

ABSTRACT
p53 is a critical host cell factor in the cellular response to a broad range of stress factors. We recently reported that p53 is required for efficient herpes simplex virus 1 (HSV-1) replication in cell culture. However, a defined role for p53 in HSV-1 replication and pathogenesis in vivo remains elusive. In this study, we examined the effects of p53 on HSV-1 infection in vivo using p53-deficient mice. Following intracranial inoculation, p53 knockout reduced viral replication in the brains of mice and led to significantly reduced rates of mortality due to herpes simplex encephalitis. These results suggest that p53 is an important host cell regulator of HSV-1 replication and pathogenesis in the central nervous system (CNS).
IMPORTANCE HSV-1 causes sporadic cases of encephalitis, which, even with antiviral therapy, can result in severe neurological defects and even death. Many host cell factors involved in the regulation of CNS HSV-1 infection have been investigated using genetically modified mice. However, most of these factors are immunological regulators and act via immunological pathways in order to restrict CNS HSV-1 infection. They therefore provide limited information on intrinsic host cell regulators that may be involved in the facilitation of CNS HSV-1 infection. Here we demonstrate that a host cell protein, p53, which has generally been considered a host cell restriction factor for various viral infections, is required for efficient HSV-1 replication and pathogenesis in the CNS of mice. This is the first report showing that p53 positively regulates viral replication and pathogenesis in vivo and provides insights into its molecular mechanism, which may suggest novel clinical treatment options for herpes simplex encephalitis.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is an etiological agent in various human mucocutaneous diseases, such as herpes labialis, genital herpes, herpetic whitlow, and keratitis. HSV-1 also causes herpes simplex encephalitis (HSE), which is sporadic and which can be lethal or result in severe neurological defects in a significant fraction of survivors, even with antiviral therapy (1).
p53 is a multifunctional host protein that plays a central role in cellular responses to a broad range of stress factors through its regulation of various cellular pathways, such as apoptosis, cell cycling, cellular senescence, DNA repair, autophagy, and innate immune control (2, 3). Since viral infection is a type of stress, it appears that viral infection activates p53 responses that trigger apoptosis of infected cells, thereby suppressing viral replication (4). Thus, when a person is infected with a DNA virus, viral genome replication induces host DNA damage responses (DDRs), which activate apoptotic p53 responses (4). In RNA virus infections, double-stranded RNAs are produced, and these double-stranded RNAs trigger antiviral responses mediated by type I interferon (IFN-I) signaling, in which p53 appears to function as both an upstream and a downstream regulator (5, 6). In agreement with these observations, the replication of various viruses in cell culture, including hepatitis C virus, vesicular stomatitis virus (VSV), Sendai virus, poliovirus, influenza A virus (IAV), vaccinia virus, human papillomavirus, and JC virus, is enhanced by the knockout or knockdown of p53 and inhibited by the overexpression of p53 (4, 6–8). Furthermore, it is well established that many viruses have acquired a variety of distinct mechanisms to counteract the negative effects of p53 in infected cells (4). Thus, p53 is generally considered a host restriction factor in a range of viral infections.
Conversely, some viruses appear to require p53 for efficient viral replication. It has been reported that knockout of p53 impairs the replication of human cytomegalovirus (HCMV) and HSV-1 (9, 10). In addition, p53 has been shown to be required for the efficient expression of the Epstein-Barr virus (EBV) protein BZLF1, a key regulator in the initiation of viral lytic infection (11). However, at later stages of lytic infection, BZLF1-mediated p53 degradation counteracts p53's inhibitory effects and prevents apoptosis of infected cells (12). Similarly, p53 promotes expression of HSV-1 ICP27, an essential regulator of viral gene expression, early in infection but at later stages of infection has the opposite effect on another critical regulator of viral gene expression, HSV-1 ICP0 (9). Interestingly, this negative effect of p53 on ICP0 expression appears to be antagonized by HSV-1 ICP22. In agreement with these observations, p53 knockout had no obvious effect on the replication of an ICP22-null HSV-1 mutant, although it significantly reduced wild-type (WT) HSV-1 replication, as described above (9). These observations suggest that these viruses have evolved multiple mechanisms to precisely (in a time- and status-dependent manner) organize p53's positive and negative effects in infected cells.
Collectively, the numerous studies described above have gradually begun to elucidate the multiple roles of p53 in the replication of a variety of viruses. However, the information provided above is mostly based on studies that used cell culture systems, and studies that have investigated whether p53 is, in fact, involved in viral replication and pathogenesis in vivo are limited. Thus far, in studies using p53-deficient (p53−/−) mice, p53 has been shown to be a critical host restriction factor in vivo for only a fraction of RNA viruses, including VSV, IAV, the polycythemia-inducing strain of Friend vius (FVP), and Abelson murine leukemia virus (5, 13–15). Furthermore, a role(s) for p53 in infection by DNA viruses in vivo has not yet been reported. In this study, we report the effects of p53 on the replication and pathogenesis of HSV-1 in vivo using p53-deficient mice.
MATERIALS AND METHODS
Mice and virus.
p53-deficient (p53−/−) ICR mice and their wild-type (p53+/+) littermates were obtained by interbreeding heterozygous (p53+/−) mice (accession number CDB 0001K) (16), provided by the RIKEN BioResource Center (BRC). The WT strain HSV-1(F) was described previously (17, 18).
Animal studies.
For intracranial infection, 5- to 7-week-old female p53−/− ICR mice and littermate control p53+/+ ICR mice were injected intracranially with 100 PFU of HSV-1(F) as described previously (17). Mice were monitored daily, and mortality was recorded from 1 to 14 days postinfection. The virus titers in the brains of the mice were determined as described previously (19). All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments of the Science Council of Japan (20). The protocol was approved by the Institutional Animal Care and Use Committee, Institute of Medical Science, the University of Tokyo (IACUC protocol approval 19-26).
Histopathology and immunohistochemistry.
Five- to 7-week-old female p53−/− ICR mice and littermate control p53+/+ ICR mice were infected intracranially with 100 PFU of HSV-1(F) and killed 6 days after infection, and their brains were perfused with 4% phosphate-buffered paraformaldehyde overnight at 4°C and then placed in 70% ethanol. The fixed brains were routinely embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Immunohistochemical detection of HSV-1 antigen was performed on paraffin-embedded sections as described previously (21). The sections were examined with a BZ-9000 fluorescence microscope (Keyence), and antigen-stained areas were quantitated using a BZ-II analyzer (version 2.1; Keyence).
Flow cytometry.
Five- to - week-old female p53−/− ICR mice and littermate control p53+/+ ICR mice were infected intracranially with 100 PFU of HSV-1(F) as described above. For isolation of white blood cells from the brains, at 6 days postinfection, the brains from infected mice were cut into small pieces, incubated in RPMI 1640 containing 2% fetal calf serum, 1 mg collagenase D (Wako)/ml, and 15 μg DNase I (Roche)/ml for 30 min at 37°C, filtered through a 70-μm-pore-size filter, suspended in 15 ml 30% Percoll (GE Healthcare) in RPMI 1640, and centrifuged at 7,800 × g for 30 min at room temperature (22). The myelin debris on the top was removed, and the layer containing white blood cells above the red blood cell layer was collected and washed. The total number of viable white blood cells was determined by the trypan blue exclusion test. The isolated white blood cells were stained with phycoerythrin (PE)-conjugated anti-CD45 (catalog number 30-F11; eBioscience), PE-Cy7-conjugated anti-Ly6G (catalog number 1A8; BD), allophycocyanin (APC)-Cy7-conjugated anti-CD11b (catalog number M1/70; BD), fluorescein isothiocyanate-conjugated anti-CD11c (catalog number N418; eBioscience), and APC-conjugated anti-F4/80 (catalog number BM8; eBioscience) antibodies at 4°C for 30 min. Immediately before flow cytometry analysis, 7-amino-actinomycin D (7-AAD; BD) was added to the cells, and 7-AAD-positive dead cells were excluded from analysis. Multiparameter analyses were performed with a flow cytometer (Verse; BD). The total number of neutrophils per brain was calculated by multiplying the fraction of CD45+ Ly6G+ CD11b+ cells (i.e., the number of CD45+ Ly6G+ CD11b+ cells divided by the number of viable 7-AAD-negative cells) by the total number of viable white blood cells isolated per brain.
RNA sequencing (RNA-seq).
Five- to 7-week-old female p53−/− and p53+/+ ICR mice were infected intracranially with 100 PFU of HSV-1(F) as described above. Total RNA was isolated at 6 days postinfection. Brains were homogenized in TriPure isolation reagent (Roche) using a disposable pestle system (Fisher), and total RNA was then isolated with a High Pure RNA tissue kit (Roche) according to the manufacturer's instructions. For RNA sequencing analysis, equal amounts of total RNA from either p53−/− or p53+/+ mice (n = 6) were pooled into one sample. Sequencing libraries were constructed with the SureSelect strand-specific RNA library (Agilent); 100-bp paired-end sequencing was performed using an Illumina HiSeq 2500 sequencer (Illumina) according to the manufacturer's instructions. The raw sequence reads were mapped to the mouse genome (mm9) or HSV-1 genome (GenBank accession number GU734771.1) using the program TopHat. The normalized transcription profiles were estimated on the basis of the mapping results using the program Cufflinks. The number of reads per kilobase of exon per million mapped reads (RPKM) was converted from the row read counts of each transcript using the program Cuffdiff. The RPKM of HSV-1 genes was multiplied by (total number of mapped reads of the HSV-1 genome)/(total number of mapped reads of the mouse genome).
Nucleotide sequence accession number.
RNA sequencing data have been registered in the DNA Data Bank of Japan under accession number DRA004742.
RESULTS AND DISCUSSION
To investigate the role of p53 in HSV-1 infection in the central nervous system (CNS), we infected p53−/− and p53+/+ mice with 100 PFU of HSV-1(F) intracranially. The survival of the infected mice was monitored for 14 days postinfection, and virus titers in the brains were assayed at 5 days postinfection. As shown in Fig. 1, most of the p53−/− mice survived, but the virus killed 77% of p53+/+ mice. Mortality in the p53+/+ mice was 5.0-fold higher than that in the p53−/− mice. Viral titers in the brains of p53+/+ mice were consistently and significantly higher (3.2-fold) than those in the brains of p53−/− mice (Fig. 2). We note that an approximately 3-fold difference in the level of HSV-1 replication in the brains of mice has been reported to lead to significant changes in the survival rates of mice (19, 23, 24).
FIG 1.
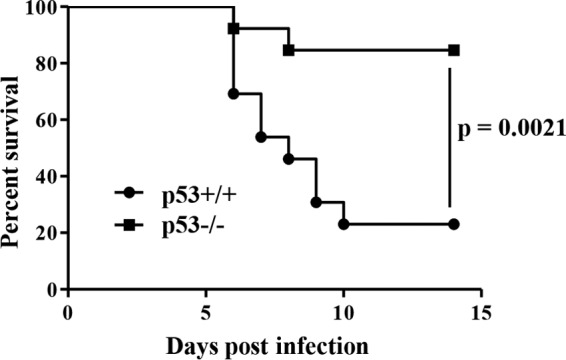
Five- to 7-week-old female p53+/+ (n = 13) and p53−/− (n = 13) ICR mice were inoculated with 100 PFU of HSV-1(F) intracranially, and survival was monitored for 14 days. Statistical significance was determined by the log-rank test.
FIG 2.
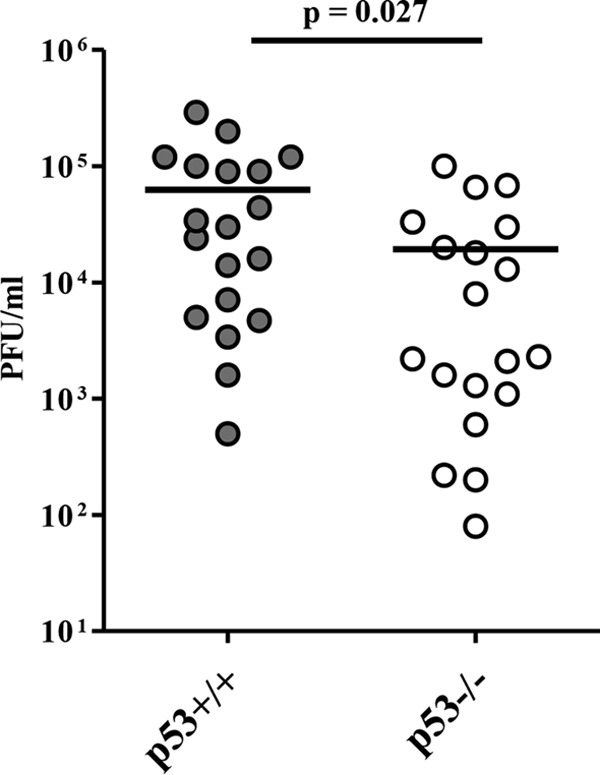
Five- to 7-week-old female p53+/+ (n = 19) and p53−/− (n = 19) ICR mice were inoculated with 100 PFU of HSV-1(F) intracranially. At 5 days postinfection, the brains of infected mice were harvested, and virus titers were assayed. Each data point is the virus titer in the brain of one mouse. The horizontal bars indicate the mean for each group. Statistical significance was determined by the two-tailed Student t test.
At 6 days postinfection we analyzed the histopathology of the brains of the p53+/+ and p53−/− mice intracranially infected with HSV-1. In agreement with the effects of p53 on viral replication and virulence in mice following intracranial infection (Fig. 1 and 2), the spread of viral antigens in the brains of p53−/− mice was apparently lower than that in the brains of p53+/+ mice (Fig. 3A, C, F, and H). The HSV antigen-positive areas in whole-brain sections of p53+/+ mice (Fig. 3A and F) were 3.8-fold larger than those in whole-brain sections of p53−/− mice. The brains of p53+/+ mice showed more edematous lesions, coincident with the spread of viral antigens, than those of p53−/− mice (Fig. 3B, D, G, and I). In addition, increased levels of neutrophil-like cells had infiltrated into sites of viral infection in p53+/+ mice relative to the levels in p53−/− mice (Fig. 3D and I). To verify this result, at 6 days postinfection we measured the recruitment of neutrophils into the brains of infected mice following intracranial inoculation. As shown in Fig. 4, the number of neutrophils (CD11b+ Ly6G+ cells) observed in the brains of p53+/+ mice was significantly higher (1.7-fold) than that observed in the brains of p53−/− mice. It has previously been reported that edematous lesions and neutrophil recruitment to mouse brain are closely associated with HSE and HSV-1 encephalitis in mice (25, 26). Furthermore, it has also been shown that wild-type mice can control murine cytomegalovirus (MCMV) brain infection but that MCMV brain infection is lethal to interleukin-10 (IL-10)-deficient mice, wherein neutrophil infiltration is increased significantly, although only about 2-fold (27). This suggests that a 2-fold difference in neutrophil infiltration in the brains of infected mice may lead to profound changes in pathogenesis. These results indicate that p53 is required for efficient viral virulence, replication, and spread and for the consequent development of viral encephalitis in the brains of mice following intracranial inoculation. This requirement for p53 for efficient viral replication in the CNS of mice is in agreement with the findings of our previous study using cell culture, as described above (9). To our knowledge, this is the first report showing that p53 plays a positive role in viral replication and pathogenesis in vivo.
FIG 3.

Histopathological features of the brains of p53+/+ and p53−/− mice following intracranial inoculation. Five- to 7-week-old female p53+/+ (n = 10) and p53−/− (n = 9) ICR mice were inoculated with 100 PFU of HSV-1(F) intracranially. At 6 days postinfection, the brains of infected mice were harvested, sectioned, and stained with hematoxylin and eosin (B, D, E, G, I, and J) or with an antibody to HSV-1 antigens (A, C, F, and H). Panels C, D, H, and I show magnified images of the regions indicated with white rectangles in panels A, B, F, and G, respectively. Panels E and J show magnified images of the uninfected regions indicated with black rectangles in panels B and G, respectively. In panels D and I, neutrophils are indicated by black arrows. Representative images are shown. Bars, 50 μm.
FIG 4.
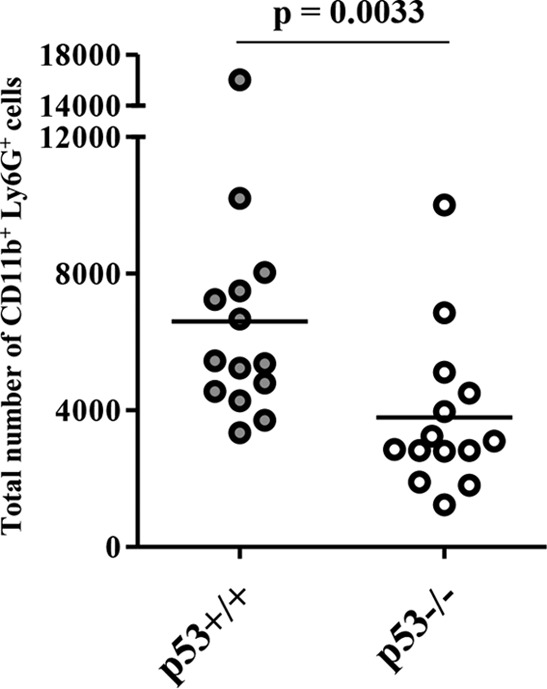
Five- to 7-week-old female p53+/+ (n = 14) and p53−/− (n = 14) ICR mice were inoculated with 100 PFU of HSV-1(F) intracranially. At 6 days postinfection, lymphocytes in the brain were harvested and CD11b+ Ly6G+ cells were quantified by flow cytometry. Each data point is the total number of CD11b+ Ly6G+ cells in the brain of one mouse. The horizontal bars indicate the mean for each group. Statistical significance was determined by the two-tailed Student t test.
Next, to investigate the effect of p53 knockout on global viral and host cellular gene expression in the brains of infected mice, we performed whole-transcriptome shotgun sequencing analysis of the brains from infected p53+/+ and p53−/− mice following intracranial infection. As shown in Table 1, p53 knockout downregulated the expression of all HSV-1 transcripts in the brains of infected mice. These results are in agreement with the results presented above showing that p53 knockout downregulated viral replication and indicate that p53 is required for global HSV-1 gene expression in the brains of mice following intracranial inoculation. Notably, although recent work has used cell culture to identify p53 response elements (p53RE) in the OriS and OriL regions of HSV-1, which appear to mediate repression of the OriL and OriS proximal genes ICP8 (UL29) and ICP4 (RS1) in a p53-dependent manner (28), p53 knockout did not increase the expression of ICP8 and ICP4 mRNAs (Table 1). In addition, p53 was previously shown to specifically upregulate expression of ICP27 mRNA early in infection (9), whereas in this study, p53 knockout decreased the expression of ICP27 (UL54) mRNA to levels similar to those of many other HSV-1 mRNAs (Table 1). These contradictions between the roles of p53 in gene expression may be due to marked differences in experimental conditions. In support of this, p53 activity has been reported to be regulated by multiple factors, and therefore, the effects of p53 may vary in different cells and tissue types and may be dependent on the expression of those factors (29–32). Furthermore, the phenotypes of p53 knockout in cell culture were investigated under relatively synchronized infection conditions at high multiplicities of infection (MOIs; 5 and 1) and/or were observed only in a specific phase of infection (9). It is possible that the phenotypes observed under such specific cell culture conditions may be difficult to detect in the brains of infected mice because the mode of infection observed in mice is very different from that observed in cell culture, and the MOIs are much lower.
TABLE 1.
HSV-1 gene expression in the brains of mice
| Gene | Normalized RPKMa |
Fold activationb | |
|---|---|---|---|
| p53+/+ mice | p53−/− mice | ||
| RL1 | 0.5 | 0.2 | 2.5 |
| RL2 | 22.0 | 3.0 | 7.3 |
| RS1 | 3.5 | 0.5 | 7.6 |
| UL1 | 72.4 | 6.7 | 10.8 |
| UL2 | 60.0 | 4.5 | 13.3 |
| UL3 | 28.9 | 2.5 | 11.6 |
| UL4 | 24.6 | 2.6 | 9.4 |
| UL5 | 19.9 | 1.7 | 11.4 |
| UL6 | 22.1 | 1.8 | 12.2 |
| UL7 | 28.5 | 3.3 | 8.7 |
| UL8 | 13.7 | 1.1 | 12.0 |
| UL9 | 20.0 | 1.8 | 11.1 |
| UL10 | 62.5 | 5.8 | 10.7 |
| UL11 | 86.1 | 7.9 | 11.0 |
| UL12 | 89.5 | 8.8 | 10.2 |
| UL13 | 35.0 | 3.9 | 9.0 |
| UL14 | 32.1 | 3.6 | 8.8 |
| UL15 | 18.8 | 2.1 | 8.9 |
| UL16 | 37.8 | 3.7 | 10.3 |
| UL17 | 12.4 | 1.3 | 9.7 |
| UL18 | 228.7 | 21.0 | 10.9 |
| UL19 | 76.4 | 6.7 | 11.4 |
| UL20 | 19.8 | 2.1 | 9.3 |
| UL21 | 28.3 | 3.1 | 9.1 |
| UL22 | 30.1 | 3.0 | 10.1 |
| UL23 | 47.7 | 3.2 | 14.7 |
| UL24 | 34.1 | 4.2 | 8.1 |
| UL25 | 36.2 | 3.0 | 12.0 |
| UL26 | 59.3 | 5.4 | 11.1 |
| UL26.5 | 16.0 | 1.0 | 15.5 |
| UL27 | 160.6 | 14.5 | 11.1 |
| UL28 | 15.4 | 1.6 | 9.9 |
| UL29 | 72.5 | 7.8 | 9.3 |
| UL30 | 35.4 | 3.3 | 10.6 |
| UL31 | 102.3 | 9.1 | 11.3 |
| UL32 | 26.4 | 2.6 | 10.3 |
| UL33 | 24.3 | 3.7 | 6.5 |
| UL34 | 158.6 | 14.0 | 11.3 |
| UL35 | 333.1 | 30.8 | 10.8 |
| UL36 | 6.6 | 0.7 | 9.8 |
| UL37 | 19.9 | 2.0 | 10.0 |
| UL38 | 17.5 | 1.6 | 11.1 |
| UL39 | 64.0 | 5.9 | 10.9 |
| UL40 | 153.1 | 14.7 | 10.4 |
| UL41 | 39.9 | 4.0 | 10.1 |
| UL42 | 139.7 | 12.7 | 11.0 |
| UL43 | 15.3 | 1.5 | 10.2 |
| UL44 | 100.7 | 9.3 | 10.8 |
| UL45 | 154.5 | 15.6 | 9.9 |
| UL47 | 32.5 | 3.1 | 10.6 |
| UL46 | 79.8 | 7.6 | 10.4 |
| UL48 | 297.6 | 29.7 | 10.0 |
| UL49 | 72.5 | 7.7 | 9.4 |
| UL49A | 41.6 | 4.0 | 10.5 |
| UL50 | 68.9 | 5.0 | 13.7 |
| UL51 | 32.6 | 3.5 | 9.3 |
| UL52 | 23.3 | 2.3 | 10.0 |
| UL53 | 40.1 | 4.7 | 8.6 |
| UL54 | 51.7 | 5.2 | 9.9 |
| UL55 | 33.4 | 3.3 | 10.0 |
| UL56 | 20.4 | 2.0 | 10.0 |
| US1 | 172.0 | 22.1 | 7.8 |
| US2 | 44.2 | 4.6 | 9.7 |
| US3 | 32.6 | 3.8 | 8.6 |
| US4 | 50.6 | 5.0 | 10.1 |
| US5 | 5.7 | 0.8 | 7.3 |
| US6 | 148.6 | 14.6 | 10.2 |
| US7 | 183.2 | 16.9 | 10.8 |
| US8 | 79.8 | 10.1 | 7.9 |
| US8A | 88.8 | 10.3 | 8.6 |
| US9 | 301.6 | 44.8 | 6.7 |
| US10 | 185.9 | 22.4 | 8.3 |
| US11 | 242.2 | 27.9 | 8.7 |
| US12 | 154.7 | 20.1 | 7.7 |
| Avg | 10.0 | ||
RPKM (the number of reads per kilobase of exon per million mapped reads) was multiplied by (total number of mapped reads of the HSV-1 genome)/(total number of mapped reads of the mouse genome).
Fold activation represents the fold increase in the level of activation in p53+/+ mice compared with the level of activation in p53−/− mice.
Many host cell factors have been reported to regulate HSV-1 replication and/or pathogenicity in the CNS, as determined using experiments performed in genetically modified mice (33–47). Notably, most of the host cell factors identified were regulators of innate and/or acquired immunity (33–37, 39–47). For instance, knockout of IPS-1, STING, TRIF, or ZDHHC impaired IFN-I responses in the brain, thereby increasing viral replication (39, 42, 43). In addition, many other host cell regulators involved in immune-related responses were shown to be protective against HSV-1 infection in the brains of mice (34–36, 41, 44, 46, 47). On the other hand, a recent report demonstrated that knockout of NLRC3, a known negative regulator of innate immune signaling induced by STING, augmented IFN-I responses, thereby decreasing HSV-1 replication and pathogenicity in the brains of mice (33). Therefore, we examined the effect of p53 knockout on the expression of reported host cell regulators of HSV-1 replication in the brains of mice using the RNA-seq data described above. As shown in Table 2, p53 knockout had no obvious effect on the expression of most of these host cell factors. However, the expression levels of gamma IFN (IFN-γ) and tumor necrosis factor alpha (TNF-α) mRNAs in p53+/+ mice were nearly 2-fold higher than those in p53−/− mice. These two host cell factors have previously been reported to be negative regulators of HSV-1 replication in the brains of mice (35, 44), casting doubt on the hypothesis that alteration of the expression of IFN-γ and TNF-α mediated by p53 knockout in mice caused the decrease in viral replication. These results suggest that p53 promotes HSV-1 replication in the brains of infected mice independently of these host cell factors. Moreover, based on the potential role of p53 in immunological responses (5, 6), one could argue that p53 is not required for efficient HSV-1 replication but just triggers an immunological response(s) to cause massive inflammation of the brains of infected mice and their consequent death. Although we cannot completely eliminate this possibility, it seems unlikely, on the basis of the observation that p53 knockout had no obvious effect on the expression of most of the immunological response genes tested and previously reported to be regulated by p53 (6) or the pro- and anti-inflammatory cytokines examined (with the exception of IL-6 and TNF-α), as shown in Table 2. The expression level of IL-6 in p53+/+ mice was more than 4-fold than higher that in p53−/− mice, and the expression level of TNF-α was nearly 2-fold higher than that in p53−/− mice (Table 2). Since IL-6 and TNF-α have previously been reported to be induced upon HSV-1 infection in the brains of mice (35, 48), it is reasonable that increased HSV-1 replication in the brains of mice results in the induction of increased amounts of these cytokines. Thus, although the host cell regulatory factors for HSV-1 infection in the CNS are gradually being defined, information on the host cell factors involved in promoting HSV-1 infection in the CNS is limited. The information included in this study will therefore make an important contribution to our understanding of the pathogenesis of HSV and offer novel therapeutic targets.
TABLE 2.
Expression profiles of host factor mRNA in the brains of mice
| Function and RefSeq accession no. | Gene name | RPKM |
Fold activationa | |
|---|---|---|---|---|
| p53+/+ mice | p53−/− mice | |||
| Regulators of HSV-1 replication within the CNS | ||||
| NM_001289591 | Tmem173 | 12.0 | 10.4 | 1.2 |
| NM_144888 | Mavs | 5.3 | 5.1 | 1.0 |
| NM_174989 | Ticam1 | 4.5 | 3.9 | 1.1 |
| NM_175160 | Zdhhc1 | 10.0 | 10.7 | 0.9 |
| NM_008361 | Il1b | 5.2 | 3.4 | 1.5 |
| NM_010508 | Ifnar1 | 10.7 | 9.5 | 1.1 |
| NM_015783 | Isg15 | 291.9 | 202.0 | 1.4 |
| NM_001205314 | Stat1 | 139.1 | 123.1 | 1.1 |
| NM_019449 | Unc93b1 | 35.7 | 32.9 | 1.1 |
| NM_010851 | Myd88 | 27.2 | 17.3 | 1.6 |
| NM_008337 | Ifnγ | 7.6 | 3.5 | 2.2 |
| NM_175547 | Nlrc3 | 0.5 | 0.5 | 1.1 |
| NM_011905 | Tlr2 | 18.6 | 12.6 | 1.5 |
| NM_013693 | Tnf | 4.2 | 2.2 | 1.9 |
| NM_009910 | Cxcr3 | 1.2 | 1.5 | 0.8 |
| p53-dependent immunological response genes | ||||
| NM_008394 | Irf9 | 44.4 | 41.1 | 1.1 |
| NM_016850 | Irf7 | 190.8 | 143.5 | 1.3 |
| NR_003520 | Mx1 | 30.3 | 16.5 | 1.8 |
| NM_172689 | Ddx58 | 28.7 | 22.2 | 1.3 |
| Pro- and anti-inflammatory cytokines | ||||
| NM_010554 | Il1a | 5.8 | 4.0 | 1.4 |
| NM_008361 | Il1b | 5.2 | 3.4 | 1.5 |
| NM_031168 | Il6 | 4.4 | 1.0 | 4.4 |
| NM_010551 | Il16 | 7.5 | 5.1 | 1.5 |
| NM_008360 | Il18 | 16.6 | 17.4 | 1.0 |
| NM_010798 | Mif | 281.2 | 274.2 | 1.0 |
| NM_010548 | Il10 | 1.6 | 1.0 | 1.6 |
| NM_011577 | Tgfb1 | 14.0 | 12.5 | 1.1 |
| NM_009367 | Tgfb2 | 6.8 | 7.0 | 1.0 |
Fold activation represents the fold increase in the level of activation in p53+/+ mice compared with the level of activation in p53−/− mice.
ACKNOWLEDGMENTS
We thank Tomoko Ando, Sachi Matsumoto, Kiyomi Imamura, Yuuta Kuze, and Terumi Horiuchi for excellent technical assistance.
This study received support from the Funding Program for Next Generation World-Leading Researchers from the Japan Society for the Promotion of Science (JSPS) to Yasushi Kawaguchi; Grants for Scientific Research from JSPS to Yasushi Kawaguchi, Yuhei Maruzuru, Akihisa Kato, and Jun Arii; Grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan to Yasushi Kawaguchi and Akihisa Kato; a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from MEXT and the Japan Agency for Medical Research and Development (AMED) to Yasushi Kawaguchi; grants from the Takeda Science Foundation to Yasushi Kawaguchi, Akihisa Kato, and Jun Arii; and a grant from the Mitsubishi Foundation to Yasushi Kawaguchi.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Menendez D, Shatz M, Resnick MA. 2013. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol 25:85–92. doi: 10.1097/CCO.0b013e32835b6386. [DOI] [PubMed] [Google Scholar]
- 3.Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato Y, Tsurumi T. 2013. Genome guardian p53 and viral infections. Rev Med Virol 23:213–220. doi: 10.1002/rmv.1738. [DOI] [PubMed] [Google Scholar]
- 5.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 6.Munoz-Fontela C, Macip S, Martinez-Sobrido L, Brown L, Ashour J, Garcia-Sastre A, Lee SW, Aaronson SA. 2008. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med 205:1929–1938. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staib C, Pesch J, Gerwig R, Gerber JK, Brehm U, Stangl A, Grummt F. 1996. p53 inhibits JC virus DNA replication in vivo and interacts with JC virus large T-antigen. Virology 219:237–246. doi: 10.1006/viro.1996.0241. [DOI] [PubMed] [Google Scholar]
- 8.Pampin M, Simonin Y, Blondel B, Percherancier Y, Chelbi-Alix MK. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J Virol 80:8582–8592. doi: 10.1128/JVI.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J Virol 87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang SS, Lo YC, Chua HH, Chiu HY, Tsai SC, Chen JY, Lo KW, Tsai CH. 2008. Critical role of p53 in histone deacetylase inhibitor-induced Epstein-Barr virus Zta expression. J Virol 82:7745–7751. doi: 10.1128/JVI.02717-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato Y, Kamura T, Shirata N, Murata T, Kudoh A, Iwahori S, Nakayama S, Isomura H, Nishiyama Y, Tsurumi T. 2009. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog 5:e1000530. doi: 10.1371/journal.ppat.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasher JM, Elenitoba-Johnson KS, Kelley LL. 2001. Loss of p53 tumor suppressor function is required for in vivo progression of Friend erythroleukemia. Oncogene 20:2946–2955. doi: 10.1038/sj.onc.1204395. [DOI] [PubMed] [Google Scholar]
- 14.Munoz-Fontela C, Pazos M, Delgado I, Murk W, Mungamuri SK, Lee SW, Garcia-Sastre A, Moran TM, Aaronson SA. 2011. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J Immunol 187:6428–6436. doi: 10.4049/jimmunol.1101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchlik E, Kalman R, Rosenberg N. 2005. Decreased virus population diversity in p53-null mice infected with weakly oncogenic Abelson virus. J Virol 79:11618–11626. doi: 10.1128/JVI.79.18.11618-11626.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokanaga T, Takeda N, Suda Y, Abe S, Matsuo I, Ikawa Y, Aizawa S. 1993. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 8:3313–3322. [PubMed] [Google Scholar]
- 17.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J Virol 85:5003–5015. doi: 10.1128/JVI.02314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Science Council of Japan. 2006. Guidelines for proper conduct of animal experiments. Science Council of Japan, Tokyo, Japan. [Google Scholar]
- 21.Koyanagi N, Imai T, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 Us3 in viral neuroinvasiveness. Microbiol Immunol 58:31–37. doi: 10.1111/1348-0421.12108. [DOI] [PubMed] [Google Scholar]
- 22.Howe CL, Lafrance-Corey RG, Sundsbak RS, Sauer BM, Lafrance SJ, Buenz EJ, Schmalstieg WF. 2012. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci Rep 2:545. doi: 10.1038/srep00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato A, Arii J, Koyanagi Y, Kawaguchi Y. 2015. Phosphorylation of herpes simplex virus 1 dUTPase regulates viral virulence and genome integrity by compensating for low cellular dUTPase activity in the central nervous system. J Virol 89:241–248. doi: 10.1128/JVI.02497-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J Virol 88:2775–2785. doi: 10.1128/JVI.03300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR. 2008. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol 181:6417–6426. doi: 10.4049/jimmunol.181.9.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundberg P, Ramakrishna C, Brown J, Tyszka JM, Hamamura M, Hinton DR, Kovats S, Nalcioglu O, Weinberg K, Openshaw H, Cantin EM. 2008. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 82:7078–7088. doi: 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mutnal MB, Cheeran MC, Hu S, Little MR, Lokensgard JR. 2010. Excess neutrophil infiltration during cytomegalovirus brain infection of interleukin-10-deficient mice. J Neuroimmunol 227:101–110. doi: 10.1016/j.jneuroim.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh JC, Kuta R, Armour CR, Boehmer PE. 2014. Identification of two novel functional p53 responsive elements in the herpes simplex virus-1 genome. Virology 460-461:45–54. doi: 10.1016/j.virol.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu GS. 2004. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther 3:156–161. doi: 10.4161/cbt.3.2.614. [DOI] [PubMed] [Google Scholar]
- 30.Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. 2016. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res 44:D313–D316. doi: 10.1093/nar/gkv1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, Sweet MJ. 2008. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res 4:5. doi: 10.1186/1745-7580-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckerman R, Prives C. 2010. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, Ouyang S, Liu ZJ, Damania B, Shu HB, Duncan JA, Ting JP. 2014. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 40:329–341. doi: 10.1016/j.immuni.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA. 2012. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J Virol 86:2273–2281. doi: 10.1128/JVI.06010-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sergerie Y, Rivest S, Boivin G. 2007. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis 196:853–860. doi: 10.1086/520094. [DOI] [PubMed] [Google Scholar]
- 36.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HW IV. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A 104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhela S, Mulik S, Reddy PB, Richardson RL, Gimenez F, Rajasagi NK, Veiga-Parga T, Osmand AP, Rouse BT. 2014. Critical role of microRNA-155 in herpes simplex encephalitis. J Immunol 192:2734–2743. doi: 10.4049/jimmunol.1302326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, Ye W, Xiong X, Zhong B, Shu HB, Wang YY. 2014. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe 16:450–461. doi: 10.1016/j.chom.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Wickham S, Lu B, Ash J, Carr DJ. 2005. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol 162:51–59. doi: 10.1016/j.jneuroim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Pasieka TJ, Collins L, O'Connor MA, Chen Y, Parker ZM, Berwin BL, Piwnica-Worms DR, Leib DA. 2011. Bioluminescent imaging reveals divergent viral pathogenesis in two strains of Stat1-deficient mice, and in αβγ interferon receptor-deficient mice. PLoS One 6:e24018. doi: 10.1371/journal.pone.0024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker ZM, Murphy AA, Leib DA. 2015. Role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with herpes simplex virus 1. J Virol 89:11080–11091. doi: 10.1128/JVI.00954-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menasria R, Boivin N, Lebel M, Piret J, Gosselin J, Boivin G. 2013. Both TRIF and IPS-1 adaptor proteins contribute to the cerebral innate immune response against herpes simplex virus 1 infection. J Virol 87:7301–7308. doi: 10.1128/JVI.00591-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansur DS, Kroon EG, Nogueira ML, Arantes RM, Rodrigues SC, Akira S, Gazzinelli RT, Campos MA. 2005. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol 166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kroll CM, Zheng M, Carr DJ. 2014. Enhanced resistance of CXCR3 deficient mice to ocular HSV-1 infection is due to control of replication in the brain ependyma. J Neuroimmunol 276:219–223. doi: 10.1016/j.jneuroim.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caignard G, Leiva-Torres GA, Leney-Greene M, Charbonneau B, Dumaine A, Fodil-Cornu N, Pyzik M, Cingolani P, Schwartzentruber J, Dupaul-Chicoine J, Guo H, Saleh M, Veillette A, Lathrop M, Blanchette M, Majewski J, Pearson A, Vidal SM. 2013. Genome-wide mouse mutagenesis reveals CD45-mediated T cell function as critical in protective immunity to HSV-1. PLoS Pathog 9:e1003637. doi: 10.1371/journal.ppat.1003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beland JL, Sobel RA, Adler H, Del-Pan NC, Rimm IJ. 1999. B cell-deficient mice have increased susceptibility to HSV-1 encephalomyelitis and mortality. J Neuroimmunol 94:122–126. doi: 10.1016/S0165-5728(98)00238-0. [DOI] [PubMed] [Google Scholar]
- 48.Dvorak F, Martinez-Torres F, Sellner J, Haas J, Schellinger PD, Schwaninger M, Meyding-Lamade UK. 2004. Experimental herpes simplex virus encephalitis: a long-term study of interleukin-6 expression in mouse brain tissue. Neurosci Lett 367:289–292. doi: 10.1016/j.neulet.2004.06.010. [DOI] [PubMed] [Google Scholar]