

ABSTRACT
Arenavirus species are responsible for severe life-threatening hemorrhagic fevers in western Africa and South America. Without effective antiviral therapies or vaccines, these viruses pose serious public health and biodefense concerns. Chemically distinct small-molecule inhibitors of arenavirus entry have recently been identified and shown to act on the arenavirus envelope glycoprotein (GPC) to prevent membrane fusion. In the tripartite GPC complex, pH-dependent membrane fusion is triggered through a poorly understood interaction between the stable signal peptide (SSP) and the transmembrane fusion subunit GP2, and our genetic studies have suggested that these small-molecule inhibitors act at this interface to antagonize fusion activation. Here, we have designed and synthesized photoaffinity derivatives of the 4-acyl-1,6-dialkylpiperazin-2-one class of fusion inhibitors and demonstrate specific labeling of both the SSP and GP2 subunits in a native-like Lassa virus (LASV) GPC trimer expressed in insect cells. Photoaddition is competed by the parental inhibitor and other chemically distinct compounds active against LASV, but not those specific to New World arenaviruses. These studies provide direct physical evidence that these inhibitors bind at the SSP-GP2 interface. We also find that GPC containing the uncleaved GP1-GP2 precursor is not susceptible to photo-cross-linking, suggesting that proteolytic maturation is accompanied by conformational changes at this site. Detailed mapping of residues modified by the photoaffinity adducts may provide insight to guide the further development of these promising lead compounds as potential therapeutic agents to treat Lassa hemorrhagic fever.
IMPORTANCE Hemorrhagic fever arenaviruses cause lethal infections in humans and, in the absence of licensed vaccines or specific antiviral therapies, are recognized to pose significant threats to public health and biodefense. Lead small-molecule inhibitors that target the arenavirus envelope glycoprotein (GPC) have recently been identified and shown to block GPC-mediated fusion of the viral and cellular endosomal membranes, thereby preventing virus entry into the host cell. Genetic studies suggest that these inhibitors act through a unique pH-sensing intersubunit interface in GPC, but atomic-level structural information is unavailable. In this report, we utilize novel photoreactive fusion inhibitors and photoaffinity labeling to obtain direct physical evidence for inhibitor binding at this critical interface in Lassa virus GPC. Future identification of modified residues at the inhibitor-binding site will help elucidate the molecular basis for fusion activation and its inhibition and guide the development of effective therapies to treat arenaviral hemorrhagic fevers.
INTRODUCTION
Arenaviruses are endemic in rodent populations worldwide, and some species can be transmitted to humans and cause severe hemorrhagic fevers with high morbidity and mortality (1, 2). Lassa virus (LASV) is prevalent in western Africa (3) and can be imported to the United States and Europe by infected travelers (4–6). Five New World (NW) species cause fatal disease in the Americas, including the Argentine hemorrhagic fever virus Junín (JUNV) (1, 7). New pathogenic species continue to emerge (8, 9), and novel arenaviruses have recently been identified in boid species of constrictor snakes (10, 11). Absent effective vaccines or therapies, hemorrhagic fever arenaviruses pose significant threats to public health and biodefense (12). Accordingly, these viruses are classified as category A priority pathogens (13).
Antiviral strategies that interfere with virus entry into the host cell have in many instances proven effective in preventing infection and treating disease. Arenaviruses enter the host cell by pH-dependent fusion of the viral and endosomal membranes (14), a process mediated by the viral envelope glycoprotein GPC. GPC is synthesized as a precursor that trimerizes and is cleaved by the cellular S1P/SKI-1 protease (15–17) to generate the receptor-binding (GP1) (18, 19) and transmembrane fusion (GP2) subunits. Unlike other viral envelope glycoproteins, GPC retains a 58-amino-acid residue signal peptide as a third, noncovalently associated subunit in the mature complex (20, 21) (Fig. 1). This stable signal peptide (SSP) contains two hydrophobic regions that span the membrane to form a hairpin structure (22), with a central ectodomain loop that interacts with GP2 to sense acidic pH and trigger membrane fusion (23, 24). As a bona fide member of the class I viral fusion proteins (25–28), the mature GPC exists in a kinetically trapped metastable state, primed by proteolytic cleavage and triggered by low pH to undergo a prescribed structural reorganization leading to formation of the thermodynamically favored trimer-of-hairpins structure and fusion of the viral and endosomal membranes (reviewed in references 29 and 30). Small-molecule compounds that interfere in this orderly process can effectively inhibit virus entry.
FIG 1.
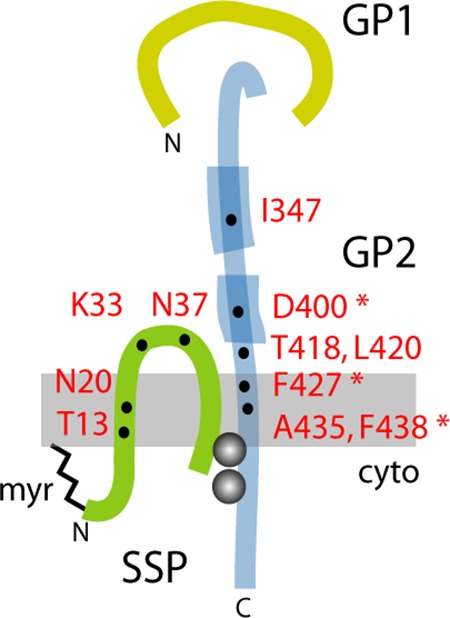
Model for the subunit organization of the tripartite GPC complex and location of resistance mutations. The SSP, GP1, and GP2 subunits are drawn schematically and are not to scale. Features include the myristate moiety at the N terminus of SSP (myr) (21), the binuclear zinc finger linking the penultimate cysteine-57 in SSP and the novel zinc-binding motif in the cytoplasmic tail of GP2 (gray balls) (54, 55), and the heptad-repeat regions in the ectodomain of GP2 that are diagnostic of class I viral fusion proteins (thickened lines) (25–28). Residues in JUNV GPC associated with resistance to or dependence on small-molecule fusion inhibitors are indicated (31, 32, 37, 40); asterisks denote mutations in GP2 that complement pH-dependent fusion defects engendered by mutations at K33 in SSP (24).
High-throughput screening campaigns at SIGA Technologies (31–34) and the Scripps Research Institute (35, 36) have recently identified six chemically distinct classes of small-molecule compounds that specifically antagonize pH-induced activation of GPC membrane fusion to prevent arenavirus entry into its host cell (37, 38). Several of these inhibitors have been shown to protect against lethal arenavirus infection in small-animal models (31, 39). Despite differences in chemistries and activity profiles against NW and/or Old World (OW) arenavirus species, these compounds appear to share a binding site on GPC (34, 37). Specifically, we have shown that all the inhibitors active against the NW JUNV compete for binding to recombinant JUNV GPC but are not displaced by those specific to the OW LASV (34). Yet, an SSP mutation in JUNV GPC that generates resistance to an NW-specific inhibitor renders the protein sensitive to inhibition by an OW-specific compound (37). Amino acid determinants of sensitivity to one or multiple classes of these inhibitors have been identified in both SSP and GP2 (Fig. 1) (31, 32, 37, 40). Interestingly, several resistance mutations also modulate the pH at which membrane fusion is triggered (23, 24). Recognizing that these inhibitors antagonize fusion activation (37, 38), we infer that they target the pH-sensing SSP-GP2 interface of GPC. However, atomic-level structural information on the intact GPC complex is not available. In this report, we describe the use of photoaffinity labeling to directly identify the inhibitor-binding site, as an initial step toward understanding the molecular basis for the pH-induced activation of GPC membrane fusion and its inhibition.
MATERIALS AND METHODS
Plasmids, cells, and recombinant baculoviruses.
The complete LASV GPC gene from the Josiah isolate (GenBank J04324) was molecularly cloned into a pcDNA3.1 plasmid vector (Life Technologies) for expression in Vero cells (40). For generating recombinant baculoviruses, the GPC open reading frame was subdivided to express SSP and the GP1-GP2 precursor (using the conventional signal peptide of human CD4) in a pFastBac Dual plasmid vector (Life Technologies) as previously described (34, 38). Expression in trans allows reconstitution of the functional tripartite GPC complex in both mammalian (21, 41) and insect (34, 38) cells and obviates concerns regarding incomplete signal peptidase cleavage of SSP (20, 42, 43). An innocuous FLAG tag appended to the C terminus of the GP1-GP2 precursor facilitates detection and affinity purification using the M2 anti-FLAG monoclonal antibody (MAb; Sigma) (34, 38). To generate a cleavage-defective (cd) LASV GPC mutant, the SKI-1/S1P cleavage site RRLL in the GP1-GP2 precursor was changed to AALL using QuikChange mutagenesis. Baculoviruses expressing wild-type and cd LASV GPC were generated using the Bac-to-Bac system (Life Technologies) (34, 38).
Expression of LASV GPC in insect cells: purification of insect cell membranes and characterization of the purified protein.
Recombinant baculoviruses were used to infect Trichoplusia ni High-Five cells, and membranes were prepared after 24 h of growth by rupturing the cells using nitrogen decompression (Parr bomb) as previously described (34, 38). Typical yields are ∼0.7 mg of LASV GPC per 3 × 109 cells per liter of culture. Washed membranes were resuspended at a concentration of ∼160 mg wet weight per ml (∼1.4 × 108 cell equivalents per ml) in buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 100 μM ZnCl2, and 20% glycerol and stored at −80°C.
LASV GPC was solubilized from the insect cell membranes by the addition of n-dodecyl β-d-maltoside (DDM; Anatrace) to 1.5% and then immobilized onto agarose beads containing the anti-FLAG M2 monoclonal antibody (Sigma) as previously described (34, 38). Beads were washed extensively in buffer containing 50 mM Tris (pH 7.5), 250 mM NaCl, 100 μM ZnCl2, and 0.1% DDM (wash buffer), and LASV GPC was eluted with the addition of 5 μM 3XFLAG peptide (Genscript). Following dialysis to remove the small peptide, the sample was subjected to size exclusion chromatography using a Superdex-200/G-75 tandem column (GE Healthcare) to determine the oligomeric state of the LASV GPC. Denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using lithium dodecyl sulfate (LDS) sample buffer with reducing agent (Life Technologies) and NuPAGE 4 to 12% bis-Tris gels (Life Technologies). Proteins were stained with Sypro Red (Life Technologies) or by Western blot analysis using an anti-FLAG horseradish peroxidase-conjugated antibody (Cell Signaling) and ECL 2 substrate (Pierce) and visualized using a Fuji FLA-3000G fluorescence imager.
Small-molecule inhibitors and photoaffinity derivatives.
Compounds 1 [lassamycin-1; (S)-16G8] and 2 [(S)-17D1] (Fig. 2) (compound numbers are in boldface in figures) were synthesized as described previously (36). ST-193 (32), ST-161 (33), and ST-294 (31) were kindly provided by SIGA Technologies, Inc. (Corvallis, OR). Chemical synthesis of photoaffinity probes 3 to 5 (Fig. 2) is outlined in Fig. 3. Briefly, probe 4 was synthesized as follows: l-tyrosinol (compound 6) was reductively alkylated using 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]benzaldehyde and sodium borohydride (NaBH4) in MeOH to provide compound 7. The published synthesis of 4-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzaldehyde provided a simple synthetic route to incorporate the diazirine (44). The secondary amine (compound 7) was then chemoselectively acylated using slow addition of the acyl chloride of N-[(2-nitrophenyl)sulfonyl]glycine to provide compound 8. Intramolecular Mitsonobu alkylation was employed using triphenylphosphine (PPh3) and diisopropyl azodicarboxylate (DIAD) in THF to yield piperazinone (compound 9). The cleavage of the (2-nitrophenyl)sulfonyl group was accomplished using thiophenol and K2CO3 in N,N-dimethylformamide (DMF) to provide secondary amine (compound 10), which was then acylated using benzofuran-2-carboxylic acid and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) in DMF to yield compound 11. The synthesis was then completed by alkylation of the phenol using propargyl bromide and K2CO3 in DMF to provide probe 4.
FIG 2.

Chemical structures of fusion inhibitors and photoaffinity derivatives. (S)-16G8 (lassamycin-1; compound 1) (36) and (S)-17D1 (compound 2) (35) provide the respective starting scaffolds for LRW1-078 (compound 4) and for LRW1-077 (compound 3) and LRW1-081 (compound 5). These photoaffinity probes include a photolabile trifluoromethyldiazirine moiety as well as a pendant alkyne for copper-catalyzed click chemistry addition of a TAMRA fluorophore.
FIG 3.

Chemical synthesis of photoaffinity probes. Synthetic routes to LRW1-078 (compound 4) (left) and LRW1-077 (compound 3) and LRW1-081 (compound 5) (right) are outlined here and described in Materials and Methods.
The synthesis of probes 3 and 5 was accomplished using a similar synthetic route. l-homophenylalaninol was reductively alkylated using 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]benzaldehyde to yield aminoalcohol 13, which was then chemoselectively acylated with the acyl chloride of N-[(2-nitrophenyl)sulfonyl]glycine to provide intermediate compound 14. Intramolecular Mitsonobu alkylation of the sulfonamide provided compound 15, and the subsequent cleavage of the (2-nitrophenyl)sulfonyl group yielded compound 16. The secondary amine 16 was then acylated using 4-(methyl-2-propyn-1-ylamino) benzoic acid to provide probe 3 or 4-(2-propynyloxy)benzoic acid to provide probe 5.
The identity of the photoaffinity probes was confirmed by 1H nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS).
Probe 3: (S)-4-(4-(methyl(prop-2-yn-1-yl)amino)benzoyl)-6-phenethyl-1-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)piperazin-2-one. 1H NMR (400 MHz, chloroform-d) δ 7.40 (d, J = 8.8 Hz, 2H), 7.32 to 7.20 (m, 4H), 7.15 to 7.01 (m, 5H), 6.81 (d, J = 8.8 Hz, 2H), 5.22 (d, J = 14.8 Hz, 1H), 4.60 (s, 2H), 4.20 to 3.99 (m, 3H), 3.76 (d, J = 14.9 Hz, 1H), 3.16 (d, J = 24.7 Hz, 2H), 3.04 (s, 3H), 2.93 to 2.62 (m, 1H), 2.52 to 2.28 (m, 1H), 2.19 (t, J = 2.3 Hz, 1H), 1.88 (td, J = 8.6, 5.1 Hz, 2H). HRMS for [C32H31F3N5O2]+: calculated, 574.2424; observed, 574.2422.
Probe 4: (S)-4-(benzofuran-2-carbonyl)-6-(4-(prop-2-yn-1-yloxy)benzyl)-1-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)piperazin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 7.68 (d, J = 7.8 Hz, 1H), 7.63 to 7.37 (m, 3H), 7.31 (dd, J = 8.5, 1.9 Hz, 3H), 7.18 (d, J = 8.1 Hz, 2H), 7.05 to 6.45 (m, 3H), 5.41 (d, J = 15.2 Hz, 1H), 5.04 (s, 1H), 4.69 (d, J = 13.6 Hz, 1H), 4.66 to 4.28 (m, 3H), 3.88 (d, J = 15.1 Hz, 1H), 3.57 to 3.36 (m, 1H), 3.14 to 2.57 (m, 3H), 2.46 (s, 1H). HRMS for [C32H26F3N4O4]+: calculated, 587.1901; observed, 587.1901.
Probe 5: (S)-6-phenethyl-4-(4-(prop-2-yn-1-yloxy)benzoyl)-1-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)piperazin-2-one. 1H NMR (400 MHz, chloroform-d) δ 7.42 (d, J = 8.6 Hz, 2H), 7.37 to 7.19 (m, 5H), 7.15 to 6.95 (m, 6H), 5.23 (d, J = 14.8 Hz, 1H), 4.74 (d, J = 2.4 Hz, 2H), 4.40 (br s, 1H), 4.12 (d, J = 17.7 Hz, 1H), 3.75 (d, J = 14.8 Hz, 1H), 3.14 (s, 2H), 2.87 (br s, 1H), 2.62 (s, 1H), 2.54 (d, J = 2.5 Hz, 1H), 1.88 (d, J = 7.4 Hz, 2H). HRMS for [C31H28F3N4O3]+: calculated, 561.2108; observed, 561.2107.
Inhibition of GPC-mediated cell-cell fusion.
The inhibitory potency of the photoaffinity derivatives was determined in a vaccinia virus-based cell-cell fusion reporter assay as previously described (23, 35, 40). In brief, Vero cells infected with a recombinant vaccinia virus encoding the bacteriophage T7 RNA polymerase (vTF7-3) and expressing LASV GPC from the minimal T7 promoter in the pcDNA3.1 vector are mixed with target cells infected with a recombinant vaccinia virus (vCB21R-lacZ) capable of expressing β-galactosidase under the control of the T7 promoter (45). GPC-mediated cell-cell fusion is induced upon exposure to acidic medium (pH 5.0) and drives expression of the β-galactosidase reporter, which is quantitated using the chemiluminescent GalactoLite Plus β-Galactosidase Reporter Gene Assay System (Applied Biosystems) in a SpectraMax L microplate luminometer (Molecular Devices). The 50% inhibitory concentrations (IC50s) were calculated using GraphPad Prism software by fitting data to single-slope dose-response curves constrained to 0% and 100% values.
Workflow for photoaffinity labeling.
Volumes of 500 μl of insect cell membranes containing an estimated 16 μg of LASV GPC were incubated with photoreactive inhibitors (typically at a final concentration of 10 μM) for 45 min at room temperature, followed by exposure to long-wavelength UV irradiation (∼366 nm) on ice for 30 min. Samples are positioned 5 cm from a Sylvania 100 W Mercury Blacklight bulb (H44GS-100) and rocked intermittently. In some experiments, nonphotoreactive fusion inhibitors were added at 50 μM with the photoaffinity probes to assess competition. Following photo-cross-linking, GPC was solubilized from the membranes by the addition of DDM to 2% and immobilized onto anti-FLAG agarose beads. Beads were washed extensively in wash buffer prior to using copper(I)-catalyzed azide-alkyne cycloaddition (click chemistry [46, 47]) to attach a tetramethylrhodamine (TAMRA) moiety to the pendant alkyne group of the photoaffinity adduct on LASV GPC. In this reaction, the beads are incubated in wash buffer containing 25 μM TAMRA-azide linker (Life Technologies catalog number T10182), 1 mM Tris(carboxyethyl) phosphine (TCEP), 1 mM CuSO4, and 100 μM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA; Sigma) at room temperature for 1 h. The reaction is stopped by extensive washing in wash buffer prior to elution of the bound LASV GPC in reducing LDS sample buffer (Life Technologies) and SDS-PAGE analysis using NuPAGE 4 to 12% bis-Tris gels (Life Technologies). TAMRA-containing protein bands were detected using a Fuji FLA-3000G imager. In some experiments, the LASV GPC was deglycosylated using peptide-N-glycosidase F (PNGase F; New England BioLabs) prior to SDS-PAGE analysis.
RESULTS
Strategy for designing inhibitor derivatives for photoaffinity labeling.
Lassamycin-1 (compound 1; Fig. 2) (36) is the active enantiomer of 16G8, a member of a class of 4-acyl-1,6-dialkylpiperazin-2-one fusion inhibitors (35). Compound 1 inhibits transduction by retroviral pseudotyped virions bearing LASV GPC with an IC50 of ∼200 nM (36). Discovery of the (S)-configuration as the active enantiomer of 16G8 led to the design and synthesis of (S)-17D1 (compound 2; Fig. 2) (35), another 4-acyl-1,6-dialkylpiperazin-2-one inhibitor that inhibits LASV GPC-mediated cell-cell fusion with an IC50 of 30 nM. Compounds 1 and 2 were subsequently used as starting scaffolds to design three alkynylated trifluoromethyldiazirine photoaffinity probes (probes 3 to 5; Fig. 2). The placement of the trifluoromethyldiazirine was guided by the known tolerance for functionality (4-OMe or 4-Me groups) at the 4-position of the N1 benzyl group (35). The optimal placement of the alkyne was then determined to be as a propargyl substituent on a heteroatom at the 4-position of the N4 benzamide group for probes 3 (LRW1-077) and 5 (LRW1-081). Insight for this alkyne placement was provided by compound 2 as well as a scanning library of derivatives of compound 2 that demonstrated that compounds with a variety of alkylated heteroatom substituents at the 4-position of the N4 benzamide group showed potent activity. In order to study probes based upon compound 1, we decided to incorporate the alkyne as an aryl propargyl ether at the 4-position of the C6 benzyl group to generate probe 4 (LRW1-078). This design was guided by our knowledge that a compound with an aryl methyl ether at this position retained potent activity (unpublished data).
Photoaffinity derivatives inhibit LASV GPC-mediated cell-cell fusion.
The potencies of compounds 3 to 5 were determined by inhibition of pH-induced cell-cell fusion using Vero cells expressing LASV GPC in a β-galactosidase reporter assay (23, 35, 40). The respective IC50s for compounds 3, 4, and 5 are 70 nM, 80 nM, and 165 nM, relative to 30 nM for 17D1 (Fig. 4). Thus, and as predicted from known structure-activity relationships, the photoaffinity derivatives maintained good potency compared to the unmodified 17D1 parent compound.
FIG 4.
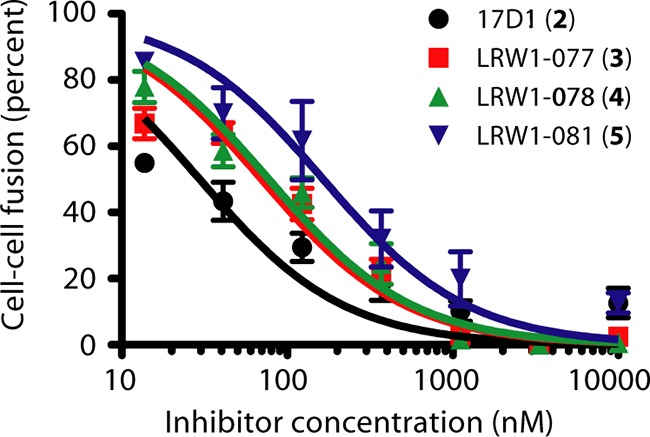
Photoaffinity derivatives potently inhibit LASV GPC-mediated cell-cell fusion. pH-induced syncytium formation was quantitated using a vaccinia virus-based β-galactosidase fusion reporter assay. Cocultures of GPC-expressing and target cells were incubated with serial dilutions of the fusion inhibitors prior to exposure to acidic medium (pH 5.0), and reporter activity was quantitated by chemiluminescence. Inhibition curves were fit to single-slope dose-response curves using GraphPad Prism software. Black circle, 17D1 (compound 2); red square, LRW1-077 (compound 3); green triangle, LRW1-078 (compound 4); blue triangle, LRW1-081 (compound 5).
Characterization of recombinant LASV GPC expressed in insect cells.
LASV GPC from the Josiah isolate was produced in insect cells and used as the substrate for photoaffinity labeling. We have previously described the purification and characterization of full-length trimeric JUNV GPC expressed in insect cells (34, 38), and similar methods were used to generate the LASV protein. In contrast to JUNV GPC, which appeared to be refractory to cleavage by the insect S1P/SKI-1 protease (38), the purified LASV GPC contained significant amounts of the mature GP1 and GP2 subunits (Fig. 5, insert). The enhanced susceptibility of LASV GPC to cleavage may reflect the presence of a preferred aromatic residue at position P7 in the S1P/SKI-1 recognition motif (48). Size exclusion chromatography demonstrated that LASV GPC formed a relatively homogeneous trimer in solution (Fig. 5), as reported for JUNV GPC (34) and consistent with the trimeric assembly of class I fusion proteins.
FIG 5.
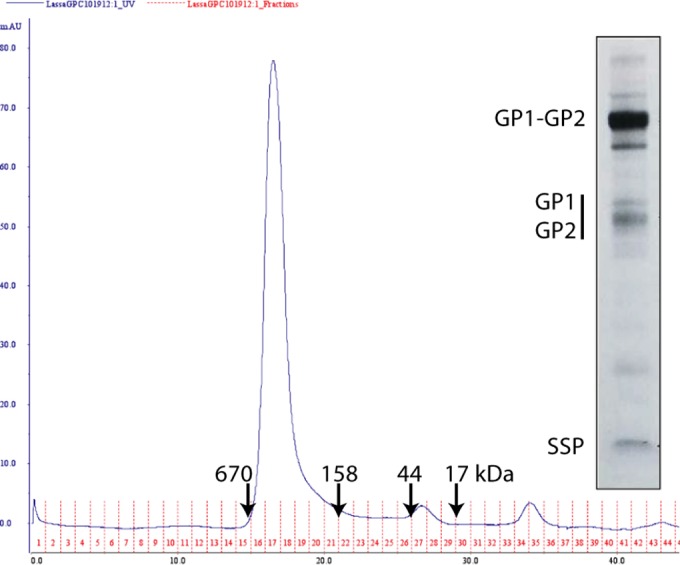
Characterization of purified LASV GPC from insect cells. LASV GPC was solubilized from insect cell membranes and purified by affinity chromatography using the C-terminal FLAG tag. Size exclusion chromatography reveals a relatively homogeneous peak consistent with a GPC trimer. Molecular size markers (in kilodaltons) are indicated by arrows. (Insert) The affinity-purified protein was resolved by SDS-PAGE and stained using Sypro Red. The heavily glycosylated GP1 subunit is heterodisperse and comigrates with GP2 in this analysis.
Photoaffinity labeling of LASV GPC.
We have previously reported that membrane reconstitution of purified JUNV GPC greatly enhances the affinity of inhibitor binding relative to the solubilized protein (34). Thus, we chose to incubate the photolabile inhibitor derivatives with LASV GPC in purified insect cell membranes. An initial titration of LRW1-077 (compound 3) revealed maximal photoaddition at 3 μM compound (not shown), and subsequent studies were performed using 10 μM the inhibitors. At this concentration, we were unable to detect any photolabeling of solubilized LASV GPC (not shown).
The standard workflow for photoaffinity labeling of LASV GPC is described in Materials and Methods. In brief, insect cell membranes containing LASV GPC were incubated with the photoreactive inhibitor and subjected to long-wavelength UV light (∼366 nm) to activate the photolabile trifluoromethyldiazirine group. The resultant short-lived and highly reactive carbene is able to insert across proximal bonds to covalently link the inhibitor to the protein. LASV GPC was then solubilized in DDM-containing buffer and bound to immobilized anti-FLAG MAb on agarose beads. Coupling of the pendant alkyne on the inhibitor adduct to an azide-containing linker bearing a fluorescent TAMRA moiety was accomplished using copper-catalyzed click chemistry. GPC was then eluted from the FLAG beads, and the fluorescently tagged subunits were identified by SDS-PAGE analysis.
By using this procedure, we could detect photoaddition by each of the modified inhibitors to SSP and either GP1 or GP2; the last two subunits are difficult to resolve using SDS-PAGE (Fig. 6A). PNGase F digestion enables clear differentiation of the GP1 and GP2 polypeptides and revealed that only GP2 was labeled (Fig. 6B). The labeling efficiencies by the three compounds were comparable, and in all cases SSP was preferentially labeled relative to GP2. However, multiple lines of existing evidence support only a single inhibitor-binding site on GPC (34, 37, 38). Therefore, we conclude that photoaddition of these probes can occur stochastically with either SSP or GP2. This result provides direct confirmation for inhibitor binding at the SSP-GP2 interface. Notably, the GP1-GP2 precursor that constitutes ∼90% of the total protein (Fig. 5, insert) was not labeled.
FIG 6.

Photoaffinity labeling of LASV GPC. LASV GPC in purified insect cell membranes was subjected to photoaffinity labeling, and the covalently linked inhibitors were subsequently modified with TAMRA using click chemistry. Subunits were resolved by SDS-PAGE analysis, and TAMRA was detected by fluorescence imaging. (A) GPC was incubated with the indicated photoreactive inhibitors alone (none) or together with an excess of nonphotoreactive competitor. ST-294 (31) is specific for NW arenaviruses, while 17D1 (36) and ST-193 (32) are broadly active against both NW and OW arenaviruses. ST-161 (33) is specific for OW arenaviruses. (B) PNGase F was used to deglycosylate GPC to demonstrate that the FLAG-tagged GP2 polypeptide (ppGP2; calculated molecular mass, 28 kDa), and not GP1 (ppGP1; calculated molecular mass, 22.7 kDa), is photoaffinity labeled.
Photoaffinity labeling of SSP and GP2 is competed by OW-active, but not NW-active, inhibitors.
The specificity of photoaddition was demonstrated by competition using 17D1 (compound 2), the parent inhibitor from which probes 3 and 5 were derived. By using a 5-fold molar excess of 17D1, labeling of both the SSP and GP2 subunits by each of the three photoreactive compounds was completely blocked (Fig. 6A). The effectiveness of competition is likely enhanced by the higher binding affinity of the parental inhibitor (Fig. 4). Furthermore, photoaddition was also blocked by the addition of excess ST-161 and ST-193, two independently identified SIGA fusion inhibitors that are respectively selective for LASV (33) or broadly active against NW and OW arenaviruses (32) (Fig. 6A). The NW-specific inhibitor ST-294 (31) failed to block photoaddition (Fig. 6A), in keeping with the inverse result that OW-specific inhibitors fail to bind JUNV GPC (34). Together, these results validate the specificity of the photoaddition reaction and the notion that these chemically distinct fusion inhibitors share a binding site on GPC (31, 32, 34, 37). The coordinate loss of SSP and GP2 labeling provides further support for a single binding site at the SSP-GP2 interface.
Neither SSP nor GP1-GP2 is labeled in the GPC precursor complex.
Despite its predominance in purified LASV GPC protein, the GP1-GP2 precursor is not labeled (Fig. 6A). To determine whether SSP associated with the (unlabeled) GP1-GP2 precursor is still susceptible to photoaddition, we generated a cleavage-defective (cd) form of LASV GPC in which the S1P/SKI-1 cleavage motif (RRLL) was mutated to AALL. As illustrated in Fig. 7A, cd-GPC is expressed well in insect cells, with no evidence of proteolytic cleavage. Photoaddition studies revealed only background labeling of SSP and the GP1-GP2 precursor in cd-GPC, none of which was competed by an excess of 17D1 or ST-193 (Fig. 7B). These results indicate that SSP in GPC containing the uncleaved GP1-GP2 precursor is not susceptible to photoaffinity labeling by these inhibitors. This result is in apparent contrast with previous surface plasmon resonance studies that demonstrated binding of NW-active inhibitors to the uncleaved JUNV GPC precursor (34, 38). It is possible that the inhibitors differ in their ability to bind their respective GPC precursors. However, the failure to label does not necessarily exclude inhibitor binding to the precursor complex. Protein bonds that are potential targets for insertion by the reactive carbene may be distant in the LASV GPC precursor and no longer accessible for reaction. In either case, the absence of labeling indicates that the environment at the inhibitor-binding site differs in precursor and mature LASV GPC and suggests that proteolytic cleavage is associated with conformational changes that alter the SSP-GP2 interface, perhaps to establish the pH-sensitive state of the mature GPC complex.
FIG 7.

Cleavage-defective LASV GPC is not susceptible to photoaffinity labeling. The SKI-1/S1P cleavage site in LASV GPC was mutated (RRLL to AALL) to prevent proteolytic maturation. (A) SDS-PAGE analysis confirms the absence of mature GP1 and GP2 subunits in cd-GPC by Sypro Red staining (left) and Western blot analysis for the C-terminal FLAG tag (right). Wild-type (wt) LASV GPC is shown for comparison. (B) Photoaffinity labeling studies using the indicated photoreactive inhibitors show that neither GP2 nor SSP is significantly labeled in the precursor GPC complex. A low level of background labeling is not competed by 17D1 or ST-193.
pH-induced fusion activation of GPC prevents labeling.
The arenavirus fusion inhibitors target the prefusion form of GPC to block low-pH triggering of membrane fusion (31, 37, 38). By virtue of controlling fusion activation, it is likely that the SSP-GP2 interface also undergoes structural reorganization during the fusion process. To investigate whether the inhibitor-binding site is retained in the postfusion complex, we pretreated insect cell membranes bearing the wild-type LASV GPC with buffer adjusted to pH 5.0, a pH that efficiently and irreversibly triggers LASV GPC-mediated cell-cell fusion. Following return to neutral pH and incubation with LRW1-077 (compound 3), we were unable to detect photolabeling of the postfusion GPC complex (Fig. 8). We conclude that the inhibitor-binding site at the SSP-GP2 interface is disrupted upon exposure to acidic pH, consistent with the notion that this intersubunit interaction maintains the prefusion GPC complex at neutral pH and responds to acidic pH to trigger the conformational changes leading to membrane fusion.
FIG 8.
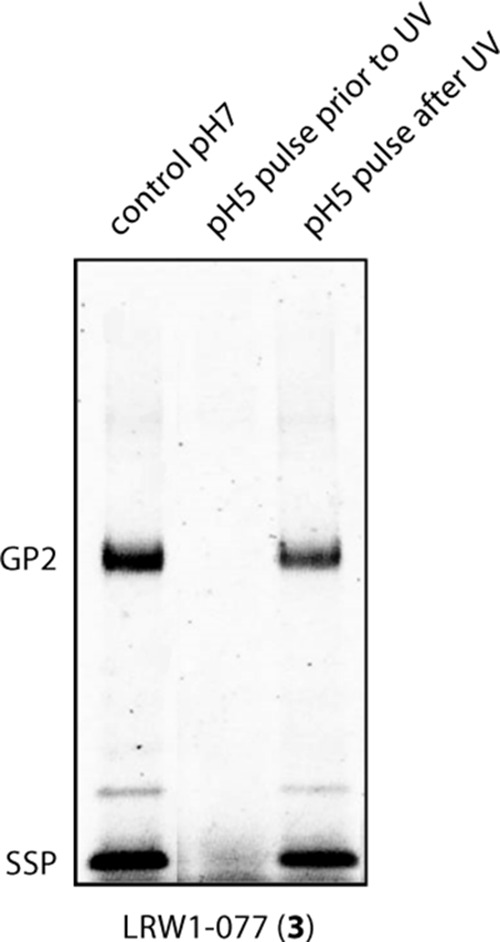
pH-triggered fusion activation abrogates photoaffinity labeling. Insect cell membranes bearing wild-type LASV GPC were incubated with LRW1-077 (3) and exposed to long-wavelength UV light at neutral pH as per the standard workflow (control pH 7) or following a 10-min pulse in buffer adjusted to pH 5.0 and subsequent neutralization (pH 5 pulse prior to UV). In order to exclude any adverse effects of the acidic buffer, labeled membranes (control pH 7) were subsequently pulsed with pH 5.0 buffer prior to SDS-PAGE analysis (pH 5 pulse after UV).
DISCUSSION
Our studies demonstrate specific photoaffinity labeling of both the SSP and GP2 subunits of LASV GPC using the novel photoreactive fusion inhibitors 3 to 5. The coordinate labeling of both subunits provides direct evidence for inhibitor binding at the SSP-GP2 interface. This result is consistent with previous genetic studies identifying determinants of inhibitor sensitivity in both of these subunits (31, 32, 37, 40). In considering the role of SSP-GP2 interactions in modulating pH-induced activation of GPC membrane fusion (23, 24), these new findings support the notion that these fusion inhibitors stabilize the SSP-GP2 interface in the prefusion GPC complex against acidic pH, thereby preventing fusion activation and virus entry (37, 38). In addition, the failure to photochemically label either subunit in the uncleaved GPC precursor reveals that proteolytic maturation of GPC is accompanied by conformational changes at the SSP-GP2 interface, changes that may be critical in establishing the pH-sensitive metastable state of the prefusion GPC complex. Similarly, the inability to label GPC after exposure to acidic pH highlights specific fusion-associated conformational changes at the SSP-GP2 interface, consistent with its role in triggering the fusogenic cascade.
Interestingly, a number of chemically diverse small-molecule fusion inhibitors of influenza virus hemagglutinin, another class I fusion protein, have been shown to act through an analogous mechanism, albeit by targeting a different intersubunit interface (49–52). Thus, stabilization of the prefusion state through the fortuitous binding of small-molecule compounds may prove to be a general mechanism for effective fusion inhibition. Indeed, a newly described inhibitor of the OW lymphocytic choriomeningitis virus GPC may act in this manner (53).
In the absence of atomic-level structural information on the tripartite GPC complex, our photoaffinity labeling studies provide an important perspective from which to better understand the structure and function of this unique viral fusion protein. Identification of SSP and GP2 residues modified by these and other photoreactive inhibitors may provide insight to guide the development of therapeutic agents to treat Lassa hemorrhagic fever.
ACKNOWLEDGMENTS
J.H.N. is grateful to Sean Amberg and Dongcheng Dai (SIGA Technologies, Inc.) for providing ST-193, ST-161, and ST-294 fusion inhibitors. We also thank Celestine J. Thomas (University of Montana) and Alex S. Evers (Washington University School of Medicine, St. Louis, MO) for constructive comments in optimizing the workflow for photoaffinity labeling.
L.R.W. is a consultant for Arisan Therapeutics in the commercial development of arenavirus fusion inhibitors.
Funding for this project was provided by National Institutes of Health grants AI074818 (to J.H.N.) and AI120490 (to J.H.N. and A. S. Evers, Washington University School of Medicine), a subaward to J.H.N. from the Rocky Mountain Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (AI065357; J. Belisle, Colorado State University) and by a National Institutes of Health grant CA042056 to D.L.B.
REFERENCES
- 1.Peters CJ. 2002. Human infection with arenaviruses in the Americas. Curr Top Microbiol Immunol 262:65–74. [DOI] [PubMed] [Google Scholar]
- 2.McCormick JB, Fisher-Hoch SP. 2002. Lassa fever. Curr Top Microbiol Immunol 262:75–109. [DOI] [PubMed] [Google Scholar]
- 3.McCormick JB, Webb PA, Krebs JW, Johnson KM, Smith ES. 1987. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis 155:437–444. doi: 10.1093/infdis/155.3.437. [DOI] [PubMed] [Google Scholar]
- 4.Haas WH, Breuer T, Pfaff G, Schmitz H, Köhler P, Asper M, Emmerich P, Drosten C, Gölnitz U, Fleischer K, Günther S. 2003. Imported Lassa fever in Germany: surveillance and management of contact persons. Clin Infect Dis 36:1254–1258. doi: 10.1086/374853. [DOI] [PubMed] [Google Scholar]
- 5.Amorosa V, MacNeil A, McConnell R, Patel A, Dillon KE, Hamilton K, Erickson BR, Campbell S, Knust B, Cannon D, Miller D, Manning C, Rollin PE, Nichol ST. 2010. Imported Lassa fever, Pennsylvania, USA, 2010. Emerg Infect Dis 16:1598–1600. doi: 10.3201/eid1610.100774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2014. Lassa fever reported in U.S. traveler returning from West Africa http://www.cdc.gov/media/releases/2014/p0404-lassa-fever.html Accessed 19 May 2016.
- 7.Delgado S, Erickson BR, Agudo R, Blair PJ, Vallejo E, Albariño CG, Vargas J, Comer JA, Rollin PE, Ksiazek TG, Olson JG, Nichol ST. 2008. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog 4:e1000047. doi: 10.1371/journal.ppat.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briese T, Paweska JT, McMullan LK, Hutchison SK, Street C, Palacios G, Khristova ML, Weyer J, Swanepoel R, Egholm M, Nichol ST, Lipkin WI. 2009. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog 5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palacios G, Savji N, Hui J, Travassos da Rosa A, Popov V, Briese T, Tesh R, Lipkin WI. 2010. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J Gen Virol 91:1315–1324. doi: 10.1099/vir.0.017798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, Dunker F, Derisi JL. 2012. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio 3:e00180-12. doi: 10.1128/mBio.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hetzel U, Sironen T, Laurinmaki P, Liljeroos L, Patjas A, Henttonen H, Vaheri A, Artelt A, Kipar A, Butcher SJ, Vapalahti O, Hepojoki J. 2013. Isolation, identification, and characterization of novel arenaviruses, the etiological agents of boid inclusion body disease. J Virol 87:10918–10935. doi: 10.1128/JVI.01123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerber R, Reindl S, Romanowski V, Gomez RM, Ogbaini-Emovon E, Gunther S, ter Meulen J. 2015. Research efforts to control highly pathogenic arenaviruses: a summary of the progress and gaps. J Clin Virol 64:120–127. doi: 10.1016/j.jcv.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 13.NIAID. 2009. NIAID category A, B, and C priority pathogens. http://www.niaid.nih.gov/topics/BiodefenseRelated/Biodefense/research/Pages/CatA.aspx Accessed 19 May 2016.
- 14.Di Simone C, Zandonatti MA, Buchmeier MJ. 1994. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology 198:455–465. doi: 10.1006/viro.1994.1057. [DOI] [PubMed] [Google Scholar]
- 15.Lenz O, ter Meulen J, Klenk H-D, Seidah NG, Garten W. 2001. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A 98:12701–12705. doi: 10.1073/pnas.221447598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beyer WR, Popplau D, Garten W, von Laer D, Lenz O. 2003. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J Virol 77:2866–2872. doi: 10.1128/JVI.77.5.2866-2872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunz S, Edelmann KH, de la Torre J-C, Gorney R, Oldstone MBA. 2003. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 314:168–178. doi: 10.1016/S0042-6822(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 18.Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, Li W, Nagel J, Schmidt PJ, Nunberg JH, Andrews NC, Farzan M, Choe H. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MBA. 1998. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 20.Eichler R, Lenz O, Strecker T, Garten W. 2003. Signal peptide of Lassa virus glycoprotein GP-C exhibits an unusual length. FEBS Lett 538:203–206. doi: 10.1016/S0014-5793(03)00160-1. [DOI] [PubMed] [Google Scholar]
- 21.York J, Romanowski V, Lu M, Nunberg JH. 2004. The signal peptide of the Junín arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1-G2 complex. J Virol 78:10783–10792. doi: 10.1128/JVI.78.19.10783-10792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agnihothram SS, York J, Trahey M, Nunberg JH. 2007. Bitopic membrane topology of the stable signal peptide in the tripartite Junín virus GP-C envelope glycoprotein complex. J Virol 81:4331–4337. doi: 10.1128/JVI.02779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.York J, Nunberg JH. 2006. Role of the stable signal peptide of the Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J Virol 80:7775–7780. doi: 10.1128/JVI.00642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.York J, Nunberg JH. 2009. Intersubunit interactions modulate pH-induced activation of membrane fusion by the Junin virus envelope glycoprotein GPC. J Virol 83:4121–4126. doi: 10.1128/JVI.02410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.York J, Agnihothram SS, Romanowski V, Nunberg JH. 2005. Genetic analysis of heptad-repeat regions in the G2 fusion subunit of the Junin arenavirus envelope glycoprotein. Virology 343:267–279. doi: 10.1016/j.virol.2005.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eschli B, Quirin K, Wepf A, Weber J, Zinkernagel R, Hengartner H. 2006. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J Virol 80:5897–5907. doi: 10.1128/JVI.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Igonet S, Vaney MC, Vonhrein C, Bricogne G, Stura EA, Hengartner H, Eschli B, Rey FA. 2011. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc Natl Acad Sci U S A 108:19967–19972. doi: 10.1073/pnas.1108910108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsy ML, Harlos K, Huiskonen JT, Bowden TA. 2013. Crystal structure of Venezuelan hemorrhagic fever virus fusion glycoprotein reveals a class 1 postfusion architecture with extensive glycosylation. J Virol 87:13070–13075. doi: 10.1128/JVI.02298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolken TC, Laquerre S, Zhang Y, Bailey TR, Pevear DC, Kickner SS, Sperzel LE, Jones KF, Warren TK, Amanda Lund S, Kirkwood-Watts DL, King DS, Shurtleff AC, Guttieri MC, Deng Y, Bleam M, Hruby DE. 2006. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antiviral Res 69:86–89. doi: 10.1016/j.antiviral.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larson RA, Dai D, Hosack VT, Tan Y, Bolken TC, Hruby DE, Amberg SM. 2008. Identification of a broad-spectrum arenavirus entry inhibitor. J Virol 82:10768–10775. doi: 10.1128/JVI.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgeson JR, Gharaibeh DN, Moore AL, Larson RA, Amberg SM, Bolken TC, Hruby DE, Dai D. 2013. Lead optimization of an acylhydrazone scaffold possessing antiviral activity against Lassa virus. Bioorg Med Chem Lett 23:5840–5843. doi: 10.1016/j.bmcl.2013.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas CJ, Casquilho-Gray HE, York J, DeCamp DL, Dai D, Petrilli EB, Boger DL, Slayden RA, Amberg SM, Sprang SR, Nunberg JH. 2011. A specific interaction of small-molecule entry inhibitors with the envelope glycoprotein complex of the Junín hemorrhagic fever arenavirus. J Biol Chem 286:6192–6200. doi: 10.1074/jbc.M110.196428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee AM, Rojek JM, Spiropoulou CF, Gundersen AT, Jin W, Shaginian A, York J, Nunberg JH, Boger DL, Oldstone MBA, Kunz S. 2008. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J Biol Chem 283:18734–18742. doi: 10.1074/jbc.M802089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitby LR, Lee AM, Kunz S, Oldstone MB, Boger DL. 2009. Characterization of Lassa virus cell entry inhibitors: determination of the active enantiomer by asymmetric synthesis. Bioorg Med Chem Lett 19:3771–3774. doi: 10.1016/j.bmcl.2009.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.York J, Dai D, Amberg SA, Nunberg JH. 2008. pH-induced activation of arenavirus membrane fusion is antagonized by small-molecule inhibitors. J Virol 82:10932–10939. doi: 10.1128/JVI.01140-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas C, Shankar S, Casquilho-Gray HE, York J, Sprang SR, Nunberg JH. 2012. Biochemical reconstitution of arenavirus envelope glycoprotein-mediated membrane fusion. PLoS One 7:e51114. doi: 10.1371/journal.pone.0051114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cashman KA, Smith MA, Twenhafel NA, Larson RA, Jones KF, Allen RD III, Dai D, Chinsangaram J, Bolken TC, Hruby DE, Amberg SM, Hensley LE, Guttieri MC. 2011. Evaluation of Lassa antiviral compound ST-193 in a guinea pig model. Antiviral Res 90:70–79. doi: 10.1016/j.antiviral.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Messina EL, York J, Nunberg JH. 2012. Dissection of the role of the stable signal peptide of the arenavirus envelope glycoprotein in membrane fusion. J Virol 86:6138–6145. doi: 10.1128/JVI.07241-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eichler R, Lenz O, Strecker T, Eickmann M, Klenk HD, Garten W. 2003. Identification of Lassa virus glycoprotein signal peptide as a trans-acting maturation factor. EMBO Rep 4:1084–1088. doi: 10.1038/sj.embor.7400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.York J, Nunberg JH. 2007. Distinct requirements for signal peptidase processing and function of the stable signal peptide (SSP) subunit in the Junin virus envelope glycoprotein. Virology 359:72–81. doi: 10.1016/j.virol.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 43.Froeschke M, Basler M, Groettrup M, Dobberstein B. 2003. Long-lived signal peptide of lymphocytic choriomeningitis virus glycoprotein pGP-C. J Biol Chem 278:41914–41920. doi: 10.1074/jbc.M302343200. [DOI] [PubMed] [Google Scholar]
- 44.Odell L, Chau N, Mariana A, Graham M, Robinson P, McCluskey A. 2009. Azido and diazarinyl analogues of bis-tyrphostin as asymmetrical inhibitors of dynamin GTPase. ChemMedChem 4:1182–1188. doi: 10.1002/cmdc.200900054. [DOI] [PubMed] [Google Scholar]
- 45.Nussbaum O, Broder CC, Berger EA. 1994. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. J Virol 68:5411–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. 2002. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41:2596–2599. doi:. [DOI] [PubMed] [Google Scholar]
- 47.Tornoe CW, Christensen C, Meldal M. 2002. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 48.Burri DJ, da Palma JR, Seidah NG, Zanotti G, Cendron L, Pasquato A, Kunz S. 2013. Differential recognition of Old World and New World arenavirus envelope glycoproteins by subtilisin kexin isozyme 1 (SKI-1)/site 1 protease (S1P). J Virol 87:6406–6414. doi: 10.1128/JVI.00072-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bodian DL, Yamasaki RB, Buswell RL, Stearns JF, White JM, Kuntz ID. 1993. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 32:2967–2978. doi: 10.1021/bi00063a007. [DOI] [PubMed] [Google Scholar]
- 50.Luo G, Torri A, Harte WE, Danetz S, Cianci C, Tiley L, Day S, Mullaney D, Yu KL, Ouellet C, Dextraze P, Meanwell N, Colonno R, Krystal M. 1997. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J Virol 71:4062–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanderlinden E, Göktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 84:4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, Skehel JJ. 2008. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U S A 105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ngo N, Cubitt B, Iwasaki M, de la Torre JC. 2015. Identification and mechanism of action of a novel small-molecule inhibitor of arenavirus multiplication. J Virol 89:10924–10933. doi: 10.1128/JVI.01587-15. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.York J, Nunberg JH. 2007. A novel zinc-binding domain is essential for formation of the functional Junín virus envelope glycoprotein complex. J Virol 81:13385–13391. doi: 10.1128/JVI.01785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Briknarova K, Thomas CJ, York J, Nunberg JH. 2011. Structure of a zinc-binding domain in the Junin virus envelope glycoprotein. J Biol Chem 286:1528–1536. doi: 10.1074/jbc.M110.166025. [DOI] [PMC free article] [PubMed] [Google Scholar]