

ABSTRACT
The mechanisms of viral control and loss of viral control in chronically infected individuals with or without protective HLA class I alleles are not fully understood. We therefore characterized longitudinally the immunological and virological features that may explain divergence in disease outcome in 70 HIV-1 C-clade-infected antiretroviral therapy (ART)-naive South African adults, 35 of whom possessed protective HLA class I alleles. We demonstrate that, over 5 years of longitudinal study, 35% of individuals with protective HLA class I alleles lost viral control compared to none of the individuals without protective HLA class I alleles (P = 0.06). Sustained HIV-1 control in patients with protective HLA class I alleles was characteristically related to the breadth of HIV-1 CD8+ T cell responses against Gag and enhanced ability of CD8+ T cells to suppress viral replication ex vivo. In some cases, loss of virological control was associated with reduction in the total breadth of CD8+ T cell responses in the absence of differences in HIV-1-specific CD8+ T cell polyfunctionality or proliferation. In contrast, viremic controllers without protective HLA class I alleles possessed reduced breadth of HIV-1-specific CD8+ T cell responses characterized by reduced ability to suppress viral replication ex vivo. These data suggest that the control of HIV-1 in individuals with protective HLA class I alleles may be driven by broad CD8+ T cell responses with potent viral inhibitory capacity while control among individuals without protective HLA class I alleles may be more durable and mediated by CD8+ T cell-independent mechanisms.
IMPORTANCE Host mechanisms of natural HIV-1 control are not fully understood. In a longitudinal study of antiretroviral therapy (ART)-naive individuals, we show that those with protective HLA class I alleles subsequently experienced virologic failure compared to those without protective alleles. Among individuals with protective HLA class I alleles, viremic control was associated with broad CD8+ T cells that targeted the Gag protein, and CD8+ T cells from these individuals exhibited superior virus inhibition capacity. In individuals without protective HLA class I alleles, HIV-1-specific CD8+ T cell responses were narrow and poorly inhibited virus replication. These results suggest that broad, highly functional cytotoxic T cells (cytotoxic T lymphocytes [CTLs]) against the HIV-1 Gag protein are associated with control among those with protective HLA class I alleles and that loss of these responses eventually leads to viremia. A subset of individuals appears to have alternative, non-CTL mechanisms of viral control. These controllers may hold the key to an effective HIV vaccine.
INTRODUCTION
HIV remains a global problem, and sub-Saharan Africa continues to bear the brunt of the epidemic, accounting for 67% of the infected people worldwide (1). Understanding the mechanisms of natural viral control in HIV infection is crucial for the identification of correlates of immune protection and for the design of an effective HIV vaccine. Previous studies have linked HIV control to a number of immunological factors, particularly HIV-specific CD8+ cytotoxic T lymphocytes (CTLs), which have been demonstrated to play an important role (2–5). However, virus-specific CD8+ T cell immune responses are not equally effective in HIV control, as the majority of infected individuals progress to disease despite the presence of these cells. HIV is able to evade immune responses by developing mutations that mediate escape from CTL recognition (6–12). In addition, the expression of certain HLA class I molecules by HIV-infected patients, such as HLA-B*27, HLA-B*57, HLA-B*58:01, HLA-B*81:01, and HLA-A*74:01, is associated with better clinical disease outcomes in some population settings (3, 13–18). HLA class I proteins present viral peptides on the surface of antigen-presenting cells to CTLs, and a major mechanism by which these protective alleles slow HIV disease progression is believed to be through CTL activity (18).
The exact mechanisms of viral control among individuals with protective HLA class I alleles are not fully understood. Analyses of HIV long-term nonprogressors (LTNPs) and elite controllers (ECs) suggest diverse and multifactorial mechanisms of natural control of viremia. Factors contributing to elite control (undetectable HIV viral load) may include attenuated viruses with slow viral evolution (19, 20), although replication-defective viruses are by no means always present (21–24). HLA-B*57-positive elite controllers maintained viral suppression despite the presence of escape mutations, with evidence of CTL responses against unmutated epitopes and de novo responses against epitopes with escape mutations (25–27).
Although the majority of HIV-1-infected individuals with controlled viremia possess protective HLA class I alleles, a substantial minority do not. For example, approximately 60% of Caucasian elite controllers express HLA-B*27 and/or HLA-B*57, and approximately 60% of South African elite controllers express HLA-B*57/58:01 and/or HLA-B*81:01 (28–30). In these cases of viremic control in the absence of protective HLA molecules, it is unclear whether immunological or other mechanisms of viral control are involved. Moreover, among controllers with or without protective HLA class I alleles, the infection is dynamic such that loss of viral control is observed at least in a subset of individuals who were previously elite controllers. The best-studied examples of progression in elite controllers are of HLA-B*27-positive subjects in whom escape within the immunodominant Gag epitope appears to precipitate loss of viremic control (10, 31). However, the mechanisms underlying the loss of viral control in previous controllers are largely undetermined, and few studies have addressed this issue. Thus, there is a clear need to identify mechanisms responsible for control in individuals with long-term chronic viral control, especially contrasting those with protective HLA class I alleles versus those with nonprotective HLA class I alleles in order to gain comprehensive understanding of viral control mechanisms that may be harnessed in prevention or therapeutic strategies.
Here, we investigated immunological and virological factors mediating control and loss of virological control in a cohort of 70 HIV-1-infected antiretroviral therapy (ART)-naive South African adults from Durban, 35 of whom possessed protective HLA class I alleles. Participants were followed longitudinally, with some maintaining virological control throughout the study duration whereas others experienced virologic failure. Specifically, we set out to test the hypothesis that progression in viremic controllers (VCs) is generally precipitated by CTL escape, in the setting of diminished CD8+ T cell breadth and polyfunctionality and lowered antiviral efficacy. To this end, we determined in these study subjects the magnitude and breadth of CTL responses, the presence or emergence of CTL escape variants, CD8+ T cell polyfunctionality and proliferation, and the ex vivo ability of CTLs to suppress HIV-1 replication before and after virologic failure.
MATERIALS AND METHODS
Study subjects.
Seventy HIV-1 subtype C-chronically infected participants from the Sinikithemba (SK) cohort in Durban, South Africa (3, 4, 32), were included in the current study. Participants were ART naive at all the time points analyzed. The date of HIV infection is unknown for all participants. Viral loads (VLs) for all subjects were measured every 6 months with the Roche Amplicor version 1.5 test, and CD4 T cell counts were measured every 3 months by TruCount technology using flow cytometry. The University of KwaZulu-Natal's Biomedical Research Ethics Committee approved the study, and all subjects provided written informed consent for participation in the study.
Definitions of study groups and subgroups.
The 70 study participants studied were divided into two groups based on their VLs at enrollment: baseline viremic controllers (bVCs) were enrolled with a VL below 2,000 HIV RNA copies/ml and baseline noncontrollers (bNCs) were enrolled with a VL above 100,000 HIV RNA copies/ml. Longitudinal follow-up of bVCs allowed us to further subdivide this group into 2 subgroups based on progression status: viremic controllers (VCs) were defined as individuals who were enrolled with a VL of less than 2,000 HIV RNA copies/ml and maintained this low viral load for the entire period of study (median of 4 years; interquartile range [IQR], 4 to 6 years), and failing viremic controllers (fVCs) were defined as bVCs who were enrolled with a VL of <2,000 HIV RNA copies/ml but subsequently lost virological control. We defined the loss of viral control as an increase in VL to more than 10,000 HIV RNA copies/ml at a minimum of 2 time points during the time of follow-up (median of 6 years; IQR, 5 to 6 years). These groups are illustrated in Fig. 1.
FIG 1.

Groups and subgroups of study participants. The 70 HIV-1 subtype C-chronically infected participants were from the Sinikithemba (SK) cohort in Durban, South Africa. Viremic controllers (VC+/−) were enrolled with a VL of below 2,000 HIV RNA copies/ml and maintained this low VL for the entire enrollment duration. Failing VCs (fVCs) were enrolled with a VL of below 2,000 HIV RNA copies/ml, and the VL increased to more than 10,000 HIV RNA copies/ml at a minimum of 2 subsequent time points.
HLA class I typing and ELISpot assay for the measurement of HIV-specific immune responses.
HLA class I typing was performed on genomic DNA samples extracted from blood at the time of enrollment as described previously (3). Of the 70 patients included in this study, 35 possessed protective HLA class I alleles (A*74:01, B*57, B*58:01, and B*81:01) (3, 13–17). These HLA alleles were selected as the principal protective alleles and are consistently the most protective that have been identified in the Durban study population (17, 30, 33). The HLAs that restricted responses seen in the nonprotective HLA group include HLAs B*15:10, B*08:01, B*44:03, B*14:02, B*42:01, B*45:01, B*39:10, and B*40:01 alleles. HIV immune responses were enumerated from frozen and freshly isolated whole peripheral blood mononuclear cells (PBMCs) by the gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay as previously described (3, 34). PBMCs were stimulated with 410 consensus clade C 18-mer overlapping peptides (OLPs) covering the entire proteome followed by confirmations with individual peptides within a reactive pool at a final concentration of 2 μg/ml and consensus clade C optimal peptides corresponding to each patient's HLA-B allele, also at a final concentration of 2 μg/ml per peptide.
Sequencing of the gag gene.
Population sequencing of the gag gene was performed as previously described (32). In brief, viral RNA was isolated from plasma samples followed by reverse transcription to generate cDNA. The gag sequences were amplified by nested PCR, purified, and directly sequenced. The sequences were then analyzed using the ABI 3130xl genetic analyzer (Applied Biosystems, Foster City, CA, USA). Sequence data were aligned to HIV-1 subtype B reference strain HXB2 (GenBank accession number K03455), and insertions with respect to HXB2 were stripped before further analysis. Editing and alignment of sequences were carried out using the Sequencher version 5 (Gene Codes Corp., Ann Arbor, MI, USA) and SeAl version 2.0a11 software, available online (http://tree.bio.ed.ac.uk/software/seal/) (A. Rambaut, Department of Zoology, University of Oxford, Oxford, United Kingdom), respectively. Some of the baseline Gag sequences used in this study were obtained from the GenBank database (accession numbers HM593195, HM593368.1, HM593206.1, HM593194.1, FJ198948.1, HM593285, HM593444, HM593233.1, HM593353.1, HM593360.1, HM593247, HM593347.1, HM593320, HM593295, and HM593217). New Gag sequences generated in this study are available in GenBank.
Flow cytometry and intracellular cytokine staining assay.
Freshly thawed cryopreserved PBMCs were left to rest at 37°C with 5% CO2 for 4 h. A total of 200,000 PBMCs per well were stimulated with consensus clade C Gag peptide pools (2 μg/ml/peptide), and phytohemagglutinin (PHA)-stimulated PBMCs (2 μg/ml) were used as a positive control. Anti-CD107a-phycoerythrin (PE)-Cy7 (BD Biosciences, San Jose, CA, USA) antibody and Golgi stop and Golgi plug (Sigma-Aldrich, St. Louis, MO, USA) were added in each well, and cells were incubated overnight at 37°C with 5% CO2.
Intracellular cytokine staining was performed as described previously (35, 36). The cells were then acquired on a flow cytometer (LSR II; BD Biosciences). The 5 functions studied were CD107a, IFN-γ, interleukin-2 (IL-2), macrophage inflammatory protein 1β (MIP-1β), and tumor necrosis factor alpha (TNF-α). Between 30,000 and 100,000 events were acquired per sample. The acquired data were analyzed by FlowJo version 9.6.2 (Tree Star, San Carlos, CA, USA). The gating strategy is shown in Fig. 2A. Boolean gating was performed in order to allow creation of a full array of possible combinations of up to 32 response patterns. Positive responses were reported after background correction, and the percentage of epitope-specific CD8+ T cell responses had to be at least two times higher than background for each tested marker. PESTLE (version 1.6.2) and SPICE 5.0 (Mario Roederer, ImmunoTechnology Section, Vaccine Research Center, NIH, Bethesda, MD, USA) were used to analyze the multifunctional data.
FIG 2.
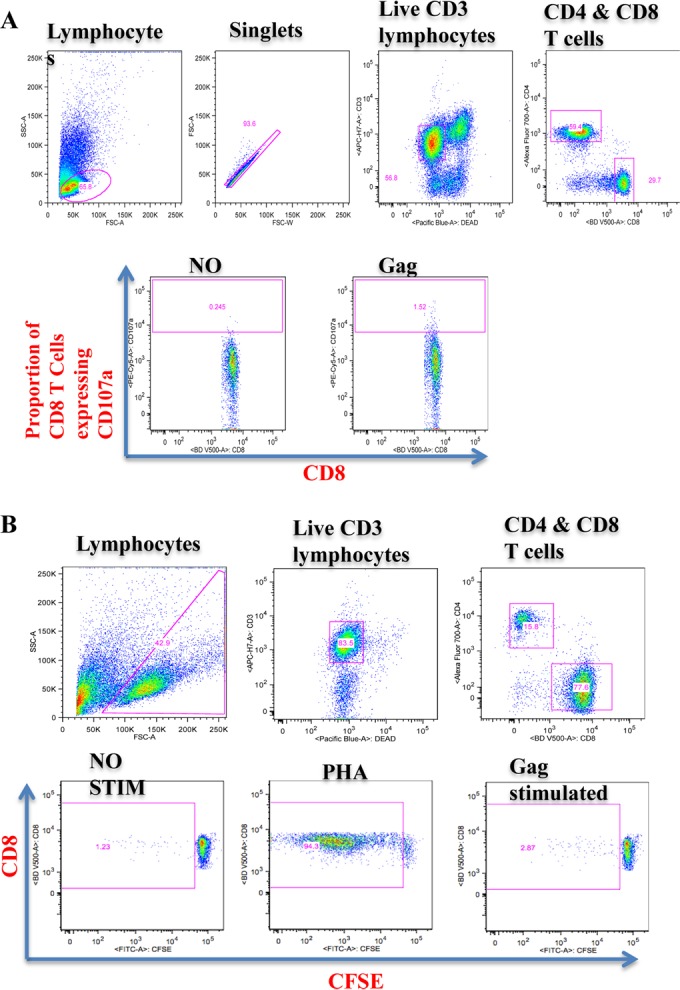
Gating strategy for polyfunctionality (A) and proliferative capacity (B) of CD8+ T cells upon stimulation with Gag peptide pools. For polyfunctionality of CD8+ T cells (A), the initial gating was on lymphocytes followed by the forward scatter height (FCS-H) versus forward scatter area (FSC-A) to eliminate the doublets. We then gated on the live CD3+ T cell population followed by gating of CD8+ and CD4+ T cell populations, followed by the gating for individual respective functions (set based on the negative control); these were used to identify positive responses. For the proliferation of CD8+ T cells (B), the initial gating was on lymphocytes followed by gating on the live CD3+ T cell population and then on CD8+ and CD4+ T cell populations. The next gates were set on the CFSE-negative CD8+ population to identify proliferated CD8+ T cell populations. The individual gating for proliferating cells was set based on the negative control (no stimulation); these were used to identify positive responses by subtraction from stimulated proliferating population. SSC, side scatter; FITC, fluorescein isothiocyanate.
CFSE proliferation assay.
Freshly thawed cryopreserved PBMCs were left to rest at 37°C with 5% CO2 for 4 h and then stained with carboxyfluorescein succinimidyl ester (CFSE) dye (Invitrogen, Paisley, United Kingdom) for 7 min at 37°C with 5% CO2. A total of 400,000 CFSE-labeled PBMCs per well were stimulated with Gag peptide pools (2 μg/ml/peptide) or PHA (2 μg/ml) as a positive control, followed by incubation at 37°C with 5% CO2 for 7 days. After 7 days, cells were then washed and stained with a live/dead marker (fixable blue dead-cell stain) (Invitrogen) for 10 min, followed by surface staining with anti-CD3 allophycocyanin (APC) H7, anti-CD4 Alexa 700, and anti-CD8 VD 500 (BD Biosciences) and a dump channel panel comprising anti-CD14 Pacific Blue, anti-CD19 Pacific Blue, and anti-CD16/56 Pacific Blue antibodies (BioLegend, San Diego CA, USA). The cells were incubated in the dark at room temperature for 20 min and then washed twice. Between 30,000 and 100,000 events were acquired per well (sample). The acquired data were analyzed by FlowJo version 9.6.2 (Tree Star). The gating strategy is shown in Fig. 2B. GraphPad Prism version 5.0a software (GraphPad Software, San Diego, CA, USA) was used to analyze group data sets.
Ex vivo viral inhibition assay.
An ex vivo viral inhibition assay was carried out to determine the ability of CD8+ T cells to suppress virus replication by measuring the amount of HIV p24 antigen as previously described (36, 37). Log inhibition values were calculated by subtracting log10 p24 values of the infected CD4+ T cells cocultured with CD8+ T cells from log10 p24 values of the infected CD4+ T cells without CD8+ T cells at day 7. GraphPad Prism version 5.0a software (GraphPad Software, San Diego, CA, USA) was used to analyze group data sets.
Statistical analysis.
GraphPad Prism version 5.0a was used to analyze the results. Statistical analysis was performed on Prism using the Mann-Whitney test, Wilcoxon matched pairs test, Fisher exact test, and Kruskal-Wallis test. Comparison between groups was corrected for multiple comparisons. To adjust for time between time points or measurements before and after loss of viral control, we used linear mixed models. A P value below 0.05 was considered significant.
Nucleotide sequence accession numbers.
New Gag sequences generated in this study are available in GenBank under accession numbers KX347069 to KX347092.
RESULTS
Loss of virological control is exclusively encountered among viremic controllers with protective HLA class I alleles.
The viral load and CD4 counts of the 70 study subjects are shown in Table 1. During the median 5-year follow-up, 7/20 of the subjects expressing protective HLA class I alleles who were viremic controllers at baseline had progressed to viral loads of >10,000 copies/ml. In contrast, 0/10 of the subjects not expressing protective HLA class I alleles who were viremic controllers at baseline had progressed to viral loads of >10,000 copies/ml (P = 0.06, Fisher's exact test) (Fig. 1 and 3). It is important to note that individuals with protective HLA class I alleles who ended up as failing viremic controllers (fVC+) had a trend toward higher baseline viral load than those who maintained control (VC+) (P = 0.06, Mann-Whitney test), suggesting that the process of control was already in motion in the former. There were some individuals whose viral load upon follow-up was above 2,000 but below 10,000 copies/ml, and these individuals (intermediates) were excluded from analysis of mechanisms of loss of control. We focused on the 2 subgroups with extreme divergent outcomes in order to better understand the factors responsible for the control among the viremic controllers with or without protective HLA class I alleles (VC+/−) and lack of viral control among fVC+/− subgroups. Overall, these data suggested that the mechanisms responsible for viral control among those without protective HLA class I alleles are more durable than the mechanisms in those with protective alleles.
TABLE 1.
Characteristics of study subjects
| Group (baseline) and protective HLA allele (+/−)a | n | Median yr of follow-up (IQR) | Median VL, HIV RNA copies/ml (IQR) | P value | Median CD4 count, cells/mm3 (IQR) | P value |
|---|---|---|---|---|---|---|
| Viremic controllers | 0.77 | 0.09 | ||||
| + | 20 | 5 (4–6) | 487 (283–1,252) | 509 (459–684) | ||
| − | 10 | 5 (4–6) | 613 (399–1,213) | 669 (550–865) | ||
| Noncontrollers | 0.26 | 0.56 | ||||
| + | 15 | 4 (3–6) | 156,000 (118,000–192,000) | 329 (257–377) | ||
| − | 25 | 3 (2.5–4) | 209,000 (132,000–330,000) | 373 (260–478) |
Protective HLA alleles: HLAs B*57, B*58:01, B*81:01, and A*74:01.
FIG 3.

Longitudinal viral load (A and C) and absolute CD4 T cell count (B and D) patterns among baseline viremic controllers with and without protective HLA alleles. fVC+, failing viremic controllers with protective HLA alleles (enrolled with a viral load of <2,000 HIV RNA copies/ml and later lost control, n = 7; loss of control is defined by an increase in viral load to >10,000 HIV RNA copies/ml at 2 or more time points during follow-up); VC+/−, viremic controllers with or without protective HLA alleles (maintained viral load of <2,000 HIV RNA copies/ml for the entire course of follow-up).
Increased breadth of CD8+ T cell responses against HIV-1 Gag in baseline viremic controllers with protective HLA class I alleles.
We next characterized HIV-specific CD8+ T cell immune responses in the study participants with the ELISpot assay using consensus subtype C overlapping peptides spanning the entire proteome followed by confirmations with individual peptides within a reactive pool. The breadth of HIV-specific responses overall did not differ significantly between the groups (Fig. 4A). However, there was a significant difference in the breadth of Gag-specific CD8+ T cell responses between the 4 groups (P value = 0.01; Kruskal-Wallis test), bVCs with protective HLA class I alleles targeting a significantly higher number of Gag epitopes than baseline noncontrollers (bNCs) with protective HLA class I alleles (P value = 0.004; Mann-Whitney test) (Fig. 4B). In contrast, among individuals without protective HLA class I alleles there were no significant differences between bVCs and bNCs in Gag-specific breadth (P value, 0.10; Mann-Whitney test). No significant differences between the groups were observed when comparing non-Gag proteins individually (data not shown). These differences are further illustrated by comparisons between Gag and Nef breadths in the four study groups (Fig. 4C to F), with bVC groups possessing significantly broader Gag than Nef responses. Taken together, these observations are consistent with previous findings indicating that broad targeting of Gag but not Nef may contribute to HIV control among viremic controllers (4, 38–40).
FIG 4.

Baseline (A to F) and longitudinal (G and H) ELISpot screening of HIV-specific T cell responses. Overall breadth to the entire HIV proteome (A) and breadth of Gag-specific immune responses (B) among individuals with or without protective HLA alleles (bVC+/− and bNC+/−). An ELISpot matrix assay followed by confirmation with overlapping peptides spanning the entire HIV-1 clade C proteome was used on thawed PBMCs. The breadth of Gag versus Nef was assessed among the study groups: bVC+ (baseline viremic controllers with protective HLA alleles, n = 18) (C), bNC+ (baseline noncontrollers with protective HLA alleles, n = 12) (E), bVC− (baseline VC without protective HLA alleles, n = 9) (D), and bNC− (baseline noncontrollers without protective HLA alleles, n = 20) (F). (G and H) Longitudinal ELISpot screening of HIV-specific T cell responses. (G) Overall breadth across entire HIV proteome. (H) Breadth of Gag among viremic controllers (VC+, n = 7), failing viremic controllers (fVC+, n = 7), and baseline noncontrollers (bNC+, n = 6) with protective HLA alleles and viremic controllers (VC−, n = 5) without protective HLA alleles. PBMCs were stimulated with optimal peptides restricted only to individuals with HLA-B alleles and spanning the whole HIV proteome. TP, time point.
Loss of virologic control is associated with the loss of CD8+ T cell responses.
In order to identify HIV-specific CD8+ T cell immune response changes associated with the loss of viral control, samples were evaluated at baseline (before loss of control) time point and at the time point when their VL first increased to above 10,000 copies/ml (after loss of virologic control).
A significant reduction in the overall breadth of CD8+ T cell responses and in Gag-specific breadth was observed among the fVC+ over time (P = 0.02 and P = 0.003; linear mixed models [Fig. 4G and H]). The group of baseline noncontrollers with protective HLA class I alleles showed a trend in reduction of overall CD8+ T cell response breadth (P = 0.06) and Gag-specific breadth (P = 0.09; linear mixed models) (Fig. 4G and H). In contrast, no changes in overall breadth or in Gag breadth were observed in VC+ or VC− groups (Fig. 4G and H) or in Nef-specific responses among the fVC+ subgroup over time (P value, 0.14; linear mixed models) (data not shown).
We next wanted to identify the specific epitopes in failing viremic controllers with protective HLA alleles (fVC+) that were lost over time. The majority (73%) of the total CD8+ T cell responses detected at baseline were no longer detectable by ELISpot assay at the time of virological failure, and their identities are shown in Table 2. Interestingly, in 3 subjects, there was an emergence of new CD8+ T cell specificities at virologic failure (Table 2). The specific epitopes targeted by VC+ and VC− subgroups are shown in Tables 3 and 4, respectively. Although we found no significant loss or gain of responses in these 2 subgroups, cumulatively VC+ lost 24 responses and gained 36 responses, while among the VC− subgroup, there were 13 responses lost and 4 gained at a later time point. Overall, these data demonstrated the maintenance of overall breadth over time in the VC+ and VC− subgroups.
TABLE 2.
Optimal HIV peptides targeted by failing viremic controllers (fVC+) before and after loss of viral controlc
| Subject no. and identifier (controller status) and peptide(s) | Allele(s) before or after loss of viremic control |
|
|---|---|---|
| Before | After | |
| 1, SK-187 (fVC+) | ||
| Gag | B*08-EI8; B*58:01-NL11; B*58:01-TW10; B*58:01-QW9; B*08-EL8 | B*08-DI8; B*58:01-TW10; B*58:01-QW9 |
| Pol | B*58:01-IAW9; B*58:01-SW10; B*08-GL9 | B*58:01-IAW9; B*08-GL9 |
| Tat/Rev/Vif/Vpr/Vpu | —b | — |
| Env | — | — |
| Nef | B*08-FL8 | B*08-FL8 |
| 2, SK-435 (fVC+) | ||
| Gag | B*57-WF9; B*57-ISW9; B*57-KF11; B*57-TW10 | B*57-ISW9; B*57-KF11 |
| Pol | B*15:10-TIL9; B*57-AF10; B*57-VI9 | B*15:10-TIL9 |
| Tat/Rev/Vif/Vpr/Vpu | B*15:10-RI11; B*57-AW9; B*57-VF9; B*57-LW9 | B*15:10-WI9a |
| Env | B*57-QL11 | — |
| Nef | B*57-KAF9 | — |
| 3, SK-224 (fVC+) | ||
| Gag | B*07-GL9; B*57-ISW9; B*57-TW10; B*57-QW9 | B*57-ISW9; B*57-QW9 |
| Pol | B*57-IW9; B*57-AF10; B*57-KI13 | B*57-IW9; B*57-AF10 |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*07-RM9; B*07-TL10 | B*07-RM9; B*07-TL10; B*57-KAF9a |
| 4, SK-242 (fVC+) | ||
| Gag | B*44-SL9 | — |
| Pol | B*58:01-SW10 | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*44-KY11; B*44-QY9; B*58:01-YY8; B*58:01-YT9; B*58:01-KY11 | — |
| 5, SK-024 (fVC+) | ||
| Gag | B*39:10-GL9; B*39:10-TL9; B*39:10-NL11; B*81:01-TL9 | B*39:10-TL9 |
| Pol | B*81:01-LI9; B*81:01-SL10 | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*81:01-RM9; B*81:01-RGF9 | — |
| 6, SK-079 (fVC+) | ||
| Gag | B*57-ISW9; B*57-KF11 | B*57-KF11 |
| Pol | B*57-AF10; B*57-FF9 | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*57-HW9 | — |
| 7, SK-188 (fVC+) | ||
| Gag | B*58:01-TW10 | |
| Pol | B*58:01-IAW9; B*58:01-SW10 | B*58:01-IAW9 |
| Tat/Rev/Vif/Vpr/Vpu | B*58:01-VF9 | B*58:01-QY10a |
| Env | B*44-MY9; B*58:01-KW11; B*58:01-QL11 | — |
| Nef | B*44-KY11; B*44-QY9; B*58:01-HW9; B*58:01-KAF9; B*58:01-QL11; B*58:01-YY8; B*58:01-YT9; B*58:01-NW9; B*58:01-KY11; B*58:01-HQ10 | — |
New response.
—, no response detected.
Longitudinal ELISpot screening of HIV-specific CD8+ T cell immune responses among failing viremic controllers (fVC+, n = 7) with protective HLA alleles. PBMCs were stimulated with optimal peptides restricted only to individual HLA-B alleles and spanning the whole HIV proteome.
TABLE 3.
Optimal HIV peptides targeted by viremic controllers with protective HLA class I alleles (VC+) at baseline and later time pointsc
| Subject no. and identifier (controller status) and peptide(s) | Allele(s) at time point: |
|
|---|---|---|
| Baseline | Latest time point | |
| 1, SK-199 (VC+) | ||
| Gag | B*57-ISW9; B*57-KF11; B*57-QW9 | B*13-YV12a; B*13-GI11a; B*13-VV9a; B*57-WF9a; B*57-ISW9; B*57-KF11; B*57-QW9 |
| Pol | B*57-IAW9 | B*13-RI10a; B*57-IAW9; B*57-SW10a; B*57-AF10a; B*57-KI13a; B*57-EY9a; B*57-FW11a |
| Tat/Rev/Vif/Vpr/Vpu | —b | B*13-LL9a; B*57-IW9a |
| Env | — | B*57-KW11a |
| Nef | — | B*13-RV9a; B*13-RI9a |
| 2, SK-089 (VC+) | ||
| Gag | — | B*58:01-WF9a; B*58:01-DW10a; B*58:01-QW9a |
| Pol | B*57-FF9; B*57-VI9 | B*15:03-FY10a; B*15:03-IY9a; B*15:03-RY9a; B*58:01-IAW9a |
| Tat/Rev/Vif/Vpr/Vpu | — | B*15:03-FY10a; B*58:01-LW9a |
| Env | — | B*58:01-KW11a; B*58:01-TWS10a |
| Nef | — | — |
| 3, SK-282 (VC+) | ||
| Gag | B*44-AW11; B*58:01-DW10; B*58:01-TW10 | B*58:01-QW9a |
| Pol | B*44-IW11; B*58:01-IAW9; B*58:01-SW10 | B*58:01-IAW9; B*58:01-SW10 |
| Tat/Rev/Vif/Vpr/Vpu | — | B*58:01-QY10a |
| Env | B*58:01-TWS10 | B*58:01-TWS10 |
| Nef | B*58:01-HW9 | — |
| 4, SK-235 (VC+) | ||
| Gag | B*39-GL9; B*39-NL11; B*81-HA9 | B*39-NL11; B*81-TL9a |
| Pol | B*81-LI9 | B*39-SL10a; B*81-SL10a |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | — | — |
| 5, SK-362 (VC+) | ||
| Gag | B*15:01-VF9; B*15:10-VL10 | B*15:03-VF9; B*15:10-GL9a |
| Pol | B*15:03-FY10; B*15:03-IY9; B*15:03-GL9; B*15:10-TL9 | B*15:03-FY10; B*15:03-GL9 |
| Tat/Rev/Vif/Vpr/Vpu | B*15:03-F10 | B*15:10-WI9a |
| Env | — | — |
| Nef | — | — |
| 6, SK-348 (VC+) | ||
| Gag | B*58:01-TW10; B*81-TL9 | B*81-TL9 |
| Pol | B*81-SL10 | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | B*58:01-KW11 | — |
| Nef | B*58:01-KAF9 | — |
| 7, SK-354 (VC+) | ||
| Gag | A*74:01-RLY10; B*81-TL9; B*81-HA9; B*35-HA9 | B*35-PY9a; B*35-HA9; B*81-HA9; B*81-TL9 |
| Pol | B*35-EY10; B*81-TL11; B*81-SL10; B*81-RQL11; B*81-TM10 | B*35-NQY9a; B*35-EY10; B*81-TL11 |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | B*35-DL9; B*81-RI10; B*81-IA9 | B*35-DL9 |
| Nef | B*35-YF9a | |
New response.
—, no response detected.
Longitudinal ELISpot screening of HIV-specific CD8+ T cell immune responses among viremic controllers (VC+, n = 7) with protective HLA alleles. PBMCs were stimulated with optimal peptides restricted only to individual HLA-B alleles and spanning the whole HIV proteome.
TABLE 4.
Optimal HIV peptides targeted by viremic controllers without protective HLA class I alleles (VC−) at baseline and later time pointsc
| Subject no. and identifier (controller status) and peptide(s) | Allele(s) at time point: |
|
|---|---|---|
| Baseline | Latest time point | |
| 1, SK-317 (VC−) | ||
| Gag | B*42-TL9; B*44-SV9; B*44-RL11; B*44-AW11 | B*42-TL9; B*44-RL11 |
| Pol | B*42-LI9; B*42-TL11; B*44-SY11; B*44-IW11 | —b |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | — | — |
| 2, SK-315 (VC−) | ||
| Gag | B*42-TL9; B*42-GL9 | B*42-TL9 |
| Pol | — | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*42-RGF9 | — |
| 3, SK-292 (VC−) | ||
| Gag | B*39-TL9 | B*39-TL9; B*39-NL11a |
| Pol | — | B*39-SL10a; B*39-IL9a |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | — | — |
| 4, SK-275 (VC−) | ||
| Gag | B*39-TL9 | B*08-EV9a |
| Pol | — | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*08-FL8 | — |
| 5, SK-209 (VC−) | ||
| Gag | B*08-DI8; B*42-GL9 | — |
| Pol | — | — |
| Tat/Rev/Vif/Vpr/Vpu | — | — |
| Env | — | — |
| Nef | B*42-FL9 | — |
New response.
—, no response detected.
Longitudinal ELISpot screening of HIV-specific CD8+ T cell immune responses among viremic controllers (VC−, n = 5) without protective HLA alleles. PBMCs were stimulated with optimal peptides restricted only to individual HLA-B alleles and spanning the whole HIV proteome.
Escape mutations within Gag only partially explain the loss of virological control in fVCs with protective HLA alleles.
We next investigated whether the observed loss of virologic control associated with loss of immune responses was primarily linked to emergence of viral escape mutations as previously suggested (6–9, 11, 12, 41, 42). Longitudinal population gag sequencing was performed from plasma samples at baseline and after loss of control for the fVC+ subjects and at baseline and available later time points for the viremic controllers with and without protective HLA class I alleles (VC+ and VC−). We assessed mutations within epitopes restricted by the patients' protective HLA class I alleles plus epitopes restricted by the other HLA B alleles that the subjects possess. The HIV-1 clade C Gag consensus sequence was used as reference for comparison in this analysis. The percentage of variant sequences was calculated by counting the number of epitopes with sequence changes divided by the cumulative number of epitopes restricted by each patient's HLA types that were tested multiplied by 100: % variant sequences = [(number of variant sequences/cumulative number of epitopes tested on all subjects) × 100].
For the fVC+ subjects, the proportion of epitopes restricted by the HLA alleles expressed, and carrying variants, did not differ before and after loss of viremic control (51% before versus 53% after [Fig. 5A]). Thus, the reduction in epitopes targeted was unrelated to escape. Further work is needed to better understand the mechanisms involved in loss of CD8+ T cell responses over time in the absence of viral escape, as demonstrated by the lack of viral escape in well-characterized Gag epitopes restricted by the protective HLA-B*57/58:01/81:01, despite the loss of responses to these epitopes (data not shown).
FIG 5.
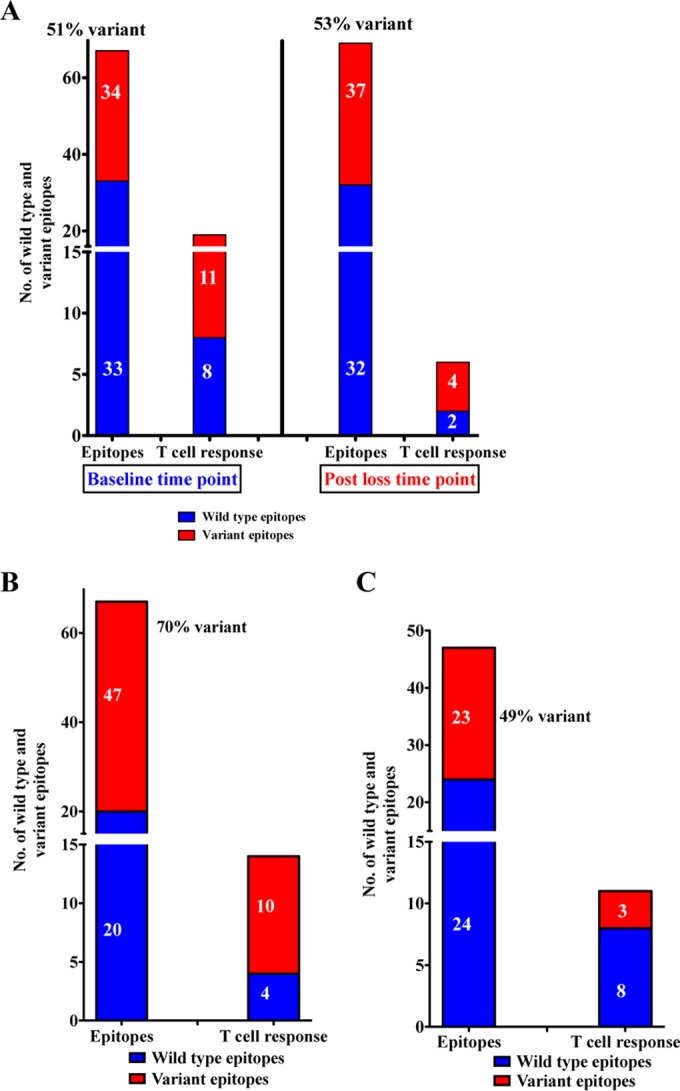
Immunogenicity and viral escape in fVC+ subjects before and after loss of viral control (A) and in VC+ (B) and VC− (C) subjects at baseline. We evaluated if the epitopes within the Gag region had variant sequences either within the epitope or within the 5 flanking amino acids. HIV-1 clade C Gag consensus sequence was used as reference for comparison in this analysis. We assessed mutations within epitopes restricted by the patients' protective HLA class I alleles plus epitopes restricted by the other HLA-B alleles that the subjects possessed. The percentage of variant sequences was calculated by counting the number of epitopes with sequence changes divided by the cumulative number of epitopes restricted by each patient's HLA types that were tested multiplied by 100 [(number of variant sequences/cumulative number of epitopes tested) × 100].
Furthermore, Gag epitope sequences of the VC+ and VC− subgroups were assessed at baseline (samples were available only at this time point). The VC+ subgroup displayed a high proportion of immune escape variants in epitopes restricted by the host HLA alleles (70% [Fig. 5B]). These data suggest that controllers with protective HLA class I alleles maintained viral suppression despite the presence of escape mutations, with evidence of CTL responses against wild-type epitopes and some epitopes with escape mutations. In contrast, the VC− subgroup displayed fewer variant epitopes (49% [Fig. 5C]) and most did not elicit detectable CD8+ T cell responses. Although the number of subjects studied here is limited, these data reveal that VC+ maintained viral control despite having the highest prevalence of variant sequences either within the epitope or in the 5 flanking amino acid residues compared to individuals lacking protective HLA class I alleles.
No significant differences in the polyfunctionality or proliferative capacity of CD8+ T cells to explain the differences in disease status.
We next studied the ability of CD8+ T cells to produce diverse cytokines and proliferate upon stimulation with Gag pool peptides at baseline and later time points. There were no significant differences in CD8+ T cell polyfunctionality to explain the divergence in disease progression among the subgroups (VC+, fVC+, VC−, and baseline noncontrollers with protective HLA class I alleles) at baseline and later time points (Fig. 6A). However, there was a nonsignificant trend toward the CD8+ T cells of viremic controllers with protective HLA class I alleles having a higher proliferative capacity than those of fVC+ at baseline (P value = 0.09, Mann-Whitney test) (Fig. 6B) and no significant differences at later time points (Fig. 6C).
FIG 6.

Polyfunctionality (A) and proliferative capacity (B and C) of CD8+ T cells upon stimulation with Gag peptide pools—among viremic controllers (VC+), failing viremic controllers (fVC+), baseline noncontrollers (bNC+) with protective HLA alleles, and viremic controllers without protective HLA alleles (VC−), at baseline and at later time points. The 5 functions studied were CD107a, IFN-γ, IL-2, MIP-1β, and TNF-α. On the pie charts (A), red represents 5 functions, orange represents 4 functions, yellow represents 3 functions, green represents 2 functions, and blue represents 1 function. The gating strategy is shown in Fig. 2A. Boolean gating was performed in order to allow creation of a full array of possible combinations of up to 32 response patterns. Positive responses were reported after background correction, and the percentage of epitope-specific CD8+ T cell responses had to be at least two times higher than background for each tested marker. PESTLE (version 1.6.2) and SPICE 5.0 (Mario Roederer, ImmunoTechnology Section, Vaccine Research Center, NIH, Bethesda, MD) were used to analyze the multifunctional data. The same subjects were assessed for the proliferation of CD8+ T cells at baseline (B) and latest time point (C). The gating strategy is shown in Fig. 2B. GraphPad Prism version 5.0a software (GraphPad Software, San Diego, CA, USA) was used to analyze group data sets.
Viremic controllers without protective HLA class I alleles display limited ex vivo CD8+ T cell inhibition capacity compared to viremic controllers with protective HLA class I alleles.
We further investigated the ability of CD8+ T cells to suppress viral replication ex vivo as a possible mechanism of viral control. We used a viral inhibition assay to directly compare the virus-inhibitory activity of CD8+ T cells ex vivo among VC+, fVC+, and VC− subgroups at baseline and later time points. Figure 7A to C shows representative data for 1 VC+, 1 VC−, and 1 fVC+ participant, respectively. We observed that viremic controllers with protective HLA class I alleles (VC+) displayed significantly higher ex vivo CD8+ T cell inhibition capacity (Fig. 7A) than did viremic controllers without protective HLA class I alleles (VC−) and failing viremic controllers with protective HLA class I alleles (fVC+) (Fig. 7B and C). VC+ subjects maintained this high ex vivo CD8+ T cell inhibitory capacity while the VC− subjects maintained the low inhibitory capacity at later time points. However, in the 1 fVC+ subject studied, despite the subject displaying a reduced ex vivo CD8+ T cell inhibitory capacity at baseline, we noted a further reduction in the ex vivo CD8+ T cell inhibitory capacity after the subject lost viral control (Fig. 7C). Interestingly, there was also a significant difference in the log10 p24 inhibition between the viremic controllers with protective HLA class I alleles and viremic controllers without protective HLA class I alleles, with the former having CD8+ T cells with greater viral inhibition capacity (P value = 0.02; Mann-Whitney test) (Fig. 7D). Taken together, these data show that CD8+ T cells from viremic controllers with protective HLA class I alleles are associated with greater viral inhibitory capacity than are those of viremic controllers without protective HLA class I alleles, suggesting an alternative mechanism of control in the latter group.
FIG 7.

Ex vivo viral inhibition of NL4-3-infected CD4+ T cells by autologous CD8+ T cells. Infected CD4+ T cells were cultured ex vivo with CD8+ T cells at a ratio of 1:1. Blue lines represent infected CD4+ T cells alone, black lines represent the negative control (uninfected CD4+ T cells), and the red lines represent ex vivo coculture of infected CD4+ T cells with autologous CD8+ T cells. (A) Representative data for 1 VC+ at baseline and later time point. (B) One VC− at baseline and later time point. (C) Representative data for 1 fVC+ at baseline and later time point. (D) Log10 p24 inhibition of individual subjects was calculated by subtracting log10 p24 values with CD8+ T cells from log10 p24 values without CD8+ T cells at day 7; here, we compared 5 VC+ with 3 fVC+ and 5 VC− individuals.
DISCUSSION
The natural control of HIV has previously been linked to several factors, including immunological factors, host genetics, and viral factors in the HIV controller groups (viremic controllers, LTNPs, and ECs). In chronically infected individuals, the evolution of immunological and viral factors and their association with divergent disease progression patterns have rarely been characterized. Additionally, the findings on mechanisms of viral control in HIV controllers using cross-sectional study design have been inconclusive and therefore need resolving. In the current study, we explored the mechanisms of viral control and lack of viral control among individuals with and without protective HLA class I alleles. Viremic controllers with similar baseline clinical characteristics but progressing to divergent viral load outcomes were analyzed. We assessed at baseline and longitudinally the CD8+ T cell responses using the ELISpot assay, analyzed the polyfunctionality and proliferative capacity of CD8+ T cells using flow cytometry, and analyzed Gag sequence evolution and the ability of CD8+ T cells to inhibit viral replication in an ex vivo viral inhibition assay.
The loss of virological control was evident among 35% of baseline viremic controllers with protective HLA class I alleles but not among baseline viremic controllers without protective HLA class I alleles. This unexpected finding may suggest that viremic controllers without HLA class I alleles are less likely to subsequently progress, suggesting a durable mechanism of viral control in these individuals. Whereas previous studies suggest that the expression of protective HLA class I alleles is a correlate of immune protection from disease progression (43, 44), our study suggests that individuals without protective HLA class I alleles who show virologic control may have more durable, yet undefined mechanisms of viral control.
We demonstrated that virologic control in subjects with protective HLA class I alleles was associated with greater breadth of CD8+ T cell responses to Gag, consistent with previous data on the importance of Gag-specific responses in chronic HIV infection (4, 38). In addition, we observed a bias toward targeting of greater breadth of HIV Gag epitopes compared to Nef independently of protective HLA class I allele expression among controllers. In contrast, baseline noncontrollers with or without protective HLA class I alleles showed no bias toward targeting Gag or Nef responses. These data further support previous work showing that Gag CD8+ T cell responses are associated with the control of viral replication (3, 4, 38) while Nef CD8+ T cell responses are associated with high viral load (39, 40). We also provide new evidence that in controllers without protective HLA class I alleles, Gag CD8+ T cell responses do not appear to play a significant role in viral control. It is possible that these individuals may have robust innate immunity or cytolytic CD4+ T cell responses (45). These alternative mechanisms will be the focus of future investigations in our cohort.
Longitudinal analysis of CD8+ T cell responses among viremic controllers with and without protective HLA class I alleles (VC+/−) over time suggests that the maintenance of CD8+ T cell responses over time is important for sustained viral control during chronic HIV infection. Further analysis of the longitudinal CD8+ T cell responses of the failing viremic controllers with protective HLA class I alleles (fVC+ subgroup) revealed that CD8+ T cell responses across the whole HIV proteome and Gag-specific responses in particular were lost over time. These observations suggest that the loss of CD8+ T cell responses may be related to the loss of viral control in these individuals. This phenomenon was also observed as a nonsignificant trend among baseline noncontrollers with protective HLA class I alleles, suggesting that this process evolves over time as the disease progresses. However, one important caveat is that we may have missed some responses due to the use of consensus peptide sequences to stimulate PBMCs in the current study. Nevertheless, our study strongly implicates diminishing CTL responses as the basis for loss of viremic control, and we further identified the specific Gag responses that were no longer detectable over time in this study. Future study will need to further characterize some of the important non-Gag responses that were also no longer detectable and contributed to loss of viremic control.
The loss of CD8+ T cell responses observed among the fVC+ subgroup may be attributed to viral escape as a result of CTL pressure. Analysis of the fVC+ subjects suggested that some of the CD8+ T cell responses were lost as a result of sequence variation (viral escape) within the cognate epitopes. This is consistent with other studies (12, 41, 42). However, some epitopes remained wild type after loss of control and yet did not elicit detectable CD8+ T cell responses. Deciphering the underlying mechanisms for loss of these responses (not explained by viral escape) will require further investigation. Nevertheless, our data unequivocally show that these CD8+ T cell responses underwent functional or phenotypic changes, and the mechanisms involved will require further investigation. Our data also reveal that the loss of viral control did not always occur as a result of escape. This is because there were no significant differences in the levels of escape at baseline versus those after the loss-of-control time point, suggesting that the loss of control is primarily related to the loss of CD8+ T cell responses.
Furthermore, previous studies have described mutations such as T242N, A163G, and T186S as reverting mutations in HIV infection, possibly due to their fitness cost (46, 47). In our study, the T242N mutation was the most common mutation and reversion to wild type occurred in 2 fVC+ subjects with no detectable CD8+ T cell response, indicating that reversion in these 2 subjects resulted from the loss of CTL pressure. However, in some individuals there was no reversion observed despite the loss of CD8+ T cell responses. Previous studies show that some mutations within Gag have a fitness cost to the virus (32, 48); however, compensatory mutations can develop and restore viral fitness, or reversion can occur, especially when the CTL pressure is lost. In our study, it is possible that the fVC+ individuals may have developed compensatory mutations that restored viral fitness, hence, the increasing viral load in these individuals.
We also showed that VC+ subjects had a high level of escape, with 70% of the epitopes at baseline having variant sequences. Viral control was sustained in these individuals despite ongoing evolution in plasma viruses, and this is in agreement with some previous studies that showed that some elite controllers maintained viral control despite ongoing viral replication and evolution in plasma viruses and that they were able to develop de novo immune responses to variant peptides (27, 49); however, we did not test for de novo immune responses in our study.
We also demonstrated here using the viral inhibition assay that viremic controllers with protective HLA class I alleles displayed significantly higher ex vivo CD8+ T cell inhibitory capacity than did those viremic controllers without protective HLA class I alleles, who displayed limited ex vivo CD8+ T cell inhibitory capacity. These data may lend further support to alternative mechanisms of HIV control in VCs without protective HLA class I alleles where CD8+ T cell responses may not be associated with virus suppression. Other intrinsic factors, such as host restriction factors or viral mutations that affect interactions with these factors, may play a role in the control of viremia in these individuals as previously shown (50, 51).
Conclusion.
Overall, our data indicate that individuals without protective HLA class I alleles were less likely to lose viral control. Furthermore, we showed that the breadth of Gag CD8+ T cell responses is the most significant correlate of viral control in individuals with protective HLA class I alleles and that the loss of virologic control in individuals with protective HLA class I alleles was related to a reduction in the total breadth of CD8+ T cell responses. The presence of escape mutations within epitopes only partially explains divergent disease progression in the subgroups studied, where the loss of some of the CD8+ T cell responses resulted from escape while the reasons for the loss of some others were not known. Additional investigation of the function and quality of the CD8+ T cells showed that these factors did not account for the divergent disease progression status observed in the subgroups studied. Furthermore, the control of viremia in individuals with protective HLA class I alleles was associated with the ability of the CD8+ T cells to inhibit viral replication. Our findings here are consistent with those of Emu et al. (44), who also demonstrated that HLA class I-restricted CD8+ T cells were responsible for viral control in elite controllers with protective HLA alleles but not in those without those alleles. We extend those findings by demonstrating that loss of CD8+ T cell breadth and viral inhibitory capacity explains loss of control in those with protective HLA alleles and that loss of the responses was not always explained by viral escape. However, further investigation will be required to characterize factors that may be associated with control in individuals without protective HLA class I alleles.
ACKNOWLEDGMENTS
We thank Denis Chopera for technical advice. We thank and acknowledge the study participants from the Sinikithemba cohort.
Funding Statement
This research was funded by grants from the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF) (64809), the Victor Daitz Foundation, and the Howard Hughes Medical Institute (HHMI) (55007427). Additional support was provided by the Organization for Women in Science for the Developing World (OWSD) and the Mark and Lisa Schwartz Foundation.
REFERENCES
- 1.UNAIDS. 2013. Global report: UNAIDS report on the global AIDS epidemic 2013. UNAIDS, Geneva, Switzerland. [Google Scholar]
- 2.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 3.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJR. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 4.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 5.Migueles SA, Connors M. 2010. Long-term nonprogressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA 304:194–201. doi: 10.1001/jama.2010.925. [DOI] [PubMed] [Google Scholar]
- 6.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CR, Rizza CR. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T-cell recognition. Nature 354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 7.Koenig S, Conley AJ, Brewah YA, Jones GM, Leath S, Boots LJ, Davey V, Pantaleo G, Demarest JF, Carter C. 1995. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat Med 1:330–336. doi: 10.1038/nm0495-330. [DOI] [PubMed] [Google Scholar]
- 8.Price DA, Goulder PJ, Klenerman P, Sewell AK, Easterbrook PJ, Troop M, Bangham CR, Phillips RE. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc Natl Acad Sci U S A 94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 10.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 11.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang S, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD, Goulder PJR. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med 199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goulder PJR, Watkins DI. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol 4:630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 13.Carrington M, O'Brien SJ. 2003. The influence of HLA genotype on AIDS. Annu Rev Med 54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 14.Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, Urban TJ, Zhang K, Gumbs CE, Smith JP, Castagna A, Cozzi-Lepri A, De Luca A, Easterbrook P, Günthard HF, Mallal S, Mussini C, Dalmau J, Martinez-Picado J, Miro JM, Obel N, Wolinsky SM, Martinson JJ, Detels R, Margolick JB, Jacobson LP, Descombes P, Antonarakis SE, Beckmann JS, O'Brien SJ, Letvin NL, McMichael AJ, Haynes BF, Carrington M, Feng S, Telenti A, Goldstein DB. 2009. Common genetic variation and the control of HIV-1 in humans. PLoS Genet 5(12):e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International HIV Controllers Study. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Bashirova A, Iversen AKN, Phair J, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Altfeld M, O'Brien SJ, Carrington M. 2005. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med 11:1290–1292. doi: 10.1038/nm1333. [DOI] [PubMed] [Google Scholar]
- 17.Matthews PC, Adland E, Listgarten J, Leslie A, Mkhwanazi N, Carlson JM, Harndahl M, Stryhn A, Payne RP, Ogwu A, Huang K-HG, Frater J, Paioni P, Kloverpris H, Jooste P, Goedhals D, van Vuuren C, Steyn D, Riddell L, Chen F, Luzzi G, Balachandran T, Ndung'u T, Buus S, Carrington M, Shapiro R, Heckerman D, Goulder PJR. 2011. HLA-A*7401-mediated control of HIV viremia is independent of its linkage disequilibrium with HLA-B*5703. J Immunol 186:5675–5686. doi: 10.4049/jimmunol.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goulder PJR, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Mikhail M, Dyer WB, Zaunders JJ, Kelleher AD, Saksena NK. 2003. First demonstration of a lack of viral sequence evolution in a nonprogressor, defining replication-incompetent HIV-1 infection. Virology 312:135–150. doi: 10.1016/S0042-6822(03)00159-4. [DOI] [PubMed] [Google Scholar]
- 20.Sandonís V, Casado C, Alvaro T, Pernas M, Olivares I, García S, Rodríguez C, del Romero J, López-Galíndez C. 2009. A combination of defective DNA and protective host factors are found in a set of HIV-1 ancestral LTNPs. Virology 391:73–82. doi: 10.1016/j.virol.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Lamine A, Caumont-Sarcos A, Chaix M-L, Saez-Cirion A, Rouzioux C, Delfraissy J-F, Pancino G, Lambotte O. 2007. Replication-competent HIV strains infect HIV controllers despite undetectable viremia (ANRS EP36 study). AIDS 21:1043–1045. doi: 10.1097/QAD.0b013e3280d5a7ac. [DOI] [PubMed] [Google Scholar]
- 22.Wong JK. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 23.Blankson JN, Bailey JR, Thayil S, Yang H-C, Lassen K, Lai J, Gandhi SK, Siliciano JD, Williams TM, Siliciano RF. 2007. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J Virol 81:2508–2518. doi: 10.1128/JVI.02165-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miura T, Brockman MA, Brumme CJ, Brumme ZL, Carlson JM, Pereyra F, Trocha A, Addo MM, Block BL, Rothchild AC, Baker BM, Flynn T, Schneidewind A, Li B, Wang YE, Heckerman D, Allen TM, Walker BD. 2008. Genetic characterization of human immunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J Virol 82:8422–8430. doi: 10.1128/JVI.00535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey JR, Williams TM, Siliciano RF, Blankson JN. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J Exp Med 203:1357–1369. doi: 10.1084/jem.20052319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailey JR, Zhang H, Wegweiser BW, Yang H, Herrera L, Ahonkhai A, Williams TM, Siliciano RF, Blankson JN. 2007. Evolution of HIV-1 in an HLA-B*57-positive patient during virologic escape. J Infect Dis 196:50–55. doi: 10.1086/518515. [DOI] [PubMed] [Google Scholar]
- 27.O'Connell KA, Brennan TP, Bailey JR, Ray SC, Siliciano RF, Blankson JN. 2010. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J Virol 84:7018–7028. doi: 10.1128/JVI.00548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis 197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 29.Leslie A, Price DA, Mkhize P, Bishop K, Rathod A, Day C, Crawford H, Honeyborne I, Asher TE, Luzzi G, Edwards A, Rousseau CM, Mullins JI, Tudor-Williams G, Novelli V, Brander C, Douek DC, Kiepiela P, Walker BD, Goulder PJ. 2006. Targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype 1. J Immunol 177:4699–4708. doi: 10.4049/jimmunol.177.7.4699. [DOI] [PubMed] [Google Scholar]
- 30.Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung'u T, Shapiro R, Frater J, Brumme ZL, Goulder PJR, Heckerman D. 2012. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J Virol 86:5230–5243. doi: 10.1128/JVI.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feeney ME, Tang Y, Roosevelt KA, Leslie AJ, McIntosh K, Karthas N, Walker BD, Goulder PJR. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol 78:8927–8930. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, Wang B, Losina E, Miura T, Chonco F, van der Stok M, Mncube Z, Bishop K, Goulder PJR, Walker BD, Brockman MA, Ndung'u T. 2010. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol 84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leslie A, Matthews P, Listgarten J, Carlson JM, Kadie C, Ndung'u T, Brander C, Coovadia H, Walker BD, Heckerman D, Goulder PJR. 2010. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol 84:9879–9888. doi: 10.1128/JVI.00320-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thobakgale CF, Ramduth D, Reddy S, Mkhwanazi N, de Pierres C, Moodley E, Mphatswe W, Blanckenberg N, Cengimbo A, Prendergast A, Tudor-Williams G, Dong K, Jeena P, Kindra G, Bobat R, Coovadia H, Kiepiela P, Walker BD, Goulder PJR. 2007. Human immunodeficiency virus-specific CD8+ T-cell activity is detectable from birth in the majority of in utero-infected infants. J Virol 81:12775–12784. doi: 10.1128/JVI.00624-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ndhlovu ZM, Proudfoot J, Cesa K, Alvino M, Mcmullen A, Vine S, Piechocka-Trocha A, Walker BD, Pereyra F. 2012. Elite controllers with low to absent effector CD8+ T cell responses maintain highly functional, broadly directed central memory responses. J Virol 86:6959–6969. doi: 10.1128/JVI.00531-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Piechocka-Trocha A, Miura T, Brockman MA, Julg BD, Baker BM, Rothchild AC, Block BL, Schneidewind A, Koibuchi T, Pereyra F, Allen TM, Walker BD. 2009. Differential neutralization of human immunodeficiency virus (HIV) replication in autologous CD4 T cells by HIV-specific cytotoxic T lymphocytes. J Virol 83:3138–3149. doi: 10.1128/JVI.02073-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. 2002. Magnitude of functional CD8+ T-cell responses to the Gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol 76:2298–2305. doi: 10.1128/jvi.76.5.2298-2305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radebe M, Nair K, Chonco F, Bishop K, Wright JK, van der Stok M, Bassett IV, Mncube Z, Altfeld M, Walker BD, Ndung'u T. 2011. Limited immunogenicity of HIV CD8+ T-cell epitopes in acute clade C virus infection. J Infect Dis 204:768–776. doi: 10.1093/infdis/jir394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novitsky V, Gilbert P, Peter T, McLane MF, Gaolekwe S, Rybak N, Thior I, Ndung'u T, Marlink R, Lee TH, Essex M. 2003. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J Virol 77:882–890. doi: 10.1128/JVI.77.2.882-890.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prado JG, Honeyborne I, Brierley I, Puertas MC, Martinez-Picado J, Goulder PJR. 2009. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J Virol 83:1018–1025. doi: 10.1128/JVI.01882-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prado JG, Prendergast A, Thobakgale C, Molina C, Tudor-Williams G, Ndung'u T, Walker BD, Goulder P. 2010. Replicative capacity of human immunodeficiency virus type 1 transmitted from mother to child is associated with pediatric disease progression rate. J Virol 84:492–502. doi: 10.1128/JVI.01743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A 97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emu B, Sinclair E, Hatano H, Ferre A, Shacklett B, Martin JN, Mccune JM, Deeks SG. 2008. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J Virol 82:5398–5407. doi: 10.1128/JVI.02176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, Streeck H. 2012. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med 4:123ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thobakgale CF, Prendergast A, Crawford H, Mkhwanazi N, Ramduth D, Reddy S, Molina C, Mncube Z, Leslie A, Prado J, Chonco F, Mphatshwe W, Tudor-Williams G, Jeena P, Blanckenberg N, Dong K, Kiepiela P, Coovadia H, Ndung'u T, Walker BD, Goulder PJR. 2009. Impact of HLA in mother and child on disease progression of pediatric human immunodeficiency virus type 1 infection. J Virol 83:10234–10244. doi: 10.1128/JVI.00921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthews PC, Leslie AJ, Katzourakis A, Crawford H, Payne R, Prendergast A, Power K, Kelleher AD, Klenerman P, Carlson J, Heckerman D, Ndung'u T, Walker BD, Allen TM, Pybus OG, Goulder PJR. 2009. HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering. J Virol 83:4605–4615. doi: 10.1128/JVI.02017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, Pereyra F, Kaufmann DE, Trocha A, Block BL, Daar ES, Connick E, Jessen H, Kelleher AD, Rosenberg E, Markowitz M, Schafer K, Vaida F, Iwamoto A, Little S, Walker BD. 2010. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J Virol 84:7581–7591. doi: 10.1128/JVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Connell KA, Hegarty RW, Siliciano RF, Blankson JN. 2011. Viral suppression of multiple escape mutants by de novo CD8+ T cell responses in a human immunodeficiency virus-1 infected elite suppressor. Retrovirology 8:63. doi: 10.1186/1742-4690-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Battivelli E, Migraine J, Lecossier D, Yeni P, Clavel F, Hance AJ. 2011. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J Virol 85:11846–11854. doi: 10.1128/JVI.05201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Granier C, Battivelli E, Lécuroux C, Venet A, Lambotte O, Schmitt-Boulanger M, Delaugerre C, Molina J-M, Chakrabarti LA, Clavel F, Hance AJ. 2013. Pressure from TRIM5α contributes to control of HIV-1 replication by individuals expressing protective HLA-B alleles. J Virol 87:10368–10380. doi: 10.1128/JVI.01313-13. [DOI] [PMC free article] [PubMed] [Google Scholar]