

ABSTRACT
Interferons (IFNs) restrict various kinds of viral infection via induction of hundreds of IFN-stimulated genes (ISGs), while the functions of the majority of ISGs are broadly unclear. Here, we show that a high-IFN-inducible gene, ISG12a (also known as IFI27), exhibits a nonapoptotic antiviral effect on hepatitis C virus (HCV) infection. Viral NS5A protein is targeted specifically by ISG12a, which mediates NS5A degradation via a ubiquitination-dependent proteasomal pathway. K374R mutation in NS5A domain III abrogates ISG12a-induced ubiquitination and degradation of NS5A. S-phase kinase-associated protein 2 (SKP2) is identified as an ubiquitin E3 ligase for NS5A. ISG12a functions as a crucial adaptor that promotes SKP2 to interact with and degrade viral protein. Moreover, the antiviral effect of ISG12a is dependent on the E3 ligase activity of SKP2. These findings uncover an intriguing mechanism by which ISG12a restricts viral infection and provide clues for understanding the actions of innate immunity.
IMPORTANCE Upon virus invasion, IFNs induce numerous ISGs to control viral spread, while the functions of the majority of ISGs are broadly unclear. The present study shows a novel antiviral mechanism of ISGs and elucidated that ISG12a recruits an E3 ligase, SKP2, for ubiquitination and degradation of viral protein and restricts viral infection. These findings provide important insights into exploring the working principles of innate immunity.
INTRODUCTION
Hepatitis C virus (HCV) presents a considerable threat to global health, with approximately 185 million people infected worldwide (1). Infection with HCV holds a high risk of causing chronic hepatitis, cirrhosis, and hepatocellular carcinoma, resulting in 350,000 human deaths each year. Although current developed direct-acting antivirals (DAAs) are capable of effectively eradicating HCV infection from patients, several hurdles remain, including high costs, emergence of drug resistance, and side effects (2). Treatment with DAAs in a portion of HCV patients suffering from advanced liver disease does not eliminate the risk of hepatocellular carcinoma. Moreover, reinfection of the cured individual is possible due to the lack of a prophylactic vaccine (3).
HCV is an enveloped positive-stranded RNA virus that belongs to the Flaviviridae family. The genome of HCV encodes a single long polyprotein precursor of about 3,000 amino acids, which is processed by cellular and viral proteases into three structural proteins, including the core, E1, and E2 proteins, and seven nonstructural proteins, including p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B. The propagation of HCV necessitates the participation of both viral proteins and host factors. Particularly, NS5A protein as a multifunctional protein is indispensable for HCV replication and virus production (4–7).
After viral infection of host cells, viral RNAs and/or DNAs are sensed as nonself by pattern recognition receptors (PRRs), resulting in activation of antiviral innate immune response to restrict virus spread (8). Activation of PRR signaling pathways leads to the production of numerous cytokines, including interferons (IFNs), interleukins, and chemokines. IFNs, including types I, II, and III, conduct functions via binding to their specific receptors, which activates the JAK-STAT pathway and induces hundreds of interferon-stimulated genes (ISGs) (9). ISGs encoding innate immune effectors are believed to inhibit almost every step of the virus life cycle (10). Lots of antiviral ISGs have been selected among the known ISG pools using different approaches, including RNA interference-based screens and overexpression screening (11–14). However, the mechanisms by which the majority of ISGs restrict virus infection are broadly unclear.
ISG12a (interferon-stimulated gene 12a), also known as IFI27 (interferon alpha-inducible protein 27), belongs to the IFI6/IFI27 family, which contains a conserved ∼80-amino-acid motif, termed the ISG12 motif (15, 16). Humans have four members of IFI6/IFI27 family, including the IFI6, ISG12a, ISG12b, and ISG12c genes; whereas, mice have only three family members, including the ISG12a, ISG12b1, and ISG12b2 genes, and do not contain an IFI6 gene ortholog (16, 17). Interestingly, the human ISG12a gene is able to be highly induced by type I IFN, while the ISG12b and ISG12c genes are not IFN inducible (16, 18). Human ISG12a and murine Ifi27 are identified as transmembrane proteins, while the exact localization of these proteins remains controversial (19–21).
Our previous studies demonstrated that ISG12a is involved in cell apoptosis triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) treatment, and Newcastle disease virus (NDV) or HCV infection (22, 23). MiR-942 targeting ISG12a is capable of regulating the TRAIL- and HCV-induced apoptosis (23, 24). More importantly, ISG12a can be markedly induced by HCV infection and mediates apoptosis of HCV-infected cells at the late stage of viral infection (6 to 9 days postinfection) in HLCZ01 cells, a novel hepatoma cell line established recently in our laboratory, which presents an innate immune mechanism for restriction of HCV propagation (24, 25). In addition, murine Ifi27 also exhibits antiviral activities on West Nile virus (WNV) and murine hepatitis virus (MHV) with unknown mechanisms (26, 27). However, the direct antiviral functions for ISG12a to restrict HCV infection, especially at the early stage of viral infection before triggering cell apoptosis, remain to be elucidated.
Ubiquitination has been known to regulate various crucial cellular events, such as cell cycle and division, DNA repair, apoptosis, and immune response, by the induction of proteasomal degradation of target proteins (28). However, whether the processes of ubiquitination are involved in the antiviral effects of the IFI6/IFI27 family members is unknown. In the present study, we report that ectopic expression of ISG12a remarkably inhibits HCV replication in both HCV infectious culture systems and replicon cells. Viral NS5A is targeted specifically by ISG12a that mediates NS5A degradation in a ubiquitination-dependent manner. K374R mutation in NS5A domain III abrogates ISG12a-induced ubiquitination and degradation of NS5A. Furthermore, ISG12a functions as a crucial adaptor that promotes an E3 ligase S-phase kinase-associated protein 2 (SKP2) to interact with and degrade viral protein. The antiviral activity of ISG12a is dependent on the E3 ligase domain of SKP2. These findings uncover an intriguing mechanism by which ISG12a restricts viral infection and provide clues for understanding the actions of innate immunity.
MATERIALS AND METHODS
Cell culture and reagents.
Huh7.5 cells and FL-neo, an Huh7-based HCV 1b full-length replicon cell line, were kindly provided by Charles M. Rice (Rockefeller University, New York, NY). The HLCZ01 cell line was established in our lab (25). HEK293T cells were purchased from Boster. HLCZ01 cells were cultured in collagen-coated tissue culture plates and cultured with Dulbecco's modified Eagle's medium (DMEM)/F-12 medium supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Gibco), 40 ng/ml of dexamethasone (Sigma), insulin-transferrin-selenium (ITS) (Lonza), penicillin, and streptomycin. Other cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, l-glutamine, nonessential amino acids, penicillin, and streptomycin. The plasmid pRL-HL, a gift from Stanley M. Lemon, is a bicistronic expression construct encoding Renilla and firefly luciferase cDNAs translated from the 5′ cap and internally from the HCV internal ribosome entry site (IRES), respectively. Cells in 12-well plates were transfected with 500 ng of pRL-HL plasmid by Lipofectamine 2000. After 24 h of transfection, the cells were harvested to assay the activities of firefly and Renilla luciferase by the Promega dual-luciferase reporter assay system (Promega).
Plasmid construction.
The short hairpin RNAs (shRNAs) targeting ISG12a and SKP2 were inserted into the pSilencer-neo plasmid (Ambion). The target sequence of ISG12a shRNA was 5′-AAGTTCATCCTGGGCTCCATT-3′. The target sequence of SKP2 shRNA was 5′-AAGTTGCAGAATCTAAGCCTG-3′. ISG12a and SKP2 cDNAs were synthesized from total cellular RNA isolated from HLCZ01 cells by standard reverse transcription-PCR (RT-PCR) and were subsequently cloned into the pcDNA3.1a vector and/or p3×FLAG-CMV vector. Multiple domains of ISG12a and SKP2 were amplified from the templates of full-length ISG12a and SKP2, respectively, and were cloned into the p3×FLAG-CMV or pcDNA3.1a vector. The primers for amplification of the genes or domains described above are shown in Table 1 and Table 2. The viral genes and regions, including the genes coding for the core, NS2, NS5A, and NS5B proteins and the different domains of NS5A, were amplified from pJFH1 and subsequently cloned into the pcDNA3.1a vector and/or p3×FLAG-CMV vector. The primers for amplification of viral genes or regions are shown in Table 3. An NS5A expression construct with the K374R mutation was generated by the QuikChange site-directed mutagenesis kits from the template of p3×FLAG-CMV-NS5A plasmid (Stratagene). The pHA-Ub (K48) plasmid was kindly provided by Zhengfan Jiang (Peking University, Beijing, China).
TABLE 1.
Primers for amplification of SKP2 domains
| Domain and primer orientation | Primer sequence |
|---|---|
| pcDNA3.1a-SKP2-N | |
| Forward | GGAATTCATGCACGTATTTAAAACTCC |
| Reverse | CCCAAGCTTCTTGGAACACTGAGACAG |
| pcDNA3.1a-SKP2-(N+L) | |
| Forward | GGAATTCATGCACGTATTTAAAACTCC |
| Reverse | CCCAAGCTTGCAATTAATCTGTAGATGAG |
| pcDNA3.1a-SKP2-DL | |
| Forward | CCGCTCGAGATGCACGTATTTAAAACTCC |
| Reverse | GGAATTCTCTTGGAACACTGAGACAGTATG |
| Forward | GGAATTCTCCCATTTCACCACCATTGC |
| Reverse | CCCAAGCTTTCATAGACAACTGGGCTTTTG |
| pcDNA3.1a-SKP2 | |
| Forward | GGAATTCATGCACGTATTTAAAACTCC |
| Reverse | CCCAAGCTTTCATAGACAACTGGGCTTTTG |
TABLE 2.
Primers for amplification of ISG12a domains
| Domain and primer orientation | Primer sequence |
|---|---|
| p3×Flag-ISG12a-DII | |
| Forward | GGGGTACCATGGTGATTGGAGGAGTTGTGGC |
| Reverse | GCTCTAGAGTAGAACCTCGCAATGACAG |
| p3×Flag-ISG12a-DII-N | |
| Forward | GGGGTACCATGGTGATTGGAGGAGTTGTGGC |
| Reverse | GCTCTAGAAACTCCACCCCCATTGGCAA |
| p3×Flag -ISG12a-DII-C | |
| Forward | GGGGTACCATGGGTGGAGTTGCCTCGGGC |
| Reverse | GCTCTAGAGTAGAACCTCGCAATGACAG |
| p3×Flag-ISG12a-DII-(106–327) | |
| Forward | GGGGTACCATGGTGATTGGAGGAGTTGTGGC |
| Reverse | GCTCTAGAAGACCCAATGGAGCCCAGGA |
| p3×Flag-ISG12a-DII-(106–297) | |
| Forward | GGGGTACCATGGTGATTGGAGGAGTTGTGGC |
| Reverse | GCTCTAGACAATCCGGAGAGTCCAGTTG |
| p3×Flag-ISG12a-DII-(106–267) | |
| Forward | GGGGTACCATGGTGATTGGAGGAGTTGTGGC |
| Reverse | GCTCTAGACTGCAGAGTAGCCACAAGGC |
| pcDNA3.1a-ISG12a | |
| Forward | GGGGTACCATGGAGGCCT CTGCTCTCAC |
| Reverse | GCTCTAGAGTAGAACCTCGCAATGACAG |
TABLE 3.
Primers for amplification of fragments of HCV
| Fragment and primer orientation | Primer sequence |
|---|---|
| p3×Flag-core | |
| Forward | GGAATTCATGAGCACCAAATCCTAAACC |
| Reverse | CGGGATCCAGCAGAGACCGGAACGGTGA |
| pcDNA3.1a-NS2 | |
| Forward | CCCAAGCTTATGTATGACGCACCTGTGCACGG |
| Reverse | GCTCTAGAAAGGAGCTTCCACCCCTTGG |
| p3×Flag-NS5A | |
| Forward | CGGAATTCATGTCCGGATCCTGGCTCCGCG |
| Reverse | GCTCTAGAGCAGCACACGGTGGTATCGT |
| p3×Flag-NS5B | |
| Forward | GGGGTACCGCCACCATGTCCATGTCATACTCCTGGA |
| Reverse | GCTCTAGACCGAGCGGGGAGTAGGAAGAGG |
| pcDNA3.1a-NS5A | |
| Forward | CGCGCGGTACCATGTCCGGATCCTGGCT |
| Reverse | CTGCAGTATAGACTGCAGCACACGTGGTAT |
| pcDNA3.1a-NS5A-DI | |
| Forward | CGCGCGGTACCATGTCCGGATCCTGGCT |
| Reverse | CTGCAGTCTAGACTAGTCTCCGCCGTGAT |
| pcDNA3.1a-NS5A-DII | |
| Forward | CGCGCGGTACCATGGACGTGGACATGGT |
| Reverse | CTGCAGTCTAGACTTGTCCGGCGTCTCCTT |
| pcDNA3.1a-NS5A-DI-DII | |
| Forward | CGCGCGGTACCATGTCCGGATCCTGGCT |
| Reverse | CTGCAGTCTAGACTTGTCCGGCGTCTCCTT |
| p3×Flag-NS5A-DIII | |
| Forward | GGGGTACCATGGTGGGTCTGAGCGAGAGCAC |
| Reverse | GCTCTAGAGGGGGGGAGAGCACAACCAG |
| p3×Flag-NS5A-DII-DIII | |
| Forward | GGGGTACCATGGACGTGGACATGT CGATGC |
| Reverse | GCTCTAGAGGGGGGGAGAGCACAACCAG |
HCV RNA and virus generation.
The pJFH1 and pJFH1/GND plasmids were gifts from Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan). The linearized DNAs from the pJFH1 and pJFH1/GND plasmids were purified and used as the templates for in vitro transcription by the MEGAscript kit (Ambion, Austin, TX). In vitro-transcribed genomic JFH1 or JFH1/GND RNA was delivered into Huh7.5 cells by electroporation. The transfected cells were transferred to complete DMEM and cultured for the indicated periods. Cells were passaged every 3 to 5 days, and the corresponding supernatants were collected and filtered with a 0.45-μm-pore filter device. The viral titers are presented as focus-forming units (FFU) per milliliter, determined by the average number of NS5A-positive foci detected in Huh7.5 cells.
Antibodies.
Monoclonal antibodies against β-actin, ISG12a, Flag tag and hemagglutinin (HA) tag were obtained from Sigma. V5 tag monoclonal antibodies were purchased from Invitrogen. Monoclonal antibodies against SKP2 were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-NS5A and anti-NS3/4A antibodies were gifts from Chen Liu (University of Florida, Gainesville, FL). Mouse monoclonal anti-NS2 antibody was kindly provided by Charles M. Rice. Goat anti-mouse and goat anti-rabbit IgG-horseradish peroxidase (HRP) secondary antibodies were purchased from Santa Cruz Biotechnology.
Real-time PCR assay.
Total cellular RNA was extracted by TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The Superscript III first-strand synthesis kit for reverse transcription of RNA was purchased from Invitrogen. After RQ1 DNase (Promega) treatment, the extracted RNA was used as the template for reverse transcription-PCR. The real-time PCR was performed as described previously (29). For absolute quantification analysis of HCV RNA, the standard curve was established by in vitro-transcribed JFH1 RNA. The primers for detection of HCV genomic RNA were described previously (25).
Western blotting.
The Western blotting procedure was reported previously (25). Briefly, cells were washed with phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris-HCl [pH 8.0] supplemented with 2 μg/ml aprotinin, 2 μg/ml leupeptin, 20 μg/ml phenylmethylsulfonyl fluoride, and 2 mM dithiothreitol [DTT]). Forty micrograms of protein was resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane, and probed with the appropriate primary and secondary antibodies. The bound antibodies were detected by Supersignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Luciferase assay.
Luciferase assays were performed with a luciferase assay kit (Promega) according to the manufacturer's instructions. The luciferase activity in cells was normalized to protein concentrations determined by Bradford assays.
IP and immunoblotting.
The cells were washed three times with ice-cold PBS and lysed in lysis buffer supplemented with protease inhibitor cocktail. The cell lysates were incubated at 4°C for 30 min and centrifuged at 12,000 × g at 4°C for 15 min. The lysates were diluted to the concentration of 2 μg/μl total cell protein with PBS before immunoprecipitation (IP). Two hundred micrograms of lysates was immunoprecipitated with the indicated antibodies. The immunocomplex was captured by adding protein G agarose bead slurry. The protein binding to the beads was boiled in 2× Laemmli sample buffer and then subjected to 10% SDS-PAGE. The protocol for immunoblotting was described above.
Bioinformatics analysis of protein-protein interaction.
In order to find the potential interacted protein of ISG12a, a biological database STRING was used to predict the interactions between ISG12a and other proteins (http://string-db.org). The top 50 output targets containing SKP2 were obtained, and functional enrichment analysis was conducted.
Statistical analysis.
All results are presented as means ± standard deviations. Comparisons between two groups were performed using Student's t test, as indicated by asterisks in some of the figures: *, P < 0.05, **, P < 0.01, and ***, P < 0.001, versus control.
RESULTS
ISG12a exhibits a nonapoptotic antiviral effect.
The proapoptotic role of ISG12a in damaging virus-infected cells for the control of viral spread was identified in our previous studies (22, 24). However, the possible nonapoptotic antiviral effect of ISG12a has not been verified yet. To avoid the potential apoptosis of cells, we conducted ectopic expression of ISG12a for up to 72 h. FL-neo cells, containing an HCV 1b full-length replicon, were transfected with pcDNA3.1a-ISG12a or pSilencer-ISG12a followed by the detection of viral protein. Ectopic expression or silencing of ISG12a in FL-neo cells significantly reduced or increased the level of NS5A protein, respectively, in a dose-dependent manner (Fig. 1A and B). Similarly, knockdown of ISG12a in HCV-infected HLCZ01 cells remarkably augmented the amount of NS5A protein compared to the control (Fig. 1C). Apart from NS5A, other viral proteins, including NS2 and NS3/4A also showed decreased or augmented expression in HCV-infected cells when we overexpressed or silenced ISG12a, respectively (Fig. 1D). Consistent with the results obtained from viral protein, silencing of ISG12a increased the level of HCV genomic RNA and overexpression of ISG12a reduced viral RNA abundance (Fig. 1E). To exclude the possibility of cell apoptosis or cell death, we measured the viability of cells treated as described above. The data demonstrated that ectopic expression (Fig. 1F) and silencing (Fig. 1G) of ISG12a, with or without HCV infection, did not trigger cell death in Huh7.5 and HLCZ01 cells. Taken together, these results indicated that ISG12a has a direct antiviral effect of restricting HCV infection.
FIG 1.

ISG12a exhibits a nonapoptotic antiviral effect. (A and B) Western blotting of lysates from FL-neo cells transfected with pcDNA3.1a-V5/His-ISG12a (upper) or pSilencer-ISG12a (lower) plasmid for a gradient dose (48 h) (A) or at different time points (B). NS5A was measured by anti-NS5A antibody. Exogenous ISG12a and endogenous ISG12a were detected with anti-V5 monoclonal antibody and anti-ISG12a polyclonal antibody, respectively. (C) HLCZ01 cells were infected by HCV (multiplicity of infection [MOI], 0.1) for 24 h followed by the transfection of pSilencer-ISG12a plasmid for 48 h. NS5A and ISG12a were detected by Western blotting. (D and E) Huh7.5 cells were infected by HCV (MOI, 0.1) for 24 h followed by the transfection of pcDNA3.1a-V5/His-ISG12a or pSilencer-ISG12a plasmid for 48 h. (D) HCV NS2, NS3/4A and NS5A proteins were detected by Western blotting. (E) HCV RNA was analyzed by real-time PCR. Error bars represented standard deviations (SD) from triplicate experiments. *, P < 0.05. (F and G) HLCZ01 and Huh7.5 cells were infected with or without HCV (MOI, 0.1) for 24 h followed by the transfection of pcDNA3.1a-V5/His-ISG12a (F) or pSilencer-ISG12a (G) plasmid for 48 h. The MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium assay] was performed, and error bars represent SD from triplicate experiments. V5 was the tag used.
ISG12a triggers the reduction of exogenous NS5A protein.
The reduction of viral proteins by ISG12a expression may be caused by the inhibition of viral RNA translation. To assess this possibility, we used a pRL-HL plasmid that contains HCV bicistronic RNAs. In the pRL-HL system, the expression of firefly and Renilla luciferase genes is driven by the HCV IRES and cap element, respectively, which allows the assay of HCV RNA translation. However, we did not observe the significant change of viral translation when we cotransfected pRL-HL plasmid with various doses of pcDNA3.1a-ISG12a (Fig. 2A) or pSilencer-ISG12a (Fig. 2B) plasmid in Huh7.5 cells and nonhepatic CHO cells. These data suggested that ISG12a is probably capable of regulating posttranslation of viral proteins.
FIG 2.

ISG12a triggers the reduction of exogenous NS5A protein. (A and B) Huh7.5 and CHO cells were cotransfected with pRL-HL plasmid and the different doses of pcDNA3.1a-V5/His-ISG12a (A) or pSilencer-ISG12a (B) plasmids for 24 h. Cells were harvested for the luciferase assay. Error bars represent SD from triplicate experiments. (C) Western blotting of lysates from HEK293T cells cotransfected with the indicated plasmids for 48 h. Exogenous core, NS2, NS3/4A, NS4B, NS5A, and NS5B proteins were detected with anti-V5 or anti-Flag antibody. V5 and Flag were the tags used.
To test the regulatory effect of ISG12a on viral proteins, we cotransfected pcDNA3.1a-ISG12a plasmid and multiple constructs designed for exogenous expression of viral proteins in HEK293T cells. Strikingly, the level of exogenous NS5A protein, not other viral proteins, including core, NS2, NS3/4A, NS4B, and NS5B, was markedly decreased in the ISG12a-expressed group compared to the control group (Fig. 2C). The data indicated the specific inhibitory regulation of the posttranslational process in NS5A protein by ISG12a.
ISG12a induces the proteasome-dependent degradation of NS5A.
Based on the above finding that ISG12a can reduce the level of exogenous NS5A protein, we speculated that ISG12a may trigger NS5A degradation via the proteasomal pathway. To verify this hypothesis, we treated the cells with proteasome inhibitor MG132 following the overexpression of ISG12a. The decrease of exogenous NS5A protein caused by ISG12a was reversed by the treatment with MG132 (Fig. 3A). Furthermore, MG132 treatment also abrogated the inhibitory effect of ISG12a on endogenous NS5A protein in FL-neo cells (Fig. 3B) and HCV-infected Huh7.5 cells (Fig. 3C). On the other hand, silencing of ISG12a increased the level of NS5A protein, which was not further affected by MG132 treatment (Fig. 3D). These results indicated that ISG12a plays a pivotal role in NS5A degradation.
FIG 3.

ISG12a induces the proteasome-dependent degradation of NS5A. (A) Western blotting of lysates from HEK293T cells cotransfected with the indicated plasmids for 18 h followed by the treatment with MG132 (25 μM) for 6 h. (B and C) FL-neo (B) and HCV-infected Huh7.5 (C) cells were transfected with pcDNA3.1a-V5/His-ISG12a or pcDNA3.1a-V5/His vector for 36 h and then treated with MG132 (25 μM) for 6 h. NS5A protein was detected by Western blotting. (D) FL-neo cells were transfected with pSilencer-ISG12a or pSilencer vector for 36 h and then treated with MG132 (25 μM) for 6 h. NS5A protein was detected by Western blotting. (E) Ubiquitination assay and Western blotting of lysates from HEK293T cells cotransfected with the indicated plasmids for 48 h. MG132 (25 μM) was added to cells for 6 h prior to harvesting of cell lysates, and IP experiments were conducted with anti-NS5A antibody followed by the ubiquitination assay of NS5A protein. (F) Cells were treated as described for panel E, and pHA-Ub (K48) plasmid was used in the ubiquitination assay of NS5A protein. (G) HLCZ01 cells were infected with or without HCV (MOI, 0.1) for the indicated time points. The ubiquitination assay was conducted as described for panel E. (H) HLCZ01 cells were infected by HCV (MOI, 0.1) for 24 h followed by the transfection of pSilencer-ISG12a or pSilencer vector plasmid for 48 h. The ubiquitination assay was conducted as described for panel (E). V5, Flag, and HA were the tags used.
In the next experiments, we observed apparent polyubiquitin chains conjugated in NS5A protein with ISG12a overexpression (Fig. 3E), which may contribute to NS5A degradation. The K48-dependent ubiquitination is a classical regulatory process for protein degradation. To explore whether ISG12a-triggered ubiquitination of NS5A belongs to a K48-mediated event, we used a mutant ubiquitin-expressed construct that only retains a K48 site with mutation of other lysine residues. The result showed that overexpression of ISG12a induced remarkable K48-linked ubiquitination of NS5A (Fig. 3F). During HCV infection, upregulation of ISG12a correlates with the increase of NS5A ubiquitination in a time-dependent manner (Fig. 3G), while the process of ubiquitination was attenuated by the knockdown of ISG12a (Fig. 3H). Collectively, these data demonstrated that ISG12a induces NS5A degradation via a ubiquitination-mediated proteasomal pathway.
ISG12a interacts with NS5A.
The result that ISG12a induces the ubiquitination of NS5A protein prompted us to assess the possibility of interaction between ISG12a and NS5A. We coexpressed ISG12a and NS5A, as well as ISG12a and other viral proteins, in HEK293T cells followed by IP analyses of the interactions between ISG12a and the individual viral protein. Consistent with our assumption, ISG12a indeed interacted with NS5A protein and had no binding affinity with other viral proteins, such as core, NS2, and NS5B (Fig. 4A). The data indicated the specific targeting property of ISG12a on viral protein.
FIG 4.

ISG12a interacts with NS5A. (A) IP analysis of ISG12a and viral protein from HEK293T cells expressing exogenous ISG12a and individual viral proteins, including NS5A, core, NS2, and NS5B. (B) Schematic representation of the domains of NS5A. (C) IP analysis of ISG12a and various domains of NS5A from HEK293T cells expressing exogenous ISG12a and different domains of NS5A, including NS5A DI, DII, DI-DII, DIII, and DII-DIII. (D) Schematic representation of the domains of ISG12a. (E) IP analysis of NS5A and various domains of ISG12a from HEK293T cells expressing exogenous NS5A and different domains of ISG12a, including ISG12a DII, DII-N, and DII-C. (F) Western blotting of lysates from HEK293T cells expressing exogenous ISG12a and different domains of NS5A for 48 h. (G) Ubiquitination assay and Western blotting of lysates from HEK293T cells cotransfected with the indicated plasmids for 48 h. MG132 (25 μM) was added to cells for 6 h prior to harvesting of cell lysates, and IP experiments were conducted with anti-Flag antibody followed by the ubiquitination assay of NS5A DIII protein. (H) Cells were treated as described for panel G, followed by the ubiquitination assay of wild-type or K374R mutant NS5A protein. V5, Flag, and HA were the tags used.
Next, we want to know which pivotal domains of ISG12a and NS5A are necessary or sufficient for their interaction. The major functional domains of NS5A are illustrated in Fig. 4B. We performed IP analyses with full-length ISG12a and various domains of NS5A. The results showed that domain III (DIII) and domain II to III (DII to DIII) of NS5A were both capable of interacting with ISG12a (Fig. 4C, right), while NS5A DI, DII, and DI-DII did not interact with ISG12a (Fig. 4C, left). Thus, we held that DIII of NS5A is the critical domain for binding to ISG12a. On the other hand, we divided the ISG12a protein into several truncations (Fig. 4D) and assessed the associations between these truncations of ISG12a and full-length NS5A protein. We did not test the domain I of ISG12a because this domain was unstable in the cells. Among other domains of ISG12a analyzed in the assay, domain II (DII) and the C-terminal domain of DII (DII-C) of ISG12a were identified to interact with NS5A, which suggested that DII-C is the key domain for ISG12a to target NS5A (Fig. 4E).
Our above findings revealed that ISG12a binds to NS5A DIII, which propelled us to test the degraded effect of ISG12a on NS5A DIII and other domains. Consistent with the properties of interactions, ISG12a did not induce the degradation of NS5A DII and DI-DII (Fig. 4F, upper) but triggered the remarkable degradation of NS5A DIII and DII-DIII (Fig. 4F, lower). In addition, ISG12a was also capable of inducing the ubiquitination of NS5A DIII (Fig. 4G). Through the analysis of distribution of lysine residues within NS5A DIII, we found only one lysine residue, K374. Ectopic expression of ISG12a did not trigger the ubiquitination of NS5A with K374R mutation, suggesting K374 within NS5A protein is a key lysine residue for ISG12a-induced degradation (Fig. 4H). These data indicated that domain III of NS5A not only contains the essential regions for ISG12a binding but also includes the functional sites, at least including the ubiquitin-conjugated K374 site, for ISG12a-mediated degradation.
The C-terminal portion of ISG12a domain II is critical for NS5A degradation.
To explore the pivotal functional region of ISG12a for degrading viral protein, we overexpressed various domains of ISG12a in FL-neo cells and assessed their inhibitory effects on viral protein. First, we found that ISG12a DII was able to degrade the NS5A protein in a dose-dependent manner (Fig. 5A). When we treated the cells with MG132, the inhibitory effect of ISG12a DII on viral protein was reversed (Fig. 5B). Moreover, ISG12a DII-C, not DII-N (the N terminal of DII), was responsible for the degradation of NS5A (Fig. 5C) and the subsequent suppression of viral replication and virus production (Fig. 5D and E). To further investigate the restricted functional region within ISG12a DII-C, we deleted the partial fragments of the C-terminal from full-length DII of ISG12a (Fig. 5F). After cotransfection of the different deletion constructs of ISG12a DII and p3×FLAG-NS5A plasmid in HEK293T cells, we found that the level of NS5A protein was reduced in the treatment of DII (amino acids [aa] 36 to 109) [DII(36–109)] and was not affected by expression of DII(36–99) and DII(36–89) (Fig. 5G). Similar results were obtained from FL-neo cells transfected with these ISG12a DII deletion constructs (Fig. 5H). These findings suggested a key region covering 10 amino acids (aa 100 to 109) with the restricted function of ISG12a. To confirm the importance of this region, we established an expression construct of ISG12a that only deletes the fragment from aa 100 to 109. Gradient transfection of this construct did not affect the amount of NS5A protein in FL-neo cells (Fig. 5I). Taken together, these results demonstrated that the DII-C of ISG12a is sufficient for triggering NS5A degradation and the fragment from aa 100 to 109 in ISG12a DII-C is required for the degraded function of ISG12a.
FIG 5.

The C-terminal domain of ISG12a domain II is critical for NS5A degradation. (A) FL-neo cells were transfected with different dose of p3×FLAG-ISG12a-DII plasmid for 48 h. NS5A was detected by Western blotting. (B) FL-neo cells were transfected with p3×FLAG-ISG12a-DII plasmid for 36 h, followed by treatment with MG132 (25 μM) for 6 h. NS5A was detected by Western blotting. (C) FL-neo cells were transfected with the indicated plasmids for 48 h. NS5A protein was detected by Western blotting. (D and E) Huh7.5 cells were infected with HCV for 6 h and then transfected with the indicated plasmids for 48 h. Intracellular HCV RNA (D) and extracellular infectious virus particles (E) were detected by real-time PCR and FFU assay, respectively. Error bars represent SD from triplicate experiments. *, P < 0.05. (F) Schematic representation of the truncations of ISG12a-DII-C. (G to I) Western blotting of lysates from HEK293T cells expressing exogenous NS5A and ISG12a domains (G) and FL-neo cells expressing exogenous ISG12a domains (H and I) for 48 h. Flag was the tag used.
SKP2 is an E3 ligase for NS5A.
ISG12a can interact with NS5A and promote the ubiquitination and degradation of NS5A via the proteasome-dependent pathway, indicating that ISG12a may participate directly in the process of NS5A ubiquitination. Because ISG12a has no activity of ubiquitin ligase (known as E3 ligase), we speculated that ISG12a is likely to recruit an E3 ligase to conduct the ubiquitination of NS5A. Using a bioinformatics search, we found a promising candidate, SKP2, which belongs to the subset of E3 ligase and is predicted to interact with ISG12a. To verify this prediction, we cotransfected the plasmids encoding SKP2 and ISG12a in HEK293T cells and analyzed the interaction between SKP2 and ISG12a. The IP assay indeed confirmed the association between ISG12a and SKP2 (Fig. 6A, left). Strikingly, we also detected the interaction between SKP2 and NS5A when we cotransfected their expression constructs in HEK293T cells (Fig. 6A, right). Additionally, HCV infection was able to enhance the interaction between ISG12a and SKP2 (Fig. 6B). These results, along with the finding of ISG12a-NS5A interaction, suggested that ISG12a, SKP2, and NS5A exist in a protein complex and the formation of this complex may lead to the ubiquitination and degradation of viral protein.
FIG 6.

SKP2 is an E3 ligase for NS5A. (A) IP analysis of SKP2 and ISG12a (left) or NS5A (right) from HEK293T cells coexpressing exogenous SKP2 and ISG12a or NS5A. (B) IP analysis of SKP2 and endogenous ISG12a from HLCZ01 cells transfected with pcDNA3.1a-myc/His-SKP2 plasmid followed by HCV infection for 48 h. (C) Ubiquitination assay and Western blotting of lysates from HEK293T cells expressing exogenous NS5A and HA-ubiquitin (HA-ub) with or without SKP2 silencing for 48 h. MG132 (25 μM) was added into cells for 6 h prior to harvesting cell lysates and IP experiments were conducted with anti-Flag antibody followed by the ubiquitination assay of NS5A protein. (D) Cells were treated as described for panel C followed by the ubiquitination assays of NS4B (left) and NS5B (right) proteins. (E) HLCZ01 cells were infected by HCV (MOI, 0.1) for 24 h followed by transfection with pSilencer-SKP2 or pSilencer vector plasmid for 48 h. The ubiquitination assay was conducted as described for panel C. (F and G) FL-neo cells were cotransfected with p3×FLAG-ISG12a and pcDNA3.1a-myc/His-SKP2 (F) or pSilencer-SKP2 (G) plasmid for 48 h followed by Western blotting of NS5A. (H) Huh7.5 and HLCZ01 cells were transfected with pcDNA3.1a-myc/His-SKP2 or pSilencer-SKP2 plasmid for 48 h. The MTS assay was performed, and error bars represent SD from triplicate experiments. V5, Flag, and HA were the tags used.
To identify the role of SKP2 in the process of NS5A ubiquitination, we overexpressed or knocked down SKP2 in the exogenous NS5A-expressed HEK293T cells. The data showed that ectopic expression of SKP2 markedly increased the level of NS5A ubiquitination (Fig. 6C). Silencing of SKP2 did not eliminate the basal ubiquitination of NS5A, indicating that the other E3 ligases may be involved in regulation of the stability of NS5A (Fig. 6C). In addition, overexpression of SKP2 had no effect on the basal ubiquitination of HCV NS4B and NS5B proteins (Fig. 6D). Importantly, HCV-induced ubiquitination of NS5A was attenuated by the knockdown of SKP2, indicating that SKP2 plays an essential role in NS5A ubiquitination (Fig. 6E). Although the individual overexpression of SKP2 had little effect on NS5A degradation, coexpression of SKP2 and ISG12a significantly enhanced the degradation of NS5A compared to the ISG12a monotreatment (Fig. 6F). On the other hand, silencing of SKP2 remarkably inhibited the degraded function of ISG12a on viral protein (Fig. 6G). Cell viability was also determined under the condition of SKP2 overexpression and knockdown. The data showed that the changes of SKP2 expression in cells did not affect cell viability (Fig. 6H). Collectively, these results indicated that SKP2 is an E3 ligase for NS5A and participates in ISG12a-mediated degradation of viral protein.
Ubiquitination of NS5A by SKP2 is dependent on ISG12a.
Although the interaction between SKP2 and NS5A was observed in our study, whether their interaction necessitates the help of ISG12a is unclear. To assess the action of ISG12a in this process, we overexpressed or knocked down ISG12a in HLCZ01 cells followed by the analysis of SKP2-NS5A interaction. The data demonstrated that ectopic expression or silencing of ISG12a significantly enhanced or attenuated the interaction between NS5A and SKP2, respectively (Fig. 7A and B). When the exogenous ISG12a, NS5A, and SKP2 were simultaneously expressed in cells, the triple interactions of them could be identified by IP analyses (Fig. 7A). Furthermore, overexpression of ISG12a also increased the level of NS5A ubiquitination by SKP2, whereas silencing of ISG12a inhibited SKP2-triggered ubiquitination of NS5A (Fig. 7C). These findings indicated that ISG12a acts as an adaptor to mediate SKP2-NS5A interaction, NS5A ubiquitination, and subsequent degradation of NS5A by SKP2.
FIG 7.

Ubiquitination of NS5A by SKP2 is dependent on ISG12a. (A and B) IP analysis of endogenous SKP2 and exogenous NS5A from HLCZ01 cells with ISG12a overexpression (A) or silencing (B). (C) Ubiquitination assay and Western blotting of lysates from FL-neo cells expressing SKP2 and HA-ubiquitin (HA-ub) with ISG12a overexpression or silencing for 48 h. MG132 (25 μM) was added to cells for 6 h prior to harvesting of cell lysates, and IP experiments were conducted with anti-NS5A antibody followed by the ubiquitination assay of NS5A protein. V5, Flag, and HA were the tags used.
Characterization of the interactions between SKP2 and ISG12a/NS5A.
To explore the interaction mechanisms between SKP2 and ISG12a, as well as SKP2 and NS5A, we divided SKP2 into several truncations (Fig. 8A). Particularly, the leucine-rich repeat (LRR) domain is implicated in substrate binding and harbors the E3 ligase activity. Using IP assays, we found that ISG12a did not interact with the N-terminal domain of SKP2 (SKP2-N) and the region containing SKP2-N and LRR, SKP2-(N+L), but could interact with the region of SKP2 with LRR deletion (SKP2-DL) (Fig. 8B). On the other hand, the tests comparing full-length SKP2 and ISG12a truncations showed that SKP2 interacted with the DII and DII-C of ISG12a and had no binding affinity with ISG12a DII-N (Fig. 8C). These results suggested that the C-terminal domain of SKP2 and the DII-C of ISG12a are the essential domains for SKP2-ISG12a interaction.
FIG 8.

Characterization of the interactions between SKP2 and ISG12a/NS5A. (A) Schematic representation of the domains of SKP2. (B and C) IP analysis of the interaction between SKP2 and ISG12a in HEK293T cells expressing exogenous ISG12a and multiple domains of SKP2 (B), or exogenous SKP2 and the different domains of ISG12a (C) for 48 h. (D and E) IP analysis of the interaction between SKP2 and NS5A in HEK293T cells expressing exogenous SKP2 and multiple domains of NS5A (D) or exogenous NS5A and the different domains of SKP2 (E) for 48 h. V5 and Flag were the tags used. SKP2-N, N-terminal of SKP2. SKP2-(N+L), SKP2 truncation containing SKP2-N and LRR. SKP2-DL, SKP2 with LRR deletion.
Next, we assessed the details of interaction between SKP2 and NS5A. After coexpression of full-length SKP2 and various domains of NS5A in HEK293T cells, the IP analyses were performed, and the data revealed that DI of NS5A did not interact with SKP2 (Fig. 8D, left), while other domains of NS5A were capable of binding to SKP2 (Fig. 8D). When the p3×FLAG-NS5A plasmid and multiple constructs of SKP2 were cotransfected in HEK293T cells, we found that both SKP2-N and SKP2-(N+L) were able to interact with NS5A (Fig. 8E). These results demonstrated that SKP2-N and NS5A DII to DIII are the pivotal domains for the interaction between SKP2 and NS5A.
ISG12a relies on the E3 ligase domain of SKP2 to restrict viral infection.
Some essential regions within ISG12a and SKP2 are identified in the above study, while the functions of these regions on viral restriction remain to be elucidated. Ectopic expression of full-length ISG12a induced abundant ubiquitination of NS5A by SKP2 in HEK293T cells (Fig. 9A). Similarly, ISG12a DII or DII-C also triggered robust ubiquitination of NS5A (Fig. 9A), whereas the DII-N of ISG12a did not employ that function like DII-C (Fig. 9A), which is consistent with the result that ISG12a DII-C, not DII-N, can interact with both SKP2 and NS5A (Fig. 4E and 8C). The experiments testing the functions of SKP2 domains revealed that only full-length SKP2 was able to promote remarkable ubiquitination of viral protein (Fig. 9B). SKP2-N and SKP2-DL, which lack the activity of E3 ligase, and SKP2-(N+L), which does not have binding affinity to ISG12a, may be responsible for their loss of function to induce NS5A ubiquitination (Fig. 8B and 9B). Moreover, SKP2-N, SKP2-(N+L), and SKP2-DL, which are unable to augment ISG12a-mediated ubiquitination of NS5A, also did not enhance the effect of ISG12a on the degradation of viral protein (Fig. 9C) and the inhibitory action of ISG12a on viral replication and virus production (Fig. 9D and E). Compared to the full-length SKP2, SKP2-DL with the ability to interact with ISG12a and NS5A still cannot amplify the antiviral effect of ISG12a (Fig. 9C to E), indicating that the E3 ligase activity within LRR of SKP2 is critical for ISG12a to restrict viral infection.
FIG 9.

ISG12a relies on the E3 ligase domain of SKP2 to restrict viral infection. (A) Ubiquitination assay and Western blotting of lysates from HEK293T cells expressing exogenous SKP2, HA-ubiquitin (HA-ub), and ISG12a domains (full-length, DII, DII-N, or DII-C). (B) Ubiquitination assay and Western blotting of lysates from HEK293T cells expressing exogenous ISG12a, HA-ub, and SKP2 domains [full-length, SKP2-N, SKP2-(N+L), or SKP2-DL]. (C) HLCZ01 cells were infected by HCV (MOI, 0.1) for 24 h followed by the transfection of the indicated plasmids for 48 h. NS5A, ISG12a, and the domains of SKP2 were analyzed by Western blotting. (D and E) Huh7.5 cells were infected by HCV (MOI, 0.1) for 24 h followed by the transfection of the indicated plasmids for 48 h. Intracellular HCV RNA (D) and extracellular infectious virus particles (E) were detected by real-time PCR and FFU assay, respectively. Error bars represented SD from triplicate experiments. *, P < 0.05; **, P < 0.01. V5, Flag, and HA were the tags used.
DISCUSSION
The innate immune response provides the first line of defense against various kinds of viral invader. IFNs induced by viral infection play an essential role in the restriction of virus propagation in host cells. Over 300 ISGs can be upregulated by IFNs and function as innate immune effectors to participate in antiviral processes, as well as acting as proviral factors for some ISGs (10). Although numerous antiviral ISGs have been identified by using different technical approaches (11–14), the functions and molecular mechanisms of the majority of ISGs in suppressing viral infection are poorly understood. To characterize further the working principles of IFN systems, we focus on elucidating the antiviral mechanisms of ISG. Our previous study revealed that HCV and NDV infection induces robustly the expression of ISG12a, which causes Noxa-dependent apoptosis of virus-infected cells (22, 24). The present study demonstrated the nonapoptotic antiviral activity of ISG12a. ISG12a targets viral NS5A protein for degradation through the proteasome-dependent pathway in HCV-infected cells (Fig. 10). The suppression of the expression of other viral proteins may be triggered by the inhibition of HCV replication that relies on the participation of NS5A.
FIG 10.
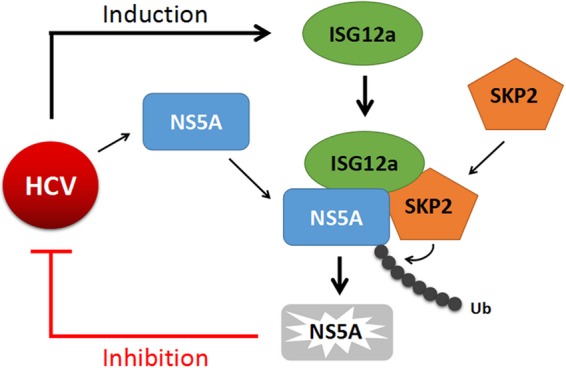
ISG12a restricts viral infection through the ubiquitination-dependent degradation pathway. ISG12a targets NS5A for ubiquitination-dependent proteasomal degradation in HCV-infected cells. SKP2 is involved in ISG12a-induced ubiquitination and degradation of viral protein. ISG12a as a crucial adaptor links SKP2 to NS5A for restriction of viral infection. Ub, ubiquitin.
The HCV NS5A protein bears pleiotropic functions, including roles in viral replication and production, and interplays with multiple cellular pathways (4–7, 30). Therefore, NS5A protein has been selected as a promising target for the development of anti-HCV inhibitors (30, 31). With this essential viral protein targeted by ISG12a, we provide a critical clue to explore the inhibitory mechanisms of innate immunity to viral infection. NS5A domains I and II are the major functional domains required for HCV replication, while the production of viral infectious particles is mainly dependent on the actions of NS5A domain III (6, 7). Moreover, viral propagation also necessitates the interaction between NS5A and numerous host proteins, such as cyclophilin A, apolipoprotein E, and phosphatidylinositol 4-kinase (32–34). The present study indicated that ISG12a binds to domain III of NS5A, which suggests ISG12a may harbor the additional functions to interrupt the interactions between NS5A and some pivotal factors of the virus or host through the degradation-independent approach. Considering NS5A functions in attenuating the activation of host antiviral pathways (such as the protein kinase R [PKR] cascade) (35), ISG12a-mediated degradation of NS5A protein is likely to improve the situation of viral evasion, contributing to effective immune responses.
SKP2 was discovered in 1995 as a protein associated with the S-phase kinase CDK2/cyclin A (36). Currently, SKP2 is known as an E3 ligase and is capable of inducing ubiquitination and subsequently proteasome-dependent degradation of target proteins (37). As many protein substrates of SKP2 involved in the processes of cancer development are identified, SKP2 is believed to play an important role in the pathogenesis of various cancers (37). However, the potential functions of SKP2 link to cellular antiviral processes are not reported yet. Our study demonstrated that SKP2 is a pivotal E3 ligase participating in the ubiquitination and degradation of viral protein, which is dependent on the modulation of ISG12a. In addition, the LRR domain of SKP2 is indispensable for this degradation process, indicating that the antiviral function of ISG12a necessitates the activity of E3 ligase of SKP2. Hence, the present study provides the first evidence for the antiviral action of SKP2. Interestingly, SKP2 functions in phosphorylation-dependent ubiquitination to specifically degrade the phosphorylated protein (37). It is widely acknowledged that HCV NS5A protein is a phosphorylated protein with abundant phosphorylation of multiple serine and threonine residues (38), which provides a potential molecular basis for understanding why SKP2 targets and ubiquitinates NS5A.
In addition to HCV or NDV infection, several infections by WNV, hepatitis E virus, influenza A virus, Japanese encephalitis virus, and Sindbis virus are capable of upregulation of human ISG12a or murine Ifi27 (17, 27, 39–41), which suggests that ISG12a/Ifi27 may play a vital role in antiviral innate immunity. Actually, we and other groups have determined the antiviral effects of ISG12a/IFI27 on HCV, NDV, WNV, and mouse hepatitis virus (MHV) infection in vitro or in vivo, though the underlying mechanisms remain unknown (22, 24, 26, 27). In the present study, we elucidate an intrinsic antiviral mechanism by which ISG12a recruits SKP2 to interact with and subsequently degrade the viral protein via a ubiquitination-dependent strategy. This novel mechanism may apply to understanding the inhibitory functions of ISG12a on the broad spectrum of various viral infections, including flavivirus (e.g., WNV), closed to HCV and other RNA viruses because of common characteristics on ISG12a induction and similar properties of viral infection between HCV and those viruses.
IFN-based therapies have been used to treat chronic hepatitis B and C viral infections for many years, and a better understanding of ISG functions will help identify some essential points that influence the outcomes of therapy and may even lead to the development of novel therapeutic approaches (42). Actually, a minority of ISGs, including PRRs, IRFs, the OAS-RNase L system, and the IFITM family members are relatively well characterized in antiviral immunity (43, 44). However, the working mechanisms of these known ISGs are entirely different from that equipped by ISG12a with functions to mediate degradation of viral protein. A novel adaptor role of ISG that triggers ubiquitination and degradation of viral protein is first exploited in present study.
Another member of the IFI6/IFI27 family, IFI6 (also known as G1P3), has been confirmed to have antiviral effects on several viruses, including HCV and yellow fever virus, by our group and others (10, 44, 45), while the underlying mechanism for IFI6 to control viral infection is yet unclear. Strikingly, human IFI6 protein shares 51% amino acid identity with human ISG12a protein and the homologous sequences are all in the domain II of ISG12a. Our data demonstrated that domain II of ISG12a is responsible for the function of ISG12a to interact with and degrade viral protein, which suggests IFI6 may use the same or similar mechanism for the suppression of viral infection.
Overall, the present study provided a novel intrinsic antiviral mechanism of host that ISG12a serves as an adaptor to recruit E3 ligase for targeting and degradation of viral protein via ubiquitination-dependent strategy. The maps of interactions between each two proteins of them revealed the pivotal domains of ISG12a and SKP2 for association with and degradation of viral protein. Particularly, the C-terminal of ISG12a domain II is essential and sufficient for its antiviral function. The LRR domain of SKP2 containing the region with E3 ligase activity is dispensable for the interactions between SKP2 and ISG12a or viral protein, while this key domain is indispensable for ISG12a-mediated restriction of HCV infection. Further investigations to explore the antiviral mechanisms of ISG12a to control other viral infection—probably according to the adaptor role of ISG12a in this study—are warranted to make full understanding of the antiviral functions of ISG12a in host innate immunity.
ACKNOWLEDGMENTS
We thank Charles M. Rice for the FL-neo and Huh7.5 cell lines and anti-NS2 antibody, Takaji Wakita for pJFH1, Stanley M. Lemon for pRL-HL plasmid, Zhengfan Jiang for pHA-Ub (K48) plasmid, and Chen Liu for sharing research materials.
This work was supported by grants from National Natural Science Foundation of China to H.Z. (81571985 and 81271885), and from National Science and Technology Major Project of the Ministry of Science and Technology of China to H.Z. (2009ZX10004-312).
REFERENCES
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.Catanese MT, Dorner M. 2015. Advances in experimental systems to study hepatitis C virus in vitro and in vivo. Virology 479-480:221–233. doi: 10.1016/j.virol.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 3.Baumert TF, Fauvelle C, Chen DY, Lauer GM. 2014. A prophylactic hepatitis C virus vaccine: a distant peak still worth climbing. J Hepatol 61(Suppl 1):S34–S44. doi: 10.1016/j.jhep.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Hughes M, Gretton S, Shelton H, Brown DD, McCormick CJ, Angus AG, Patel AH, Griffin S, Harris M. 2009. A conserved proline between domain II and III of hepatitis C virus NS5A influences both RNA replication and virus assembly. J Virol 83:10788–10796. doi: 10.1128/JVI.02406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Welsch C, Yi M, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J Virol 85:6645–6656. doi: 10.1128/JVI.02156-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tellinghuisen T, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol 82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horner SM. 2014. Activation and evasion of antiviral innate immunity by hepatitis C virus. J Mol Biol 426:1198–1209. doi: 10.1016/j.jmb.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Lin W, Kumthip K, Cheng D, Fusco DN, Hofmann O, Jilg N, Tai AW, Goto K, Zhang L, Hide W, Jang JY, Peng LF, Chung RT. 2012. A functional genomic screen reveals novel host genes that mediate interferon-alpha's effects against hepatitis C virus. J Hepatol 56:326–333. doi: 10.1016/j.jhep.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, Binder M, Frese M, Bartenschlager R. 2012. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 14.Fusco DN, Brisac C, John SP, Huang YW, Chin CR, Xie T, Zhao H, Jilg N, Zhang L, Chevaliez S, Wambua D, Lin W, Peng L, Chung RT, Brass AL. 2013. A genetic screen identifies interferon-α effector genes required to suppress hepatitis C virus replication. Gastroenterology 144:1438–1449. doi: 10.1053/j.gastro.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheriyath V, Leaman DW, Borden EC. 2011. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J Interferon Cytokine Res 31:173–181. doi: 10.1089/jir.2010.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker N, Porter A. 2004. Identification of a novel gene family that includes the interferon-inducible human genes 6-16 and ISG12. BMC Genomics 5:8. doi: 10.1186/1471-2164-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrada L, Liang X, Zheng W, Johnston C, Levine B. 2002. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol 76:11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu C, Zhu H, Subramanian MG, Moore PA, Xu Y, Nelson DR. 2007. Anti-hepatitis C virus activity of albinterferon alfa-2b in cell culture. Hepatol Res 37:941–947. doi: 10.1111/j.1872-034X.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 19.Rosebeck S, Leaman DW. 2008. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis 13:562–572. doi: 10.1007/s10495-008-0190-0. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Shin J, Lee K. 2009. Interferon-stimulated gene ISG12b1 inhibits adipogenic differentiation and mitochondrial biogenesis in 3T3-L1 cells. Endocrinology 150:1217–1224. doi: 10.1210/en.2008-0727. [DOI] [PubMed] [Google Scholar]
- 21.Papac-Milicevic N, Breuss JM, Zaujec J, Ryban L, Plyushch T, Wagner GA, Fenzl S, Dremsek P, Cabaravdic M, Steiner M, Glass CK, Binder CJ, Uhrin P, Binder BR. 2012. The interferon stimulated gene 12 inactivates vasculoprotective functions of NR4A nuclear receptors. Circ Res 110:e50–e63. doi: 10.1161/CIRCRESAHA.111.258814. [DOI] [PubMed] [Google Scholar]
- 22.Liu N, Long Y, Liu B, Yang D, Li C, Chen T, Wang X, Liu C, Zhu H. 2014. ISG12a mediates cell response to Newcastle disease viral infection. Virology 462-463:283–294. doi: 10.1016/j.virol.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Liu N, Zuo C, Wang X, Chen T, Yang D, Wang J, Zhu H. 2014. miR-942 decreases TRAIL-induced apoptosis through ISG12a downregulation and is regulated by AKT. Oncotarget 5:4959–4971. doi: 10.18632/oncotarget.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D, Meng X, Xue B, Liu N, Wang X, Zhu H. 2014. MiR-942 mediates hepatitis C virus-induced apoptosis via regulation of ISG12a. PLoS One 9:e94501. doi: 10.1371/journal.pone.0094501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang D, Zuo C, Wang X, Meng X, Xue B, Liu N, Yu R, Qin Y, Gao Y, Wang Q, Hu J, Wang L, Zhou Z, Liu B, Tan D, Guan Y, Zhu H. 2014. Complete replication of hepatitis B virus and hepatitis C virus in a newly developed hepatoma cell line. Proc Natl Acad Sci U S A 111:E1264–E1273. doi: 10.1073/pnas.1320071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucas TM, Richner JM, Diamond MS. 2015. The Interferon-stimulated gene Ifi27l2a restricts West Nile virus infection and pathogenesis in a cell-type and region-specific manner. J Virol 90:2600–2615. doi: 10.1128/JVI.02463-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen M, Schmitt S, Buac D, Dou QP. 2013. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets 17:1091–1108. doi: 10.1517/14728222.2013.815728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu R, Yang D, Lei S, Wang X, Meng X, Xue B, Zhu H. 2015. HMGB1 promotes hepatitis C virus replication by interaction with stem-loop 4 in the viral untranslated region. J Virol 90:2332–2344. doi: 10.1128/JVI.02795-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pawlotsky JM. 2013. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 59:375–382. doi: 10.1016/j.jhep.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 31.Yu X, Gao Y, Xue B, Wang X, Yang D, Qin Y, Yu R, Liu N, Xu L, Fang X, Zhu H. 2014. Inhibition of hepatitis C virus infection by NS5A-specific aptamer. Antiviral Res 106:116–124. doi: 10.1016/j.antiviral.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol 85:7460–7464. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol 84:1532–1541. doi: 10.1128/JVI.01021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delang L, Paeshuyse J, Neyts J. 2012. The role of phosphatidylinositol 4-kinases and phosphatidylinositol 4-phosphate during viral replication. Biochem Pharmacol 84:1400–1408. doi: 10.1016/j.bcp.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demetrick DJ, Zhang H, Beach DH. 1996. Chromosomal mapping of the genes for the human CDK2/cyclin A-associated proteins p19 (SKP1A and SKP1B) and p45 (SKP2). Cytogenet Cell Genet 73:104–107. doi: 10.1159/000134318. [DOI] [PubMed] [Google Scholar]
- 37.Bochis OV, Irimie A, Pichler M, Berindan-Neagoe I. 2015. The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer. J Gastrointestin Liver Dis 24:225–234. doi: 10.15403/jgld.2014.1121.242.skp2. [DOI] [PubMed] [Google Scholar]
- 38.Ross-Thriepland D, Harris M. 2015. Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol 96:727–738. doi: 10.1099/jgv.0.000009. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Qi Y, Harrison TJ, Luo B, Zhou Y, Li X, Song A, Huang W, Wang Y. 2014. Hepatitis E genotype 4 virus from feces of monkeys infected experimentally can be cultured in PLC/PRF/5 cells and upregulate host interferon-inducible genes. J Med Virol 86:1736–1744. doi: 10.1002/jmv.24014. [DOI] [PubMed] [Google Scholar]
- 40.Pommerenke C, Wilk E, Srivastava B, Schulze A, Novoselova N, Geffers R, Schughart K. 2012. Global transcriptome analysis in influenza-Infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS One 7:e41169. doi: 10.1371/journal.pone.0041169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clarke P, Leser JS, Bowen RA, Tyler KL. 2014. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio 5:e00902-14. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li K, Lemon SM. 2013. Innate immune responses in hepatitis C virus infection. Semin Immunopathol 35:53–72. doi: 10.1007/s00281-012-0332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu H, Zhao H, Collins CD, Eckenrode SE, Run Q, McIndoe RA, Crawford JM, Nelson DR, She JX, Liu C. 2003. Gene expression associated with interferon alfa antiviral activity in an HCV replicon cell line. Hepatology 37:1180–1188. doi: 10.1053/jhep.2003.50184. [DOI] [PubMed] [Google Scholar]
- 45.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]