the effect of paramagnetic ions. 1D 1H NMR experiments “zg” pulse sequence were performed with 64 scans and an acquisition time of 3.8 sec. 1D 13C NMR experiments “zgdc30” pulse sequence were performed with 30000 scans, 1.5 sec relaxation delay, and 1.0 sec acquisition time. 2D 1H-1H COSY experiments “cosygpmfphpp” pulse sequence were performed with 48 scans, 512 increments, 1.5 sec relaxation delay, and 120 msec acquisition time. 2D 1H-1H NOESY experiments “noesygpph” pulse sequence were performed with 48 scans, 512 increments, 1.5 sec relaxation delay, 120 msec acquisition time, and 300 msec mixing time. 2D 1H-13C HSQC experiments “hsqcgpph” pulse sequence were performed with 48 scans, 512 increments, 1.5 sec relaxation delay, and 120 msec acquisition time. 2D 1H-13C HMBC experiments “hmbcgpndqf” pulse sequence were performed with 72 scans, 512 increments, 1.5 sec relaxation delay, and 120 msec acquisition time. 2D 1H-13C HSQC-TOCSY experiments “hsqcetgpml” pulse sequence were performed with 48 scans, 512 increments, 80 msec TOCSY mixing time, 1.5 sec relaxation delay, and 120 msec acquisition time. 2D spectra were recorded with GARP carbon decoupling. 48 dummy scans were used prior to the start of acquisition. 2048 total points were collected in f2. 13C transmitter offset was set at 90.0 ppm. The polarization transfer delay CNST2 was set with a 1JC-H coupling value of 145 Hz. The delay for evolution of long-range couplings CNST13 in HMBC was set with Jlr coupling value of 7.4 Hz.
The structural characterization were also performed by Varian Inova 500 MHz spectrometer equipped with 5 mm triple resonance XYZ or broadband PFG probe, and processed by VnmrJ 2.2D software. The constructs were analyzed by NMR experiments, including 1D- (“s2pul” pulse sequence 1H or 13C), 2D- (1H-1H “gCOSY” pulse sequence COSY, “TOCSY” pulse sequence TOCSY, 1H-13C “gHSQC” pulse sequence HSQC, “gHMBC” pulse sequence HMBC, and “gHSQCTOXY” pulse sequence HSQC-TOCSY) NMR. NMR experiments were performed at 298 K. Samples (5.0 to 10.0 mg) were each dissolved in 0.5 mL D2O (99.994%, Sigma, Co.) and lyophilized three times to remove the exchangeable protons.
Molecular dynamics pre-processing
The hexasaccharides were created with the GLYCAM06 (version j-1) parameter set39 and sulphate parameters29, neutralized with Na+ ions, and solvated by the TIP5P water model in a truncated octahedral box 12 Å around the ligand40. Ensemble charges were developed for the GlcN residue according to the standard GLYCAM protocol39. Briefly, electrostatic potentials were calculated by GAUSSIAN09 at the HF/6-31G(*) level of theory for 100 snapshots at 0.5 ns intervals (50 ns total). Partial atomic charges were computed using the RESP module of AMBER14 by fitting to these electrostatic potentials with a restraint weight of 0.0141.
Additional Information
How to cite this article: Hsieh, P.-H. et al. Uncovering the Relationship between Sulphation Patterns and Conformation of Iduronic Acid in Heparan Sulphate. Sci. Rep. 6, 29602; doi: 10.1038/srep29602 (2016).
Supplementary Material
Acknowledgments
This work is supported in part by grants from National Institutes of Health (GM102137, HL094463 and P41 GM103390) and by a grant from Eshelman Institute for Innovation.
Footnotes
Author Contributions P.-H.H. synthesized the hexasaccharides and conducted NMR analysis. D.F.T. and R.J.W. conducted MD analysis. M.G. helped NMR data analysis. P.-H.H. and J.L. designed the project. All authors participated in writing the manuscript.
References
- Xu D. & Esko J. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 83, 129–157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. et al. heparan sulfate modulates neutrophil and endothelial function in antibacterial innate immunity. Infect. Immun. 83, 3648–3656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V. N. et al. 3-O-sulfated heparan sulfate controls epithelial Kit + FGFR2b + progenitor expansion via rapid autocrine regulation of 3-O-sulfotransferases. Develop. Cell 29, 662–673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. & Linhardt R. J. Chemoenzymatic synthesis of heparan sulfate and heparin. Nat. Prod. Rep. 31, 1676–1685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J., Spillmann D., Li J.-p. & Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J. Cell Biol. 174, 323–327 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P., Xu Y., Keire D. A. & Liu J. Chemoenzymatic synthesis and structural characterization of 2-O-sulfated glucuronic acid containing heparan sulfate hexasaccharides. Glycobiology 24, 681–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattelle B. M. & Almond A. Is N-acetyl-D-glucosamine a rigid 4C1 chair? Glycobiology 21, 1651–1662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. et al. Synthesis of conformationally locked L-iduronic acid derivatives: direct evidence for a critical role of the skew-boat 2S0 conformer in the activation of antithrombin by heparin. Chemistry 7, 4821–4834 (2001). [DOI] [PubMed] [Google Scholar]
- Raman R., Venkataraman G., Ernst S. & Sasisekharan R. Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc. Natl. Acad. Sci. 100, 2357–2362 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro D. R. et al. Evidence for conformational equilibrium of the sulfated L-iduronate residues in Heparin and in synthetic heparin Mono- and Oligosaccharides: NMR and forced-field studies. J. Am. Che. Soc. 108, 6773–6778 (1986). [Google Scholar]
- Ferro D. R. et al. Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 195, 157–167 (1990). [DOI] [PubMed] [Google Scholar]
- van Boeckel C. A. A., van Aelst S. F., Wagenaars G. N. & Mellema J.-R. Conformational analysis of synthetic heparin-like oligosaccharides containing a-L-idopyranosyluronic acid. Recl. Trav. Chim. Pays-Bas. 106, 19–29 (1987). [Google Scholar]
- de paz J.-L. et al. The activation of fibroblast growth factors by heparin: synthesis, structure, and biological activity of heparin-like oligosaccharides. ChemBiochem 2, 673–685 (2001). [DOI] [PubMed] [Google Scholar]
- Ojeda R., Angulo J., Nieto P. M. & Martin-Lomas M. The activation of fibroblast growth factors by heparin: Synthesis and structural study of rationally modified heparin-like oligosaccharides. Can. J. Chem. 80, 917–936 (2002). [Google Scholar]
- Lucas R., Angulo J., Nieto P. M. & Martin-Lomas M. Synthesis and structural study of two new heparin-like hexasaccharides. Org. Biomol. Chem. 1, 2253–2266 (2003). [DOI] [PubMed] [Google Scholar]
- Angulo J. et al. The activation of fibroblast growth factors (FGFs) by glycosaminoglycans: Influence of the sulfation pattern on the biological activity of FGF1. Chembiochem 5, 55–61 (2004). [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia J. C., Corzana F., de Paz J. L., Angulo J. & Nieto P. M. Conformations of the iduronate ring in short heparin fragments described by time-averaged distance restrained molecular dynamics. Glycobiology 23, 1220–1229 (2013). [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia J. C. et al. Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chem. Eur. J. 18, 16319–16331 (2012). [DOI] [PubMed] [Google Scholar]
- Xu Y. et al. Homogeneous and reversible low-molecular weight heparins with reversible anticoagulant activity. Nat. Chem. Biol. 10, 248–250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. Chemoenzymatic synthesis of homogeneous ultra-low molecular weight heparin. Science 334, 498–501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulloy B. & Forster M. Conformation and dynamics of heparin and heparan sulfate. Glycobiology 10, 1147–1156 (2000). [DOI] [PubMed] [Google Scholar]
- Liu C. et al. Molecular mechanism of substrate specificity for heparan sulfate 2-O-sulfotransferase. J. Biol. Chem. 289, 13407–13418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattelle B. M., Hansen S. U., Gardiner J. M. & Almond A. Free energy landscapes of iduronic acid and related monosaccharides. J. Am. Chem. Soc. 132, 13132–13134 (2010). [DOI] [PubMed] [Google Scholar]
- Angulo J., Nieto P. M. & Martin-Lomas M. A molecular dynamics description of the conformational flexibilit of the L-iduronate ring in glycosaminoglycans. Chem. Commun. 13, 1512–1513 (2003). [DOI] [PubMed] [Google Scholar]
- Sattelle B. M. et al. Dependence of pyranose ring puckering on anomeric configuration: methyl idopyranosides. J. Phys. Chem. B. 116, 6380–6386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hricovini M. Solution structure of heparin pentasaccharide: NMR and DFT analysis. J. Phys. Chem. B. 119, 12397–12409 (2015). [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia J. C. et al. 3D structure of a heparin mimetic analogue of a FGF-1 activator. A NMR and molecular modelling study. Org. Biomol. Chem. 11, 8269–8275 (2013). [DOI] [PubMed] [Google Scholar]
- Angulo J. et al. Dynamic properties of biologically active synthetic heparin-like hexasaccharides. Glycobiology 15, 1008–1015 (2005). [DOI] [PubMed] [Google Scholar]
- Singh A. et al. Extension and vilidation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem., 10.1139/cjc-2015-0606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. The effect of precursor structures on the action of glucosaminyl 3-O-sulfotransferase-1 and the biosynthesis of anticoagulant heparan sulfate. J. Biol. Chem. 276, 28806–28813 (2001). [DOI] [PubMed] [Google Scholar]
- Atha D. H., Lormeau J.-C., Petitou M., Rosenberg R. D. & Choay J. Contribution of monosaccharide residues in heparin binding to antithrombin III. Biochemistry 24, 6723–6729 (1985). [DOI] [PubMed] [Google Scholar]
- Jin L. et al. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. 94, 14683–14688 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini M., Mourier P. A., Torri G. & Viskov C. Antithrombin-binding oligosaccharides: structural diversities in a unique function? Glycoconj. J. 31, 409–416 (2014). [DOI] [PubMed] [Google Scholar]
- Guerrini M. et al. An unusual antithrombin-binding heparin octasaccharide with an additional 3-O-sulfated glucosamine in the active pentasaccharide sequence. Biochem. J. 449, 343–351 (2013). [DOI] [PubMed] [Google Scholar]
- Guerrini M. et al. Effects of molecular conformation and anticoagulant activities of 1,6-anhydrosugars at the reducing terminal of antithrombin-binding octasaccharides isolated from low-molecular weight heparin enoxaparin. J. Med. Chem. 53, 8030–8040 (2010). [DOI] [PubMed] [Google Scholar]
- Liu J., Moon A. F., Sheng J. & Pedersen L. C. Understanding the substrate specificity of the heparan sulfate sulfotransferases by a combined synthetic crystallographic approach. Curr. Opin. Struct. Biol. 22, 550–557 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhardt R. J. 2003. Claude S. Hudson Award address in carbohydrate chemistry. Heparin: structure and activity. J. Med. Chem. 46, 2551–2564 ( 2003). [DOI] [PubMed] [Google Scholar]
- Lindahl U., Kusche-Gullberg M. & Kjellen L. Regulated diversity of heparan sulfate. J. Biol. Chem. 273, 24979–24982 (1998). [DOI] [PubMed] [Google Scholar]
- Kirschner K. N. et al. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M. W. & Jorgensen W. L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112, 8910–8922 (2000). [Google Scholar]
- Bayly C. I., Cieplak P., Cornell W. D. & Kollman P. A. A well-behaved electrostatice potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 97, 10269–10280 (1993). [Google Scholar]
- Murphy K. J., McLay N. & Pye D. A. Structural studies of heparan sulfate hexasaccharides: New insights into iduronate conformational behavior. J. Am. Chem. Soc. 130, 12435–12444 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.