

Abstract
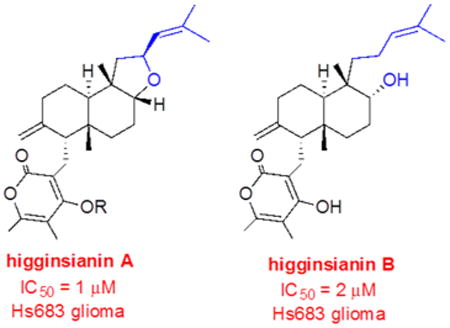
Two new diterpenoid α-pyrones, named higginsianins A (1) and B (2), were isolated from the mycelium of the fungus Colletotrichum higginsianum grown in liquid culture. They were characterized as 3-[5a,9b-dimethyl-7-methylene-2-(2-methylpropenyl)dodecahydronaphtho[2,1-b]-furan-6-ylmethyl]-4-hydroxy-5,6-dimethylpyran-2-one and 4-hydroxy-3-[6-hydroxy-5,8a-dimethyl-2-methylene-5-(4-methylpent-3-enyl)decahydronaphthalen-1-ylmethyl]-5,6-dimethylpyran-2-one, respectively, by using NMR, HRESIMS, and chemical methods. The structure and relative configuration of higginsianin A (1) were confirmed by X-ray diffractometric analysis, while its absolute configuration was assigned by electronic circular dichroism (ECD) experiments and calculations using a solid-state ECD/TDDFT method. The relative and absolute configuration of higginsianin B (2), which did not afford crystals suitable for X-ray analysis, were determined by NMR analysis and by ECD in comparison with higginsianin A. 1 and 2 were the C-8 epimers of subglutinol A and diterpenoid BR-050, respectively. The evaluation of 1 and 2 for antiproliferative activity against a panel of six cancer cell lines revealed that the IC50 values, obtained with cells reported to be sensitive to proapoptotic stimuli, are by more than 1 order of magnitude lower than their apoptosis-resistant counterparts (1 vs >80 μM). Finally, three hemisynthetic derivatives of 1 were prepared and evaluated for antiproliferative activity. Two of these possessed IC50 values and differential sensitivity profiles similar to those of 1.
Graphical abstract
Fungal secondary metabolites have been an excellent source of new antibiotics, antifungals, herbicides, and antitumor compounds.1,2 They belong to different classes of organic compounds such as aromatics, amino acids, coumarins, isocoumarins, cytochalasans, ethanones, furopyrans, nonenolides, oxazatricycloalcalenones, and terpenes, among many others. Therefore, the continued exploration of fungi in search of structurally new products with promising bioactivities is an important area of research.
Colletotrichum is a fungal genus comprising a large number of endophytic, saprophytic, and plant pathogenic species. It is one of the most economically important plant pathogenic genera that causes anthracnose disease of fruits and leaves in a wide range of hosts, resulting in severe crop reduction and conspicuous postharvest losses of tropical and subtropical cereals, grasses, legumes, vegetables, and tree fruits.3–11 For this reason species belonging to this genus have been the subject of extensive studies involving their pathogenesis, morphology, multigene analysis, physiology, host range, and disease life cycle. However, because of the large size of the genus, the production of secondary metabolites by various species is only partially explored (for a review, see Carcía-Paión and Collado, 2003).12 Thus, we evaluated the culture filtrates and mycelia of ca. 100 strains belonging to many species of the genus Colletotrichum in order to find novel metabolites with potential phytotoxic, antibiotic, antifungal, and anticancer activities. One of the evaluated species was a strain of Colletotrichum higginsianum, a fungus that belongs to C. destructivum species complex13 and causes anthracnose leaf spot disease on many cultivated forms of Brassica. The present study reveals that this fungus produces a mycelium, whose extract displays in vitro antitumor activity. Bioactivity-guided fractionation of this extract resulted in the isolation of two novel diterpenoid α-pyrones, named higginsianins A and B. These compounds were chemically characterized and evaluated for antiproliferative activity against a panel of six cancer cell lines.
Results and Discussion
The EtOAc organic extract was obtained from the maceration of the lyophilized C. higginsianum mycelium. The extraction was carried out at room temperature in the dark. The extract was subjected to bioactivity-guided fractionation by evaluating the separated fractions for antiproliferative activity against a number of cancer cell lines as detailed in the Experimental Section. This process yielded two main bioactive metabolites as homogeneous solids, which were named higginsianins A and B (1 and 2, 13.3 and 4.4 mg/L, respectively). While 1 crystallized as white needles, 2 proved to be not crystallizable.
Compound 1 had a molecular formula of C27H38O4 as deduced from its HRESIMS spectrum and consistent with nine hydrogen deficiencies. The preliminary investigation of its 1H and 13C NMR spectra (Table 1) showed signals for carbonyl, olefinic, and hydroxy groups consistent with the bands observed in the IR spectrum,14 while its UV spectrum showed a band typical of an extended conjugated ester.15 A further investigation of its 1H NMR spectrum (Table 1) showed the presence of a broad doublet (J = 9.0 Hz, H-13) at δ 5.14 and two broad singlets (H2-19) at δ 4.47 and 4.22, belonging to a trisubstituted and a 1,1-disubstituted double bond. Further, a triplet of doublets (J = 9.0 and 6.8 Hz, H-12) at δ 4.78 and a broad singlet (H-8) at δ 3.64 were observed and assigned to the two oxygenated methine groups. The 1H NMR spectrum also contained two singlets (Me-26 and Me-27) at δ 2.09 and 1.83 and two broad singlets (Me-15 and Me-16) at δ 1.64 and 1.63, attributed to four vinylic methyls, as well as two singlets (Me-17 and Me-18) at δ 0.88 and 0.86 due to two quaternary methyl groups.16 On the basis of the couplings observed in the HSQC spectrum,17 the signals observed in the 13C NMR spectrum at δ 127.3, 111.1, 82.9, 73.0, 25.8, 21.6, 20.7, 18.2, 17.3, and 9.9 were assigned to carbons C-13, C-19, C-8, C-12, C-15, C-18, C-17, C-16, C-26, and C-27.18 The 13C NMR spectrum also contained a singlet typical of a carbonyl of an α-pyrone ring, six singlets of olefinic carbons, one of which was oxygenated, two quaternary carbons, two methines, and six methylene groups for a total of 27 carbons, consistent with a polycyclic diterpene ring system joined to a tetrasubstituted α-pyrone ring. Indeed, in the COSY spectrum17 the olefinic proton H-13 was coupled with the two methyl groups (Me-15 and Me-16) and with the proton of the adjacent oxygenated methine (H-12). The methine proton (H-12) was, in turn, coupled with the protons of the adjacent methylene group (H2-11), which resonated as two double doublets (J = 12.8 and 6.6 Hz and J = 12.8 and 9.0 Hz, respectively) at δ 2.15 and 1.17. The olefinic protons (H2-19) were coupled with the protons of the methylene group (H2-2), which resonated as a broad doublet of double doublets (J = 17.9, 13.5, and 4.8 Hz) and a multiplet at δ 2.39 and 2.10, respectively. The olefinic protons (H2-19) were also coupled with the methine proton (H-4), which appeared as a double doublet (J = 10.9 and 5.3 Hz) at δ 2.03 through coupling with the adjacent methylene protons (H2-20). The methylene protons (H2-20) resonated as two double doublets (J = 13.7 and 10.9 Hz and J = 13.7 and 5.3 Hz) at δ 2.61 and 2.55. The methylene protons (H2-2) were coupled with those of the adjacent methylene group (H2-1), which appeared as a broad doublet (J = 13.5) and a double triplet (J = 13.5 and 4.3 Hz) at δ 1.45 and 1.30, respectively. The methylene group (H2-1) was, in turn, coupled with the adjacent methine proton (H-10), which was observed as a multiplet at δ 1.65. The proton (H-8) of the oxygenated methine carbon appearing as a broad singlet at δ 3.64 was coupled with the protons of the adjacent methylene group (H2-7), which resonated as two multiplets at δ 1.74 and 0.78. The methylene protons (H2-7), in turn, were coupled with the protons of the adjacent methylene group (H2-6), which appeared as two multiplets at δ 1.94 and 0.92. The couplings observed in the HSQC spectrum allowed us to assign the signals at δ 56.1, 48.9, 38.5, 32.0, 29.4, 24.6, 22.7, and 21.4 to their corresponding methine and methylene carbons C-4, C-11, C-10, C-2, C-6, C-1, C-7, and C-20. In addition, the carbonyl, six quaternary olefinic, and two aliphatic carbons observed δ 165.1, 163.7, 155.7, 148.5, 134.6, 127.3, 111.1, 105.8, 102.9, and 44.1 and 37.6 were assigned to C-25, C-22, C-24, C-3, C-14, C-13, C-19, C-23, C-21, and C-9 and C-5 on the basis of the couplings observed in the HMBC spectrum. In particular, the couplings between C-25 and H2-20, C-22 and H2-20 and Me-27, C-24 and Me-26, C-3 and H-4 and H-2A, C-14 and Me-15 and Me-16, C-23 and Me-26 and Me-27, C-21 and H2-20, and C-9 and H2-7, H-11A and Me-17 and C-5 with H-7A, H-10 and Me-18 were detected. Thus, the chemical shifts of all the protons and corresponding carbons of 1 were assigned as shown in Table 1. Some of the HMBC couplings allowed us to identify the substituents and their locations on the α-pyrone residue, the location of the 2,2-dimethylethenyl side chain, and that of the exocyclic methylene. In particular, the 3-(5,6-dimetyl-4-hydroxy)-α-pyronyl residue appeared linked to the methylene H2C-20, which, in turn, was linked to C-4 because of the coupling of C-4 with H2-20. The dimethylethenyl was determined to be attached to C-12 by the coupling observed between its geminal proton (H-12) and C-13. The carbon C-3 of the exocyclic methylene group was included in a six-membered ring due to the coupling observed between C-3 and H-4 and H-2A, C-4 and Me-18, C-5 and H-10, C-2 and H2-19, and C-1 and H-10. Ring A appeared to be joined with ring B through the carbons C-5 and C-10 because of the couplings observed between C-5 and H-7A and between C-6 and Me-18. The Me-18 was located at C-5 because of the couplings of Me-18 with C-5 and C-6. Finally, the residual unsaturation was due to a 2,3,3,5-tetrasubstituted tetrahydrofuran ring, which as mentioned above bears the dimethylethenyl group at C-12. This ring was determined to be joined to the decahydronaphthalene system through carbons C-9 and C-8 due to the couplings observed between C-6 and H-8, C-8 and Me-17, and C-9 and H2-7 and Me-17. The latter coupling also allowed us to establish that Me-17 was attached to C-9.
Table 1. 1H and 13C NMR Data of Higginsianins A and B (1 and 2)a.
| position | 1 | 2 | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| δCb | δH (J in Hz) | HMBC | δCb | δH (J in Hz) | HMBC | |
| 1 | 24.6 | 1.45 br d (13.5) | H-10 | 25.5 | 1.84 m | H-10 |
| 1.30 dt (13.5, 4.3) | 1.53 m | |||||
| 2 | 32.0 | 2.39 br ddd (17.9, 13.5, 4.8) | H2-19 | 31.7 | 2.10 m | H-4, H-1A |
| 2.10 m | ||||||
| 3 | 148.5 | H-4, H-2A | 148.5 | H2-20, H-4 | ||
| 4 | 56.1 | 2.03 dd (10.9, 5.3) | H2-20, Me-18 | 55.0 | 2.12 m | H2-20, Me-18 |
| 5 | 37.6 | H-7A, H-10, Me-18 | 37.7 | Me-18, H2-20, H-4, H-6 | ||
| 6 | 29.4 | 1.94 m | H-8, Me-18 | 28.1 | 2.09 m | H-8, Me-18 |
| 0.92 m | 0.90 m | |||||
| 7 | 22.7 | 1.74 m | 22.5 | 1.50 m | H2-6 | |
| 0.78 m | 1.21 dd (12.7, 4.8) | |||||
| 8 | 82.9 | 3.64 br s | H-11A, Me-17 | 72.2 | 3.57 br s | H2-11, Me-17 |
| 9 | 44.1 | H2-7, H-11A, Me-17 | 39.0 | Me-17 | ||
| 10 | 38.5 | 1.65 m | H2-11 | 40.1 | 1.68 dd (12.7, 2.6) | H2-11 |
| 11 | 48.9 | 2.15 dd (12.8, 6.6) | H-10, Me-18 | 39.6 | 1.27 m | H-10, Me-18 |
| 1.17 dd (12.8, 9.0) | ||||||
| 12 | 73.0 | 4.78 td (9.0, 6.6) | H-11B | 21.3 | 2.11 m | H2-11, H-13 |
| 1.85 m | ||||||
| 13 | 127.3 | 5.14 br d (9.0) | H-12, H-11B, Me-15, Me-16 | 125.2 | 5.04 br t (7.4) | Me-15, Me-16, H2-11 |
| 14 | 134.6 | Me-15, Me-16 | 131.0 | Me-15, Me-16 | ||
| 15 | 25.8 | 1.64 br s | Me-16 | 25.8 | 1.60 br s | Me-16, H-13 |
| 16 | 18.2 | 1.63 br s | Me-15 | 17.3 | 1.54 br s | Me-15 |
| 17 | 20.7 | 0.88 s | 18.8 | 0.75 s | ||
| 18 | 21.6 | 0.86 s | 22.5 | 0.88 s | ||
| 19 | 111.1 | 4.47 br s | 109.9 | 4.45 br s | H-4 | |
| 4.22 br s | 4.28 br s | |||||
| 20 | 21.4 | 2.61 dd (13.7, 10.9) | H-4 | 21.8 | 2.55 d (8.3) | H-4, H2-2 |
| 2.55 dd (13.7, 5.3) | ||||||
| 21 | 102.9 | H2-20 | 102.5 | H2-20 | ||
| 22 | 163.7 | H2-20, Me-27 | 165.4 | Me-26, Me-27 | ||
| 23 | 105.8 | Me-26, Me-27 | 107.4 | Me-26, Me-27 | ||
| 24 | 155.7 | Me-26 | 154.9 | Me-26 | ||
| 25 | 165.1 | H2-20 | 166.2 | H2-20 | ||
| 26 | 17.3 | 2.09 s | Me-27 | 16.9 | 2.09 s | Me-26 |
| 27 | 9.9 | 1.83 s | Me-26 | 9.9 | 1.81 s | Me-27 |
The chemical shifts are in δ values (ppm) from TMS. 2D 1H,1H (COSY) and 13C,1H (HSQC) NMR experiments delineated the correlations of all protons and their corresponding carbons.
Multiplicities were assigned by the DEPT spectrum.
On the basis of the above analysis higginsianin A (1) was determined to be 3-[5a,9b-dimethyl-7-methylene-2-(2-methylpropenyl)dodecahydronaphtho[2,1-b]furan-6-ylmethyl]-4-hydroxy-5,6-dimethylpyran-2-one. The structure assigned to 1 was supported by all of the couplings observed in the HMBC spectrum (Table 1) and by its HRESIMS spectrum, which gave the sodium cluster [M + Na]+ at m/z 449.2665 and, when the same spectrum was recorded in negative mode, the pseudomolecular ion [M – H]− at m/z 425.2665.
The structure was unambiguously confirmed by the X-ray analysis of suitable crystals obtained as white needles by slow evaporation of a MeOH–EtOAc mixture (1:3). An ORTEP view of the molecular structure is shown in Figure 2. Compound 1 crystallizes in the triclinic P1 space group with two independent molecules (named A and B) in the unit cell. Bond lengths and angles in 1 are in the normal range. In particular, bond distances at the tetrasubstituted heterocyclic six-membered ring confirm the α-pyrone identity of the group. The two independent molecules A and B differ only by the orientation of the α-pyrone due to its rotation by 180° around the C(20)–C(21) bond. The molecule of compound 1 consists of a polycyclic ring system incorporating the trans-decahydronaphthalene system fused to the tetrahydrofuran ring. The energetically more stable chair conformation is observed for the six-membered rings A and B, while the five-membered ring C adopts an envelope conformation with C-9 at the flap position. In ring A, the geometry at C-3 and the C(3)–C(19) bond distance clearly indicate the presence of an exocyclic double bond. The molecular geometry of 1 features trans A/B and cis B/C junctions within the polycyclic ring system, with the α-pyrone-CH2– group in the axial position at C-4 and the dimethylethenyl group in the equatorial position at C-12. In this way the molecule assumes an overall folded shape.
Figure 2.
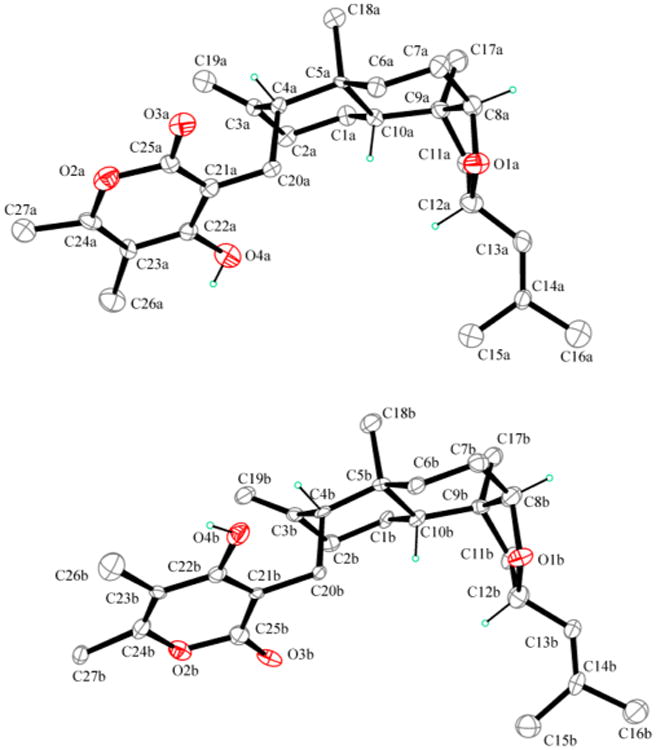
ORTEP view of higginsianin A (1). The two crystallographically independent molecules, molecule A (up) and molecule B (down), are drawn in the same perspective view. Displacement ellipsoids are drawn at the 30% probability level. Only the methine hydrogen atoms are reported for clarity.
In the absence of a strong anomalous scatterer, it was possible to determine only the relative and not the absolute configuration by X-ray analysis. Six chirality centers are present in the molecule at C-4/C-5/C-8/C-9/C-10/C-12, whose relative configuration is R/R/R/S/R/S. In the crystal packing, the chains of molecules A and chains of molecules B running along the a-axis are formed via intermolecular O–H….O=C hydrogen bonds. The relative configuration determined by X-ray analysis was in agreement with the correlations observed in the NOESY spectrum (Table 2),17 in particular with the cross-peak between H-8 and Me-17, which confirmed the cis stereochemistry of the junction between B and C rings.
Table 2. NOESY Data of Higginsianins A and B (1 and 2).
| 1 | 2 | ||
|---|---|---|---|
|
|
|
||
| irradiated | observed | irradiated | observed |
| H-8 | Me-17, H-7A | H-8 | Me-17 |
| H-12 | Me-15, Me-16 | H-19A | H-19B, H-2B, H-1B |
| H-19A | H-19B, H-2B | H-19B | H-19A, H-2A, H-4 |
| H-19B | H-19A, H-4 | ||
The structure assigned to 1 is closely related to those of subglutinols A and B (6 and 7, Figure 1), two epimeric diterpenoid pyrones isolated from Fusarium subglutinans and manifesting immunosuppressive activity.19 When the set of data obtained for 1 was compared with the literature data reported for subglutinol A,19 the physical properties (mp and [α]25D) were different, whereas the spectroscopic data (IR, UV, and 1H and 13C NMR) were similar. This is in agreement with the structure assigned to 1, which appeared to be an epimer of subglutinol A differing by the cis stereochemistry at the B/C ring junction. Indeed, as expected, the 1H NMR spectra of 1 and subglutinol A differed by the 3JH,H coupling constants measured for protons H-8 and H2-7. For subglutinol A, one of the two 3JH,H's amounted to 11.5 Hz,19 indicating an anti arrangement, while for 1 both 3JH,H's were small, indicating a double gauche orientation between H-8 and H2-7. Furthermore, H-12 had the α-orientation in 1, similar to that reported for subglutinol A, but opposite to the β-position found in its 12-epimer subglutinol B (7). Consequently, the couplings between H-12 and H2-11 in 1 are similar to those observed in subglutinol A.
Figure 1.

Structures of higginsianins A and B (1 and 2), higginsianin A derivatives (3–5), subglutinols A and B (6 and 7), and diterpenoid BR-050 (8).
The absolute configuration of 1 was investigated by means of electronic circular dichroism (ECD) spectroscopy. Figures 3 and 4 show the absorption and ECD spectra measured for (−)-1 in methanol solution and in the solid state (dispersed in KCl matrix). The two ECD spectra are consistent with each other and show two major negative bands, namely, a very broad weaker band in the 250–350 nm region and a stronger one centered around 205 nm. The two ECD bands correspond to the two major absorption bands mainly associated with the transitions of the substituted α-pyrone chromophore and, at short wavelengths, of the two alkenes. The solid-state ECD spectrum shows minor additional features in the 215–240 nm region, whereas only a shoulder is visible in the solution spectrum. The consistency between the solution and solid-state spectra demonstrates that the effects of intermolecular exciton couplings in the crystals are negligible.20 The availability of the solid-state X-ray structure of 1 allowed its utilization as input geometry for calculating the ECD spectrum with time-dependent DFT21 for comparison with the solid-state experimental spectrum. This technique, known as the solid-state ECD/TDDFT approach, avoids the uncertainty associated with a conformational analysis and the use of optimized geometries22,23 and has allowed reliable configurational assignments of several natural products in recent years.24,25 In the current case, the two independent molecules found in the unit cell were considered together in the TDDFT calculation (inset in Figure 4), which was run at the CAM-B3LYP/TZVP level on (4R,5R,8R,9S,10R,12S)-1. The calculated spectrum (Figure 4) compares well with the experimental solid-state CD spectrum. In particular, the negative sign, relative intensity, and position of the two major ECD bands are reproduced. The unusually broad first ECD band is probably related to an effect intrinsic to the solid state, possibly including the distortion of the pyrone ring. In fact, the band is less broad in the solution spectra, and a single π–π* transition centered on the α-pyrone is calculated in this region. The agreement between the spectra shown in Figure 4 allowed us to assign the absolute configuration of higginsianin A as (−)-(4R,5R,8R,9S,10R,12S)-1. This assignment matches that of subglutinol A (6), whose absolute configuration was determined by total synthesis.26 The two natural products differ, however, in the configuration at C-8, leading to their epimeric relationship.
Figure 3.
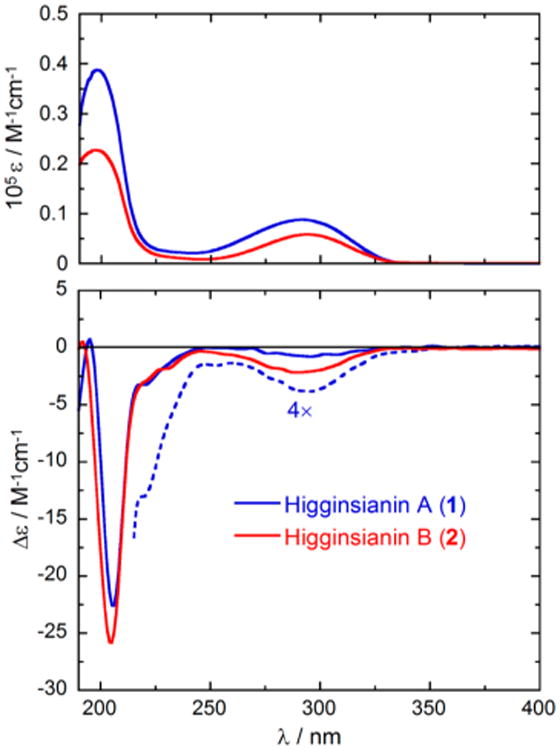
Absorption (top) and ECD spectra (bottom) of higginsianins A (1, blue) and B (2, red) in methanol solution. Sample concentration: 1, 2.9 mM and 11.7 mM (expansion, dashed blue line); 2, 2.6 mM.
Figure 4.
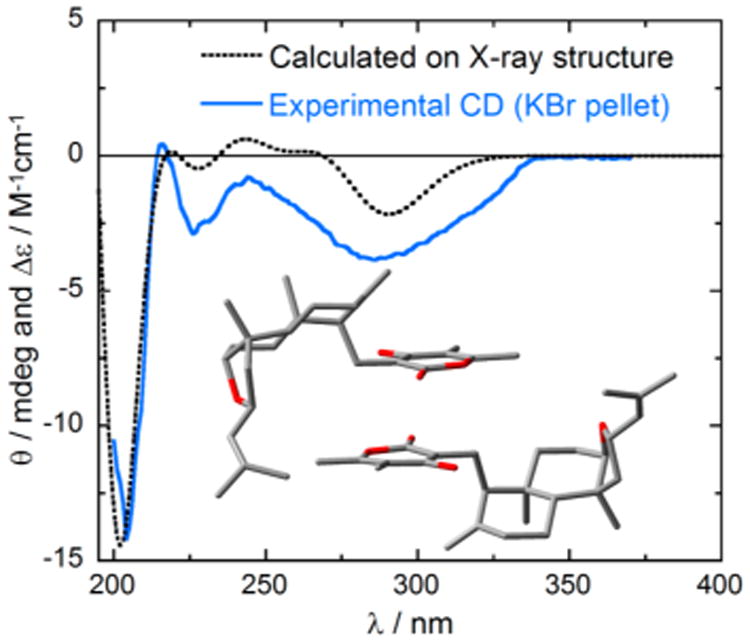
Solid blue line: Solid-state ECD spectrum of higginsianin A, (−)-1, as KCl pellet. Dotted black line: TDDFT-calculated ECD spectrum of (4R,5R,8R,9S,10R,12S)-1 at the CAM-B3LYP/TZVP level using the X-ray geometry as the input structure, including two molecules (as in the unit cell, see inset).
To obtain the initial structure–activity relationship, 1 was converted to three semisynthetic derivatives (3–5). Thus, acetylation of 1 with acetic anhydride in pyridine gave the corresponding 22-O-acetyl derivative (3, Figure 1). Its IR spectrum did not contain the hydroxy band, and the 1H NMR spectrum differed from that of 1 by the presence of the singlet at δ 2.31 due to the acetyl group. The ESIMS spectrum gave the ions corresponding to [2M + K]+, [2M + Na]+, [M + Na]+, and [M + H]+ at m/z 974, 958, 491, and 469. Furthermore, by treatment with an ethereal solution of diazomethane, 1 gave a mixture of two derivatives, 4 and 5 (Figure 1). Derivative 4 was the expected C-22 methyl ether27 whose IR spectrum did not show any hydroxy band. Its 1H NMR spectrum differed from that of 1 by the presence of the singlet of the OMe group at δ 3.86. Its EIMS spectrum contained the ions corresponding to [2M + K]+, [2M + Na]+, [2M + H]+, [M + K]+, [M + Na]+, and [M + H]+ at m/z 919, 903, 881, 479, 463, and 441. The structure of derivative 5 was determined by IR, NMR, and MS techniques and is shown in Figure 1. Its IR spectrum still contained the hydroxy band, and its 1H NMR spectrum differed from that of 1 by the presence of a broad singlet at δ 3.72 due to the H2-28 protons. The ESIMS contained the ions corresponding to [2M + K]+, [2M + Na]+, [M + K]+, [M + Na]+, and [M + H]+ at m/z 919 and 903, 479 and 463, and 441. The interesting formation of 5 can be explained with the proposed mechanism shown in Figure 5. Specifically, the mechanism involves the oxonium ylide intermediate I, which undergoes a rearrangement through the expansion of ring C, yielding 5. The alternative derivative showing the insertion of a methylene group between C-12 and O atoms of the furane ring was ruled out by the spectroscopic data of 5. In fact, the 1H NMR spectrum of 5 showed for H-12 and H2-28 signals typical for oxygenated methine (td, J = 8.3 and 7.2 Hz) and methylene (br s) at δ 4.81 and 3.72, respectively, very similar as in 1. Instead, with respect to 1, H-8 appeared upfield shifted (Δδ 2.07) at δ 1.57.
Figure 5.
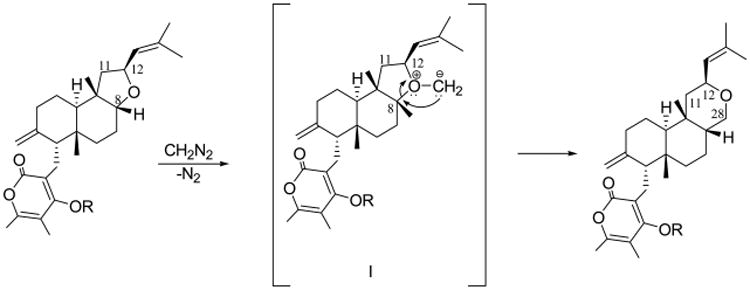
Proposed mechanism for the rearrangement 1 → 5.
Higginsianin B (2) had a molecular formula of C27H40O4 as deduced from its HRESIMS, consistent with eight hydrogen deficiencies, one less than was observed with 1. When its 1H and 13C NMR spectra (Table 1) were compared to those of 1, they differed only by the signal of C-12 bearing the dimethylethenyl side chain. The 1H NMR spectrum of 2 contained the signal of a new methylene group (H2-12), whose protons appeared both as multiplets at δ 2.11 and 1.85, both upfield shifted with respect to those of 1 (Δδ 2.67 and 2.93). As expected, in the 13C NMR spectrum the signal for C-12, found at δ 21.3, appeared as a triplet instead of a doublet and was also upfield shifted (Δδ 51.7). These results indicated that the 2,3,3,5-tetrasubstituted tetrahydrofuran ring found in 1 was open in 2, resulting in the methylene group (H2-12) and the secondary hydroxy at C-8. The chemical shifts of all protons and the corresponding carbons of 2, which were assigned on the basis of the couplings observed in the COSY, HSQC, and HMBC spectra, are shown in Table 1. Thus, higginsianin B (2) was determined to be 4-hydroxy-3-[6-hydroxy-5,8a-dimethyl-2-methylene-5-(4-methylpent-3-enyl)decahydronaphthalen-1-yl-methyl]-5,6-dimethylpyran-2-one. This structural assignment was confirmed by the HRESIMS spectrum, which contained the [M + Na]+ ion at 451.2820. When the same spectrum was recorded in negative-mode HR, the pseudomolecular ion [M – H]− was recorded at m/z 427.2826.
The relative configuration of 2 appeared to be the same as that of 1, as deduced from the correlations observed in the NOESY spectrum (Table 2). The absolute configuration of 2 was established by comparison of its ECD spectrum with that of 1 (Figure 3). Not surprisingly, the two compounds have very similar spectra in the entire measured range with the close similarity of all major bands. Moreover, the structural difference between 1 and 2 was observed in the region far from the main α-pyrone chromophore, whereas the environment in the immediate proximity to this chromophore was exactly the same. Therefore, 2 was assigned the same absolute configuration as 1, which is (−)-(4R,5R,8R,9S,10R)-2. Higginsianin B (2) appeared to be the C-8 epimer of the diterpenoid BR-050 (8) isolated from the insect pathogenic fungus Torrubiella luteorostrata together with the new macrocyclic metabolites named torrubiellutins A–C.28
The in vitro antiproliferative activity of compounds 1–5 was determined by means of the MTT colorimetric assay using five human and one murine cancer cell line (Table 3). The five human cancer cell lines included the Hs683 oligodendroglioma, the U373 glioblastoma (GBM), the A549 non-small-cell lung cancer (NSCLC), the MCF-7 breast carcinoma, and the SKMEL-28 melanoma models. The murine cell line was the B16F10 melanoma model. Our previous studies showed that the Hs68329–31 and B16F10 cells32,33 are sensitive to pro-apoptotic stimuli, whereas the U37329–31 and SKMEL2833 cell lines are more resistant to pro-apoptotic stimuli. The A549 cell line also displays some levels of resistance to pro-apoptotic stimuli,34 while the MCF-7 cells have inefficient caspase-3, which plays a major role in apoptotic cell death.35 The data in Table 3 clearly show that the apoptosis-resistant cancer cell subpanel, which included U373 GBM, SKMEL-28 melanoma, A549 NSCLC, and MCF-7 breast cancer cells, was significantly less sensitive (double-digit micromolar IC50's) to the antiproliferative effects induced by compounds 1–4 than Hs683 oligodendroglioma and B16F10 melanoma cells, lacking such apoptosis resistance (single-digit and submicromolar IC50's). In contrast, all cell lines had similar sensitivities to compound 5. Thus, the differential sensitivities in this cancer panel were independent of whether the compounds contained the closed (1, 3, and 4) or open (2) tetrahydrofuran ring C. In contrast, such cell-line-dependent selectivity was lost in compound 5, containing the tetrahydropyran moiety as ring C. It is likely that the substitution of the pyran for the furan ring fundamentally changes the overall shape of the molecule and thus alters its biological properties. The absence of any difference between 1 and its 22-O-acetyl derivative (3) could be due to a facile hydrolysis of 3 into 1 by intracellular esterases. However, the similar activity profile of the 22-O-methyl derivative (4) suggests that perhaps structural alterations in this part of the molecule are not important for activity.
Table 3. Determination of the Antiproliferative Activity of Compounds 1–5 Using the MTT Colorimetric Assaya.
| compound | glioma | carcinoma | melanoma | mean ± SEMe | mean of means ± SDe | |||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| Hs683f | U373 | A549 | MCF7 | SKMEL28 | B16F10 | |||
| 1 (exp 1)b | 1 | 63 | 38 | 19 | 73 | 1 | 33 ± 13 | 39 ± 8 |
| 1 (exp 2) | 1 | NDc | 33 | 82 | > 100 | 3 | >44 ± 20 | |
| 2 (exp 1) | 2 | 84 | 42 | 2 | 88 | 25 | 41 ± 16 | 38 ± 4 |
| 2 (exp 2) | 2 | ND | 29 | 53 | 89 | 3 | 35 ± 16 | |
| 3 | 1 | ND | 47 | 41 | 84 | 2 | 35 ± 16 | –d |
| 4 | 0.4 | ND | 16 | 38 | 48 | 1 | 21 ± 10 | – |
| 5 | 49 | ND | 35 | 24 | 51 | 25 | 37 ± 6 | – |
| mean ± SEM | 7 ± 5 | 81 ± 8 | 34 ± 4 | 37 ± 10 | 76 ± 8 | 9 ± 4 | ||
The data represent the IC50 concentration (μM), i.e., the compound concentration that reduces by 50% the growth of a given cell line (as compared to the control value) after culturing the cells for 72 h with the compound of interest.
Each experiment (“exp”) was carried out in sextuplicate. Two independent experiments (two times sextuplicates) were carried out for compounds 1 and 2, while one experiment (one time sextuplicate) was carried out for compounds 3, 4, and 5.
“ND”, not determined.
Not calculated.
SEM, means standard error of the mean; SD, standard deviation. SEMs were calculated for n > 3, while SDs were calculated for n < 3.
The origin and histological type of each cell line analyzed are as follows. Human glioma model lines included the Hs683 oligodendroglioma (ATCC code HTB-138) and the U373 glioblastoma (ECACC code 08061901) cell lines. Melanoma models included the human SKMEL-28 (ATCC code HTB-72) and the mouse B16F10 (ATCC code CRL-6475) cell lines. Human carcinoma models included the A549 NSCLC (DSMZ code ACC107) and the MCF-7 breast (DSMZ code ACC115).
Morphological analyses of the antiproliferative effects associated with 1 in Hs683 oligodendroglioma cells suggest that at the IC50 concentration (∼1 μM; Table 3) this compound induces cytostatic rather than cytotoxic effects (Figure 6). However, at a much higher concentration, i.e., 50 μM, the antiproliferative effects in Hs683 cancer cells are clearly cytotoxic (Figure 6). In addition, the quantitative video-microscopy (QVM) analysis performed on the Hs683 GBM cell line (Figure 7) indicates that growth of the cells is inhibited by ca. 50% after a 72 h treatment with 1 used at 1 μM. This is consistent with the MTT measurements (Table 3), revealing an IC50 of 1 μM after a similar treatment of Hs683 cells for 72 h with 1. The MTT colorimetric assay determines the levels of metabolically active cells, while the QVM analyses permit the direct visual observation of cells in cell culture. Thus, the two techniques are complementary, and the similar IC50 values determined using these two methods significantly increase the level of confidence in the results shown in Table 3. Finally, the QVM data also indicate that 1 is neither photosensitive nor photoactivable, which is an important attribute of a potential anticancer agent.
Figure 6.
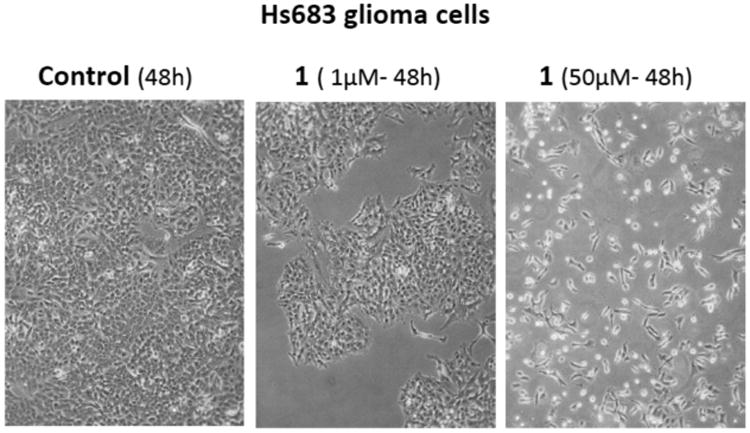
Morphological illustrations (phase-contrast microscopy) of the antiproliferative effects induced by higginsianin A (1) in Hs683 glioma cells at 1 versus 50 μM.
Figure 7.
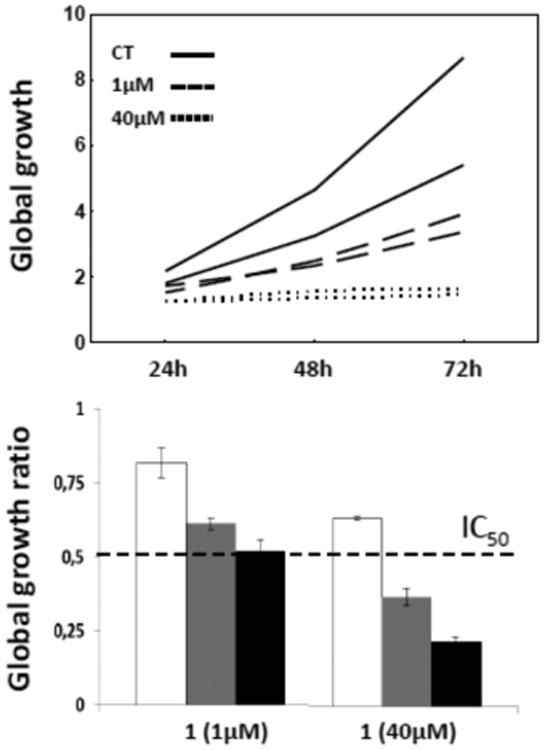
Quantitative effects of higginsianin A (1) on the Hs683 glioma cell line. Digitized images were obtained by means of computer-assisted phase-contrast microscopy (quantitative video-microscopy). A global growth ratio (the GGR index) was calculated, resulting in a value that can be directly compared to the IC50 value determined by MTT assay (the hatched horizontal line in the bottom chart, i.e., 1 μM (see Table 3). First, the global growth (GG) is calculated for each control and for each treated condition at 24, 48, and 72 h by dividing the number of cells on the last image (at 24, 48, and 72 h) by the number of cells on the first image (upper panel; full line: control condition; hatched line: 1 μM; dotted line: 40 μM). The GGR index is obtained by dividing the GG values calculated for cancer cells treated with 1 by the GG values calculated for the control. The experiment was performed once in duplicate, and the data represent the mean ± SEM values. White, gray, and black bars represent the data obtained at 24, 48 and 72 h, respectively.
In conclusion two new diterpenoid α-pyrones, named higginsianins A (1) and B (2), were isolated from the mycelium of the fungus C. higginsianum grown in liquid culture. Their structures, including relative and absolute configurations of all stereocenters, were fully elucidated using NMR techniques, X-ray crystallography, and electronic circular dichroism experiments. Higginsianin A (1) is the C-8 epimer of subglutinol A (6), previously isolated from F. subglutinans, while higginsianin B (2) is the C-8 epimer of diperpenoid BR-050 (8), previously isolated from Torrubiella luterostrata. These latter compounds did not show antiproliferative activity, but 6 exhibited anti-inflammatory activity with 8, which also showed antimalarial property. Instead, higginsianins A and B, as well as a number of semisynthetic analogues, were found to possess promising antiproliferative effects in a panel of six cancer cell lines, as determined independently with the MTT colorimetric assay and quantitative videomicroscopy. This supports the importance of the stereochemical differences in determining the biological activity. Furthermore, the promising cytostatic, rather than cytotoxic, activity found with the new diterpenoids warrants their further study as potential anticancer agents.
Experimental Section
General Experimental Procedures
Melting points were measured on a Gallenkamp melting point apparatus; optical rotations were measured in a MeOH solution on a Jasco P-1010 digital polarimeter; ECD spectra were recorded on a JASCO J-815 spectropolarimeter in MeOH. Solid-state ECD spectra were measured with the KCl pellet technique, described in Pescitelli et al., 2009.24 IR spectra were recorded as a glassy film on a PerkinElmer Spectrum One FT-IR spectrometer, and UV spectra were recorded in MeOH solution on a PerkinElmer Lambda 25 UV/vis spectrophotometer. 1H and 13C NMR spectra were recorded at 500 and 400 and at 125 and 100 MHz, respectively, in CDCl3 on Bruker spectrometers. The same solvent was used as an internal standard. Carbon multiplicities were determined by DEPT spectra.17 DEPT, COSY-45, HSQC, HMBC, and NOESY experiments17 were performed using Bruker microprograms. HRESIMS spectra were recorded on Thermo LTQ Velos, and ESI and APCIMS spectra were recorded on Agilent Technologies 6120 Quadrupole LC/MS instruments. Analytical and preparative TLC were performed on silica gel plates (Merck, Kieselgel 60, F254, 0.25 and 0.5 mm, respectively) or on reversed-phase (Whatman, KC18 F254, 0.20 mm) plates; the compounds were visualized by exposure to UV light and/or iodine vapors and/or by spraying first with 10% H2SO4 in MeOH and then with 5% phosphomolybdic acid in EtOH, followed by heating at 110 °C for 10 min. CC: Silica gel (Merck, Kieselgel 60, 0.063–0.200 mm).
Fungal Strain
The C. higginsianum isolate used in this study is IMI 349063 (CABI Culture Collection). The strain was originally collected in 1991 in Trinidad from spot symptoms on a living leaf of Brassica rapa subsp. chinensis (common name: pak-choi). This strain was selected because it is the most commonly used in research laboratories as a reference for plant pathology, evolutionary analyses, and phylogenetics. 36,37 The genome of C. higginsianum IMI 349063 has also been sequenced (GenBank accession no. CACQ00000000.2), and expression resources are available.36
Extraction and Purification of Higginsianis A and B
Small fragments of mycelium obtained by colonies of C. higginsianum actively growing on PDA were used for seeding Roux bottles containing 200 mL of a sterile defined liquid medium named M1-D.38 Bottles were kept in still condition at 25 °C in the dark in an incubator for 4 weeks, then filtered by Whatman no. 4 filter paper. The harvested mycelium was lyophilized (28.2 g from 8.37 L of culture filtrate) and macerated with EtOAc (3 × 1 L) for 24 h at room temperature in the dark. The organic extracts were combined, dehydrated with anhydrous Na2SO4, and evaporated under reduced pressure, yielding a brown oil (3.7 g), showing in vitro antiproliferative activity (<30 μg/mL; data not shown). This oil was purified by CC eluted with n-hexane–acetone (7:3), yielding 10 groups of homogeneous fractions. The residue of the fourth fraction was purified by CC eluted with CHCl3–i-PrOH (95:5), yielding eight groups of homogeneous fractions. The residue of the first fraction (245 mg), of this latter column, was crystallized using EtOAc–n-hexane, obtaining higginsianin A (1, 78.9 mg, Rf 0.80, Figure 1) as white crystals. The residues of the fifth and sixth fraction (174.2 and 260.0 mg, respectively) of the first column were combined and further purified by CC, eluted with CH2Cl2–acetone (9:1), giving 11 groups of homogeneous fractions. The residue of the fifth fraction (40.7 mg) was crystallized using EtOAc–n-hexane, obtaining further amounts of higginsianin A (1, 32.7 mg, for a total amount of 111.6 mg, 13.3 mg/L, Rf 0.80). 1 was recrystallized using MeOH–EtOAc (1:3) to obtain crystals suitable for X-ray analysis. The eighth fraction of the first column was obtained as a homogeneous solid and identified as higginsianin B (2, 37.4 mg, 4.4 mg/L, Rf 0.50) as reported below.
Higginsianin A (1), 3-[5a,9b-dimethyl-7-methylene-2-(2-methyl-propenyl)dodecahydronaphtho[2,1-b]furan-6-ylmethyl]-4-hydroxy-5,6-dimethylpyran-2-one
white crystals; mp 160 °C; [α]25D −67.3 (c 0.3); IR νmax 3225, 1666, 1642, 1563 cm−1; UV λmax nm (log ε) 293 (3.9); 1H and 13C NMR see Table 1; HRESIMS (+) m/z 449.2665 [calcd for C27H38NaO4 449.2668, M + Na]+; HRESIMS (−) m/z 425.2665 [calcd for C27H37O4 425.2671, M – H]−.
Higginsianin B (2), 4-hydroxy-3-[6-hydroxy-5,8a-dimethyl-2-methylene-5-(4-methylpent-3-enyl)decahydronaphthalen-1-yl-methyl]-5,6-dimethylpyran-2-one
amorphous solid; [α]25D −68.8 (c 0.25); IR νmax 3255, 1667, 1644, 1571 cm−1; UV λmax nm (log ε) 293 (3.8); 1H and 13C NMR see Table 1; HRESIMS (+) m/z 451.2820 [calcd for C27H40NaO4 451.2824, M + Na]+; HRESIMS (−) m/z 427.2826 [calcd for C27H39O4 427.2833, M – H]−.
22-O-Acetyl Derivative of 1 (3)
Higginsianin A (1, 4.0 mg) dissolved in pyridine (10 μL) was acetylated with acetic anhydride (10 μL) at room temperature for 1 h. The reaction was stopped by addition of MeOH, and the azeotrope, obtained by the addition of benzene, was evaporated by an N2 stream. The oily residue (4.3 mg) was purified by preparative TLC eluted with CHCl3–i-PrOH (95:5), to give 22-O-acetylhigginsianin A as a homogeneous compound (3, Rf 0.80, 3.8 mg). Derivative 3: IR νmax 1723, 1642, 1627, 1580, 1181 cm-1; UV λmax nm (log ε) 302 (3.5); 1H NMR, δ 5.22 (1H, br d, J = 9.1, H-13), 4.77 (1H, td, J = 9.1 and 7.2 Hz, H-12), 4.59 (1H, br s, H-19), 4.32 (1H, br s, H-19′), 3.71 (1H, br s, H-8), 2.65 (1H, m, H-20), 2.55 (2H, m, H-20′ and H-2), 2.31 (3H, s, MeCO), 2.21 (3H, br s, Me-26), 2.17 (1H, m, H-4), 2.10 (1H, m, H-2′), 1.87 (1H, m, H-11′), 1.79 (3H, br s, Me-27), 1.71 (3H, s, Me-15), 1.70 (3H, s, Me-16), 1.65 (1H, m, H-6), 1.57 (2H, m, H-10 and H-7), 1.40 (1H, m, H-1), 1.28 (1H, m, H-1′), 1.25 (1H, m, H-11′), 0.98 (3H, s, Me-17), 0.96 (3H, s, Me-18), 0.88 (2H, m, H-6′ and H-7′); ESIMS (+) m/z 974 [2M + K]+, 958 [2M + Na]+, 491 [M + Na]+, 469 [M + H]+.
Reaction of Higginsianin A with Diazomethane
To higginsianin A (1, 6.0 mg) dissolved in MeOH (2 mL) was added an ethereal solution of diazomethane (200 μL). The reaction was carried out overnight at room temperature in the dark. The reaction was stopped by evaporation under a N2 stream. The residue (6.2 mg) was purified by TLC, eluted with n-hexane–acetone (8:2), yielding derivatives 4 and 5, both as homogeneous compounds (2.9 and 2.1 mg, Rf 0.28 and 0.39). Derivative 4: IR νmax 1671, 1644, 1601 cm−1; UV λmax nm (log ε) 302 (3.5); 1H NMR δ 5.23 (1H, br d, J = 8.3, H-13), 4.77 (1H, td, J = 8.3 and 7.2 Hz, H-12), 4.51 (1H, br s, H-19), 4.19 (1H, br s, H-19′), 3.86 (3H, s, MeO), 3.71 (1H, br s, H-8), 2.62 (1H, dd, J = 13.5 and 3.5 Hz, H-20), 2.54 (1H, dd, J = 13.5 and 11.4 Hz, H-20′), 2.47 (1H, m, H-2), 2.23 (3H, br s, Me-26), 2.20 (1H, m, H-11), 2.15 (1H, m, H-2′), 1.95 (1H, dd, J = 11.4 and 3.5 Hz, H-4), 1.89 (3H, br s, Me-27), 1.79 (1H, m, H-6), 1.71 (6H, s, Me-15 and Me-16), 1.50 (2H, m, H-10 and H-7), 1.41 (1H, m, H-1), 1.28 (1H, m, H-1′), 1.25 (1H, m, H-11′), 0.95 (3H, s, Me-17), 0.93 (3H, s, Me-18), 0.82 (2H, m, H-6′ and H-7′); ESIMS (+) m/z 919 [2M + K]+, 903 [2M + Na]+, 881 [2M + H]+, 479 [M + K]+, 463 [M + Na]+, 441 [M + H]+. Derivative 5: IR νmax 3225, 1670, 1647, 1566 cm−1; UV λmax nm (log ε) 301 (3.3); 1H NMR, δ 5.23 (1H, br d, J = 8.3, H-13), 4.81 (1H, td, J = 8.3 and 7.2 Hz, H-12), 4.54 (1H, br s, H-19), 4.28 (1H, br s, H-19′), 3.72 (2H, br s, H2-28), 2.71 (1H, t, J = 12.2 Hz, H-20), 2.67 (1H, dd, J = 12.2 and 3.8 Hz, H-20′), 2.46 (1H, br t, J = 12.6 Hz, H-2), 2.21 (1H, m, H-11), 2.19 (1H, m, H-2′), 2.18 (3H, br s, Me-26), 2.12 (1H, m, H-6), 2.07 (1H, m, H-4), 1.85 (1H, m, H-1), 1.71 (6H, s, Me-15 and Me-16), 1.70 (2H, m, H-10 and H-7), 1.57 (1H, m, H-8), 1.42 (2H, m, H2-1), 1.23 (1H, m, H-11′), 0.96 (3H, s, Me-17), 0.95 (3H, s, Me-18), 0.80 (2H, m, H-6′ and H-7′); ESIMS (+) m/z 919 [2M + K]+, 903 [2M + Na]+, 479 [M + K]+, 463 [M + Na]+, 441 [M + H]+.
Crystal Structure Determination of Higginsianin A (1)
Colorless single crystals were obtained at ambient temperature by slow evaporation of a MeOH–EtOAc (1:3) solution. X-ray data collection was performed at 173 K on a Bruker-Nonius KappaCCD diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.710 73 Å, CCD rotation images, thick slices, φ and ω scans to fill the asymmetric unit). Cell parameters were obtained from a least-squares fit of the θ angles of 201 reflections in the range 3.404° ≤ θ ≤ 21.613°. A semiempirical absorption correction (multiscan, SADABS) was applied. The structure was solved by direct methods (SIR97 package)39 and refined by the full matrix least-squares method on F2 against all independent measured reflections (SHELXL program of SHELX97 package).40 All H atoms were placed in calculated positions and allowed to ride on carrier atoms (C–H in the range 0.95–1.00 Å; U = 1.2Uiso or 1.5 for methyl C); 16 324 intensities were collected in the range 3.04° ≤ θ ≤ 25.00°, 8274 independent reflections and 573 parameters. In the absence of strong anomalous scatterers it was not possible to establish the absolute configuration, the Flack parameter41 being meaningless. Voids of 206.7 Å3, containing disordered MeOH solvent crystallization molecules, were found in the unit cell. It was not possible to model the disordered solvent, and Platon/SQUEEZE procedure was used to obtain solvent-free reflection data. The final refinement converged to R1 = 10.26 for 3310 observed reflections having [I > 2σ(I)] and wR2 = 0.2219 for all data. Final minimum and maximum residual electronic density was −0.357 and 0.456 e/Å3. The presence of disordered crystallization solvent in the structure accounts for the rather high values for R and the residual electronic density. Crystal data: formula C27H38O4; formula weight 426.57 g/mol; triclinic P1; a = 7.2340(1) Å, b = 10.321(1) Å, c = 19.308(2) Å, α = 85.37(1)° β = 79.85(2)°, γ = 69.87(1) 120.0°; V = 1332.0(3) Å3; Z = 2.
CCDC-1057132 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Computational Section
DFT and TDDFT calculations were run with Gaussian'09,42 with default grids and convergence criteria. Starting from the X-ray structure of 1, the two independent molecules of the unit cell were considered together. Missing hydrogen atoms were added, and all hydrogen atoms were optimized with the DFT method at the B3LYP/6-31G(d) level. TDDFT calculations were run using various functionals (B3LYP, CAM-B3LYP, PBE0, M06) and basis sets (SVP, TZVP). The CAM-B3LYP/TZVP combination was employed for the final calculation, including 48 excited states (roots). CD spectra were generated by applying a Gaussian band shape with a 2250 cm−1 exponential half-width, from dipole-length rotational strengths. The difference from dipole-velocity values was negligible. The calculated spectrum in Figure 4 is red-shifted by 10 nm and scaled by a factor of 4 to compare with the experimental spectrum.
Cancer Cell Lines
Human and murine cancer cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), the European Collection of Cell Culture (ECACC, Salisbury, UK), and the Deutsche Sammlung von Mikroorganismen and Zellkulturen (DSMZ, Braunschweig, Germany). The code number and histological type of each of the cell lines used in the current study are detailed in Table 3. All the details relating to the culture media used for each cell line are provided in Lefranc et al. 2013.31
Determination of the IC50 Growth Inhibitory Concentrations in Vitro
The MTT colorimetric assay was used as detailed previously.31 The MTT assay measures the number of metabolically active (thus living) cells that are able to transform the yellow substrate 3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into the blue formazan dye via a mitochondrial reduction involving succinate dehydrogenase.31 The amount of formazan obtained at the end of the experiment (measured by spectrophotometry) is directly proportional to the number of living cells. The determination of the optical density in the control compared to the treated cells therefore enables quantitative measurements of the effects of compounds on the growth of cell lines in vitro. Each experimental condition was assessed in six replicates.
Computer-Assisted Phase-Contrast Microscopy (Quantitative Videomicroscopy) Analysis
The direct visualization of the in vitro antiproliferative effects induced by 1 (higginsianin A) at 1 and 40 μM in human Hs683 and U373 glioma cells was performed as detailed elsewhere.31
Supplementary Material
Acknowledgments
The authors V.M., M.F., R.L., and R.K. thank the Belgian Brain Tumor Support (BBTS), the National Cancer Institute (CA186046-01A1), and the Welch Foundation (AI-0045) for financial support. A.E. is associated with the Istituto di Chimica Biomolecolare del CNR, Pozzuoli, Italy. R.K. is a director of research with the Fonds National de la Recherche Scientifique (FRS-FNRS, Belgium).
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.5b00779.
Notes: The authors declare no competing financial interest.
References
- 1.Evidente A, Kornienko A, Cimmino A, Andolfi A, Lefranc F, Mathieu V, Kiss R. Nat Prod Rep. 2014;31:617–627. doi: 10.1039/c3np70078j. [DOI] [PubMed] [Google Scholar]
- 2.Kornienko A, Evidente A, Vurro M, Mathieu V, Cimmino A, Evidente M, van Otterlo WAL, Dasari R, Lefranc F, Kiss R. Med Res Rev. 2015;35:937–967. doi: 10.1002/med.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutton BC. In: Colletotrichum, Biology, Pathlogy and Control. Bailey JA, Jeger MJ, editors. CAB International; Walliford: 1992. pp. 1–26. [Google Scholar]
- 4.Hyde KD, Cai L, McKenzie EHC, Yang YL, Zhang JZ, Prihastuti H. Fungal Diversity. 2009;39:96–113. [Google Scholar]
- 5.Hyde KD, Chomnuti P, Crous PW, Groenewald JZ, Damm U, Ko TW, Shivas RG, Summerell BA, Tan YPA. Personia. 2010;25:50–60. doi: 10.3767/003158510X548668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lubbe CM, Demman S, Lamprechi SC, Crous PW. Australas Plant Pathol. 2006;35:37–41. [Google Scholar]
- 7.Abang MM, Abraham WR, Asiedu R, Hoffmann P, Wolf G, Winter S. Mycol Res. 2009;113:130–140. doi: 10.1016/j.mycres.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Crouch JA, Beirn LA, Cortese LA, Bonos SB, Clarke BB. Mycol Res. 2009;113:1411–1421. doi: 10.1016/j.mycres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Kim HR, Lim TH, Kim J, Kim YH, Kim HT. Plant Pathol J. 2009;25:13–20. [Google Scholar]
- 10.Masyahit M, Kamaruzaman S, Yahya A, Ghazali M. Am J Appl Sci. 2009;6:902–912. [Google Scholar]
- 11.Phoulivong S, Cai L, Chen H, McKenzie EHC, Abdelsalam K, Chukeatirote E, Hyde KD. Fungal Divers. 2010;44:33–43. [Google Scholar]
- 12.García-Paión CM, Collado IG. Nat Prod Rep. 2003;20:426–431. doi: 10.1039/b302183c. [DOI] [PubMed] [Google Scholar]
- 13.Damm U, O'Connell RJ, Groenewald ZJ, Crous PW. Stud Mycol. 2014;79:49–84. doi: 10.1016/j.simyco.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakanishi K, Solomon PH. Infrared Absorption Spectroscopy. 2nd. Holden Day; Oakland: 1977. pp. 17–44. [Google Scholar]
- 15.Scott AI. Interpretation of Ultraviolet Spectra of Natural Products. Pergamon Press LTD; Oxford: 1964. pp. 135–164. [Google Scholar]
- 16.Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds – Tables of Spectral Data. 3rd. Springer-Verlag; Berlin: 2000. pp. 161–243. [Google Scholar]
- 17.Berger S, Braun S. 200 and More Basic NMR Experiments: a Practical Course. 1st. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 18.Breitmaier E, Voelter W. Carbon-13 NMR Spectroscopy. VCH; Weinheim: 1987. pp. 183–280. [Google Scholar]
- 19.Lee JC, Lobkovsky E, Pliam NB, Strobel G, Clardy J. J Org Chem. 1995;60:7076–7077. [Google Scholar]
- 20.Padula D, Di Pietro S, Capozzi MAM, Cardellicchio C, Pescitelli G. Chirality. 2014;26:462–470. doi: 10.1002/chir.22270. [DOI] [PubMed] [Google Scholar]
- 21.Autschbach J, Nitsch-Velasquez L, Rudolph M. Top Curr Chem. 2010;298:1–98. doi: 10.1007/128_2010_72. [DOI] [PubMed] [Google Scholar]
- 22.Pescitelli G, Di Bari L, Berova N. Chem Soc Rev. 2011;40:4603–4625. doi: 10.1039/c1cs15036g. [DOI] [PubMed] [Google Scholar]
- 23.Polavarapu P. Chirality. 2012;24:909–920. doi: 10.1002/chir.22015. [DOI] [PubMed] [Google Scholar]
- 24.Pescitelli G, Kurtán T, Flörke U, Krohn K. Chirality. 2009;21:E181–E201. doi: 10.1002/chir.20795. [DOI] [PubMed] [Google Scholar]
- 25.Pescitelli G, Kurtáan T, Krohn K. In: Comprehensive Chiroptical Spectroscopy. Berova N, Woody RW, Polavarapu P, Nakanishi K, editors. Wiley; New York: 2012. pp. 145–176. [Google Scholar]
- 26.Kim H, Baker JB, Park Y, Park HB, DeArmond PD, Kim SH, Fitzgerald MC, Lee DS, Hong J. Chem - Asian J. 2010;5:1902–1910. doi: 10.1002/asia.201000147. [DOI] [PubMed] [Google Scholar]
- 27.Carey FA, Sandberg RJ. Advanced Organic Chemistry Part B Reactions and Synthesis. Kluver Academic/Plenum; New York: 2001. pp. 152–152.pp. 608–609. [Google Scholar]
- 28.Pittayakhajonwut P, Usuwan A, Intaraudom C, Khoyaiklang P, Supothina S. Tetrahedron. 2009;65:6069–6073. [Google Scholar]
- 29.Branle F, Lefranc F, Camby I, Jeuken J, Geurts-Moespot A, Sprenger S, Sweep F, Kiss R, Salmon I. Cancer. 2002;95:641–655. doi: 10.1002/cncr.10710. [DOI] [PubMed] [Google Scholar]
- 30.Mathieu V, De Nève N, Le Mercier M, Dewelle J, Gaussin JF, Dehoux M, Kiss R, Lefranc F. Neoplasia. 2008;10:1383–1392. doi: 10.1593/neo.08928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefranc F, Nuzzo G, Hamdy NA, Fakhr I, Moreno Y, Banuls L, Van Goietsenoven G, Villani G, Mathieu V, van Soest R, Kiss R, Ciavatta ML. J Nat Prod. 2013;76:1541–1547. doi: 10.1021/np400107t. [DOI] [PubMed] [Google Scholar]
- 32.Mathieu V, Le Mercier M, De Neve N, Sauvage S, Gras T, Roland I, Lefranc F, Kiss R. J Invest Dermatol. 2007;127:2399–23410. doi: 10.1038/sj.jid.5700869. [DOI] [PubMed] [Google Scholar]
- 33.Van Goietsenoven G, Hutton J, Becker JP, Lallemand B, Robert F, Lefranc F, Pirker C, Vandenbussche G, Van Antwerpen P, Evidente A, Berger W, Prévost M, Pelletier J, Kiss R, Kinzy TG, Kornienko A, Mathieu V. FASEB J. 2010;24:4575–4584. doi: 10.1096/fj.10-162263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieu A, Remmelink M, D'Haene N, Penant S, Gaussin JF, Van Ginckel R, Darro F, Kiss R, Salmon I. Cancer. 2004;101:1908–1918. doi: 10.1002/cncr.20571. [DOI] [PubMed] [Google Scholar]
- 35.Dumont P, Ingrassia L, Rouzeau S, Ribaucour F, Thomas S, Roland I, Darro F, Lefranc F, Kiss R. Neoplasia. 2007;9:766–776. doi: 10.1593/neo.07535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Connell R, Herbert C, Sreenivasaprasad S, Khati M, Esquerré-Tugayé MT, Dumas B. Mol Plant-Microbe Interact. 2004;17:272–282. doi: 10.1094/MPMI.2004.17.3.272. [DOI] [PubMed] [Google Scholar]
- 37.Baroncelli R. PhD Thesis. University of Warwick; Coventry, United Kingdom: 2012. Colletotrichum acutatum sensu lato: from diversity study to genome analysis. [Google Scholar]
- 38.Pinkerton F, Strobel G. Proc Natl Acad Sci U S A. 1976;73:4007–4011. doi: 10.1073/pnas.73.11.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R. J Appl Crystallogr. 1999;32:115–119. [Google Scholar]
- 40.Sheldrick GM. Acta Crystallogr, Sect A: Found Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 41.Flack HD. Acta Crystallogr, Sect A: Found Crystallogr. 1983;A39:876–881. [Google Scholar]
- 42.Frisch MJ, et al. Gaussian'09, Revision D.01. Gaussian, Inc.; Wallingford, CT: 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.