

ABSTRACT
Novel influenza viruses often cause differential infection patterns across different age groups, an effect that is defined as heterogeneous demographic susceptibility. This occurred during the A/H2N2 pandemic, when children experienced higher influenza attack rates than adults. Since the recognition of conserved epitopes across influenza subtypes by CD8+ cytotoxic T lymphocytes (CTLs) limit influenza disease, we hypothesized that conservation of CTL antigenic peptides (Ag-p) in viruses circulating before the pH2N2-1957 may have resulted in differential CTL immunity. We compared viruses isolated in the years preceding the pandemic (1941 to 1957) to which children and adults were exposed to viruses circulating decades earlier (1918 to 1940), which could infect adults only. Consistent with phylogenetic models, influenza viruses circulating from 1941 to 1957, which infected children, shared with pH2N2 the majority (∼89%) of the CTL peptides within the most immunogenic nucleoprotein, matrix 1, and polymerase basic 1, thus providing evidence for minimal pH2N2 CTL escape in children. Our study, however, identified potential CTL immune evasion from pH2N2 irrespective of age, within HLA-A*03:01+ individuals for PB1471-L473V/N476I variants and HLA-B*15:01+ population for NP404–414-V408I mutant. Further experiments using the murine model of B-cell-deficient mice showed that multiple influenza infections resulted in superior protection from influenza-induced morbidity, coinciding with accumulation of tissue-resident memory CD8+ T cells in the lung. Our study suggests that protection against H2N2-1957 pandemic influenza was most likely linked to the number of influenza virus infections prior to the pandemic challenge rather than differential preexisting CTL immunity. Thus, the regimen of a CTL-based vaccine/vaccine-component may benefit from periodic boosting to achieve fully protective, asymptomatic influenza infection.
IMPORTANCE Due to a lack of cross-reactive neutralizing antibodies, children are particularly susceptible to influenza infections caused by novel viral strains. Preexisting T cell immunity directed at conserved viral regions, however, can provide protection against influenza viruses, promote rapid recovery and better clinical outcomes. When we asked whether high susceptibility of children (compared to adults) to the pandemic H2N2 influenza strain was associated with immune evasion from T-cell immunity, we found high conservation within T-cell antigenic regions in pandemic H2N2. However, the number of influenza infections prior to the challenge was linked to protective, asymptomatic infections and establishment of tissue-resident memory T cells. Our study supports development of vaccines that prime and boost T cells to elicit cross-strain protective T cells, especially tissue-resident memory T cells, for lifelong immunity against distinct influenza viruses.
INTRODUCTION
Respiratory infections caused by influenza viruses are responsible for approximately half a million deaths worldwide annually (1), representing a significant health and economic burden (2). The emergence of new influenza A virus (IAV) lineages, as observed during 1918, 1957, 1968, and 2009, as well as the frequent zoonosis of diverse avian and swine IAVs, represents a persistent pandemic threat (3). Neutralizing antibody (nAb)-based vaccines are currently the most effective means to control influenza. Nonetheless, since these vaccines are directed against the rapidly changing surface glycoproteins, protection is predominantly strain specific (4). The ability of IAVs to elude immune surveillance by antigenic drift makes effective protection against seasonal influenza short-lived, resulting in susceptibility to reassortant or newly emerged influenza viruses.
In contrast, CD8+ T lymphocytes (CTLs) recognize major histocompatibility complex class I presenting viral peptides primarily derived from the internal influenza proteins that exhibit greater conservation across influenza subtypes (5, 6). Thus, preexisting CTL memory can mediate heterologous immunity among H1N1, H2N2, H3N2, H5N1, and H7N9 influenza A viruses (7–10), providing a potential for the highly sought “universal” influenza vaccine. CTL-mediated heterosubtypic protection against influenza viruses has been demonstrated in mice (11–13) and supported in humans (14–17), although immune evasion from immunodominant influenza CTL epitopes also occurs (18).
Newly emerged influenza viruses may be associated with differential infection patterns in age groups, due to heterogeneous demographic susceptibility. For example, epidemiological studies on susceptibility to pandemic influenza viruses have highlighted a disproportionately high attack rate in children during the H2N2 “Asian” pandemic in 1957 (19). This intriguing susceptibility pattern to a supposedly ‘novel’ virus was revealed in a unique retrospective analysis of archived active surveillance data, collected during the period from 1947 to 1957, culminating with the emergence of the pandemic 1957 H2N2 virus (pH2N2-1957) (19). Among individuals reported to have experienced influenza infections prior to emergence of pH2N2-1957, 55% of children had influenza symptoms during the pandemic compared to only 5% of adults (19).
It is thought that in the absence of pH2N2-specifc nAbs, preestablished CD8+ T cell memory contributed to asymptomatic disease during the H2N2 pandemic; however, no immunological evidence for preexisting CD8+ T cell immunity toward pH2N2-1957 has yet been provided. Here, we investigated how infection history may have shaped CD8+ T cell memory directed against pH2N2-1957 and hypothesized two scenarios that could account for differential age susceptibility toward this virus as follows.
(i) Early H1N1 influenza strains (circulating from ca. 1918 to 1940, to which only adults would have been exposed) comprised CTL antigenic peptides closely related to those found in pH2N2-1957. Conversely, H1N1 viruses circulating closer to 1957 (1941 to 1956, to which both adults and children may have been recently exposed) shared fewer CTL immunogenic peptides with pH2N2-1957, resulting in reduced CTL reactivity and protection from disease.
(ii) Alternatively, differences in the number of prior influenza virus exposures had an impact on pH2N2-1957 differential susceptibility among the age groups. In particular, the severe influenza outbreaks recorded in 1947 (20) and 1950–1951 (21), associated with hemagglutinin (HA) intrareassortment events (22) raised the possibility that adults may have experienced more than one influenza virus infection (pre- and post-1940) that could have generated a boosting effect on CD8+ T cell memory. This is in contrast to children born since 1941 (19), who were less likely to have had the opportunity for infection with serologically distinct influenza viruses for CTL memory boosting.
Importantly, reemergence of an H2 virus in humans remains a significant concern due to the lack of nAbs against this hemagglutinin in individuals under 50 years of age. H2N2 viruses that circulate in birds (23) show genetic similarities with the ancestors of the 1957 pandemic virus (24–26), are replicatively fit, can induce contact transmission, and are pathogenic in mammals (27). Since influenza virus-specific CD8+ T cell memory can promote recovery from new influenza viruses, they have potential to reduce pathogenicity of reemerging H2 viruses (28). We therefore investigated preexisting CTL immunity in the context of the 1957 H2N2 pandemic.
Using an evolutionary and functional approach, we analyzed the viral sequences of several immunogenic influenza proteins—nucleoprotein (NP), matrix 1 (M1), and polymerase basic 1 (PB1) (8)—and found a high level of conservation (∼70%) in CTL peptides across 40 years of influenza virus evolution (1918 to 1957). Furthermore, consistent with phylogenetic models, influenza viruses circulating from 1940 to 1957 shared the majority (∼89%) of CTL peptides with pH2N2-1957, indicating minimal viral escape from preexisting CTL immunity in children. Experiments using B-cell-deficient (μMT) mice showed that in the absence of antibodies, repeated influenza virus infections consolidate protection from influenza disease, associated with an increase in the number of tissue-resident CD8+ memory T (CD8+ TRM) cells in the lung. Thus, our data suggest that population protection against pH2N2-1957 was more likely related to the number of influenza virus infections prior to pandemic challenge rather than to the differential preexisting CTL immunity.
MATERIALS AND METHODS
Ethics statement.
All animal experimentation was conducted according to the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes guidelines for housing and care of laboratory animals and performed in accordance with institutional regulations after pertinent review and approval by the University of Melbourne Animal Ethics Experimentation Committee (no. 1312880.3) and the Walter and Eliza Hall Institute Ethics Committee (no. 2011.031) in Melbourne. Human peripheral blood was collected from healthy volunteers, with written informed consent, and approved by the University of Melbourne Human Ethics Committee (no. 1443389.3).
Donors and PBMC isolation.
Cryopreserved peripheral blood mononuclear cells (PBMCs) from healthy donors expressing relevant human leukocyte antigen (HLA) alleles (HLA-A*02:01+, -A*03:01+, -B*15:01+, and -A*11:01+; 12 donors [see Table 2]) was used in the study. To isolate PBMCs, blood was collected in heparinized tubes for PBMC isolation using Ficoll-Paque (GE Healthcare, Sweden) density gradient centrifugation. Cryopreserved PBMCs were used throughout the study. HLA genotyping was performed by the Victorian Transplant and Immunogenetics Service (Parkville, Victoria, Australia).
TABLE 2.
Conservation of CTL peptides (relative to pH2N2-1957) from 1918 to 1940 and from 1941 to 1956
| Protein | No. of peptides | No. of conserved Ag-p |
% conserved Ag-p |
||
|---|---|---|---|---|---|
| 1918–1940 | 1941–1956 | 1918–1940 | 1941–1956 | ||
| NP | 71 | 35 | 57 | 49 | 80 |
| M1 | 39 | 27 | 39 | 69 | 100 |
| PB1 | 32 | 28 | 28 | 88 | 88 |
| Total | 142 | 90 | 124 | 69 | 89 |
Influenza A protein and CTL antigenic regions conservation analysis.
We obtained influenza protein sequences from the NCBI Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html) and performed data searches in the Immune Epitope Database (http://www.iedb.org/) to compile a list of experimentally tested CTL antigenic regions in the influenza proteome (data not shown). Based on the immunogenicity, we focused on the proteins NP (71 epitopes), M1 (39 epitopes), and PB1 (32 epitopes). To analyze conservation across interpandemic periods, we aligned protein sequences pertaining to all reported human influenza A isolates in circulation in the following data sets: (i) from 1918 to 1957 (A/Brevig Mission/1918 [H1N1], used as a reference sequence), (ii) from 1918 to 1940, and (iii) from 1941 to 1956 (A/Singapore/1/1957 [H2N2], used as a reference for the latter two data sets). The conservation of whole proteins was calculated as a proportion of the number of mutated residues with respect to the total number of residues in each protein (see Fig. 2). For the conservation analysis of CTL antigenic regions, we compared the corresponding region in the reference sequences against the human isolates during the relevant interpandemic period (from 1918 to 1957: n = 97 strains for NP, n = 99 strains for M1, and n = 98 for PB1). The analytic data in Tables 1 and 2 and Fig. 2A were determined using the IEDB conservancy tool (tools.immuneepitope.org/tools/conservancy/iedb_input) at a 100% cutoff match. Sporadic changes were discarded, i.e., CTL antigenic regions that were >80% during the time period analyzed were classified as conserved (Tables 1 and 2).
FIG 2.
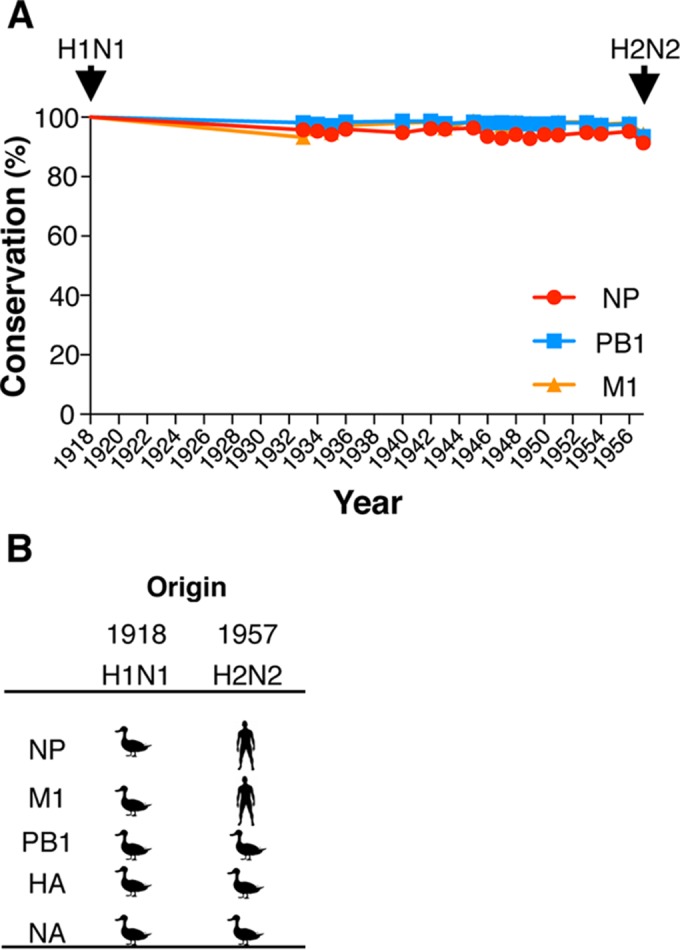
CD8+ T cell immunogenic NP, M1, and PB1 proteins remain largely conserved across the 1918-1957 interpandemic period. Influenza viral proteins were aligned using the Influenza Virus Resource tools (www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). (A) Conservation from 1918 to 1957 was determined with respect to the reference sequences of A/H1N1/Brevig Mission/1918; arrows depict year of emergence of a novel pandemic viruses. (B) Origin of the emerging pandemic viruses from 1918 to 1957; 1918-“Spanish” influenza virus was likely introduced from an avian source. The H2N2 “Asian” influenza pandemic resulted from introduction of avian H2, N2, and PB1 from birds (77).
TABLE 1.
Conservation of CTL peptides (relative to pH1N1-1918) from 1918 to 1957
| Protein | Total no.a | H1N1 and H2N2 virusesb |
|
|---|---|---|---|
| Cons (no.) | Cons (%) | ||
| NP | 71 | 36 | 51 |
| M1 | 39 | 28 | 72 |
| PB1 | 32 | 28 | 88 |
| Inclusivec | 142 | 92 | 70 |
That is, the total number of Ag-p.
Cons, conserved Ag-p.
That is, the sum or mean percentage of Ag-p.
CTL restimulation and intracellular cytokine assay.
Peptides used for functional analysis were purchased from GenScript (Piscataway, NJ) and dissolved in Hanks balanced salt solution (HBSS). Polyclonal T cell lines were established by pulsing PBMCs with peptide at 10 μM in RPMI (Gibco/Invitrogen) for 1.5 h at 37°C (29). After incubation with the peptide, the PBMCs were washed twice and cocultured with the unpulsed fraction at a 1:2 peptide-pulsed/peptide-unpulsed ratio. Cultures were maintained for 10 days in cRPMI (RPMI supplemented with 1 mM sodium pyruvate, 5 mM HEPES buffer, 2 mM l-glutamine, 100 mM nonessential amino acids, 55 mM 2-mercaptoethanol, streptomycin at 100 mg/ml, and penicillin at 100 U/ml [all from Gibco]) and 10% (vol/vol) heat-inactivated fetal calf serum (Thermo Scientific), with rhIL-2 (Apollo, Australia) at 10 U/ml added at 2-day intervals from day 4 onward. On day 10 after peptide stimulation, intracellular cytokine staining (ICS) was performed. Briefly, C1R cells transfected with HLA-A*02:01 were pulsed with cognate peptide at 10 μM peptide for 1.5 h at 37°C. For HLA-A*03:01-, -A*11:01-, and -B*15:01-restricted peptides, freshly thawed autologous PBMCs were incubated with 10 μM peptide as target antigen-presenting cells (APCs). The corresponding peptide-pulsed APCs (1 × 105 cells) were cocultured with the relevant polyclonal T cell line (2 × 105 cells, at day 10) for 5 h. The cells were stained for 30 min on ice in the dark with anti-CD3-PE-Cy7 (BioLegend), anti-CD4-Pacific Blue and anti-CD8α-PerCP-Cy5.5 (both of these monoclonal antibodies were from BD Biosciences), and Live/Dead-NIR fixable stain (Life Technologies). Cells were then fixed and permeabilized with Cytofix/Cytoperm reagent (BD Biosciences) for 20 min on ice in the dark. Intracellular staining with anti-gamma interferon (IFN-γ)-FITC and anti-tumor necrosis factor alpha (TNF-α)-APC (BD Biosciences) diluted in Permwash (BD Biosciences) was performed for 30 min on ice in the dark. After a washing step, the cells were analyzed by flow cytometry on an LSR II or BD FACSCanto II instrument (BD Biosciences). Cytokine background from polyclonal T cell lines incubated with no peptide control APCs was subtracted.
Isolation of epitope-specific IFN-γ-producing cells and single-cell TCR repertoire analysis.
Day 10 polyclonal T cell lines (2 × 105 cells) were expanded in cRPMIh (RPMI supplemented as described above except the FCS was replaced with 10% heat-inactivated human male AB plasma-serum [Sigma-Aldrich], as indicated by the manufacturer) and restimulated with 1 μM peptide for 2 h in a 96-well cell-culture plate for 2 h. IFN-γ-producing CD8+ T cells were detected using an IFN-γ secretion assay (Miltenyi Biotec), as described previously (30), and surface staining was performed. CD3+ CD4− CD8+ IFN-γ+ live lymphocytes were single cell sorted using a FACSAria III (BD Biosciences) into 96-well-PCR plates (Qiagen). T-cell-receptor (TCR) sequence determination was performed as described previously (30, 31).
Phylogenetic analysis.
The full protein coding genes of the M1, NP, and PB1 from human influenza H1N1 and H2N2 viruses isolated from 1918 to 1967 were acquired from the Global Initiative of Sharing Avian Influenza Data (GISAID) database and analyzed using the maximum-likelihood method. Analysis was performed using a general-time-reversible substitution model with a γ-shaped rate variation in RaXML v8 (30). The final data set of 43 sequences (Fig. 2B to D) was assembled after the removal of duplicate strains and sequences that contained incorrect sampling years or those that were overly divergent indicating extensive laboratory passage using the root-to-tip regression in Path-O-Gen v1.4 (now TempEst; http://tree.bio.ed.ac.uk/software/tempest/).
Mice and influenza virus infection.
Female μMT (H-2b) mice (32) were bred and housed under pathogen-free conditions at the Walter Eliza Hall Institute, Melbourne, Australia. Mice were lightly anesthetized by the inhalation of methoxyflurane and then infected intranasally (i.n.) with 103 PFU of H3N2-HKx31 influenza A virus dissolved in 30 μl of phosphate-buffered saline (PBS). Sequential infections were performed in the same manner at intervals of ≥30 days, and animals were culled at day 51 after the last infection. All experiments were approved and conducted under guidelines set by the Walter Eliza Hall Institute Animal Ethics Committee.
Tissue sampling and cell preparation.
Lungs were recovered from mice 51 days after primary, secondary, and tertiary influenza virus infection. The lungs were treated with 3 mg of type III collagenase (Worthington)/ml supplemented with 5 mg of DNase (Invitrogen)/ml in RPMI (33). Cell suspensions from lungs were incubated for 45 min at 37°C in petri dishes to remove adherent cells and resuspended in fluorescence-activated cell sorting (FACS) buffer for flow cytometric analysis. Cells were stained with Live/Dead Aqua fixable stains (Life Technologies) for 15 min in PBS at room temperature. The cells were washed once with FACS buffer and stained with anti-CD45.2-BV711, anti-CD8a-Pacific Blue, anti-CD62L-PE-Cy7, anti-CD103-APC, and anti-CD69-PE for 30 min on ice. The cells were washed twice with FACS buffer for FACS analysis on an LSR Fortessa (BD Biosciences).
RESULTS
Conservation of CTL antigenic peptides from 1918 to 1957 and the evolution of human influenza viruses.
We first considered whether heterogeneous demographic influenza susceptibility toward pH2N2-1957 may have resulted from differential CTL reactivity due to previous exposure with viruses displaying distinct CTL antigenic regions (Fig. 1A). We analyzed the conservation of protein and CTL peptides across human influenza strains isolated between 1918 and 1957, when there was a shift from H1N1 to H2N2. Our focus was on the most immunogenic NP, M1, and PB1 viral proteins (8, 17, 34). At the protein level, we found that these remained largely conserved across the interpandemic 1918-1957 period (Fig. 2). M1 and PB1 were equivalently conserved (mean conservation for 1918 to 1957 of ∼98%), followed by NP (mean conservation for 1918 to 1957 of ∼95%) (Fig. 2). The overall level of conservation relative to 1918-H1N1 remained well above 90% (range, 91 to 100%). High protein conservation is indicative of shared CTL antigenic peptides among H1N1 viruses circulating before pH2N2-1957.
FIG 1.
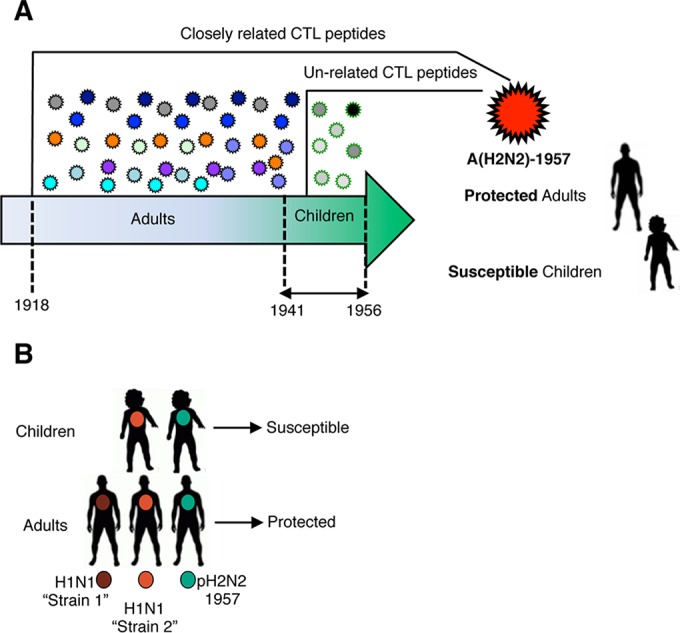
Hypothesized scenarios that could influence CTL immunity to pH2N2-1957 and heterogeneous demographic susceptibility. (A) H1N1 viruses circulating from 1918 to 1941 that could only infect people who reach adulthood by 1957 comprised CTL peptides closely related to pH2N2-1957, resulting in strong heterosubtypic immunity. Children (born from 1941 to 1956) were infected with strains circulating immediately prior to pH2N2-1957 that had less conserved immunogenic peptides relative to pH2N2-1957, resulting in diminished CTL immunity. (B) Intrareassortment events lead to the emergence of multiple H1N1 viruses (“Strain 1” and “Strain 2”) circulating prior to pH2N2-1957. In contrast to children, adults would have experience more than one influenza infection before pH2N2-1957 challenge that could have boosted CTL memory increasing their protective potential.
Despite the high level of conservation within the NP, M1, and PB1 proteins, mutations occurred at a range of 4 to 49 substitutions in any given year from 1918 to 1957 (Fig. 2A). Thus, we next analyzed whether the variations occurred within 142 functionally tested CTL antigenic regions presented by 51 HLA-A and -B molecules (data not shown). Consistent with the conservation at the protein level, ∼70% of the Ag-p remained conserved from 1918 to 1957 (Table 1). Despite its introduction from avian reservoirs (35) (Fig. 2B), CTL antigenic regions in the PB1 were the most highly conserved (28 of 32 conserved Ag-p, ∼88%), followed by M1 (28 of 39 conserved Ag-p, ∼72%) and NP (36 of 71 Ag-p, 51%). This conservation of CTL antigenic regions would mean that existing CTL-mediated immunity should have provided protection against H2N2-1957 pandemic viruses.
Subsequently, to address whether heterogeneous demographic susceptibility to pH2N2-1957 was due to previous exposures with influenza viruses comprising distinct CTL peptides (Fig. 1A), we analyzed conservation of antigenic regions during 1918–1940 and 1941–1957 relative to pH2N2-1957 (Table 2 and Fig. 3A). Influenza viruses circulating during the 1918-1940 period (exposure of adults only) shared 69% conserved CTL peptides with pH2N2-1957 (35 of 71 in the NP, 27 of 39 in the M1, and 28 of 32 in the PB1; Table 2 and Fig. 3A). Strikingly, viruses that circulated during 1941 to 1956 (to which children would have been exposed to) shared the vast majority (89%) of conserved CTL peptides with pH2N2-1957 (57 of 71 in the NP; 39 of 39 in the M1, and 28 of 32 in the PB1; Table 2 and Fig. 3A). Our data thus demonstrate that differences in susceptibility of children and adults to pH2N2-1957 were not due to differential CTL reactivity resulting from exposure to influenza viruses bearing distinct CTL antigenic regions (Fig. 3A).
FIG 3.

H1N1 viruses circulating from 1941 to 1956 shared more Ag-p with pH2N2-1957 reflective of the evolutionary relationships of the NP, M1, and PB1 human influenza viruses. (A) Proportion of shared Ag-p with pH2N2-1957 (y axis) in H1N1 viruses circulating from 1918 to 1940 (exposure of adults only) and from 1941 to 1956 (children exposure window) across the M1, NP, and PB1. (B to D) M1 (B) and NP (C) genes of pH2N2-1957 were derived from preceding H1N1 viruses, in contrast to PB1 genes (D), which were acquired through reassortment from avian ancestors.
To verify our results in an evolutionary context, we applied phylogenetic analysis to visualize the evolution of the NP, M1, and PB1 gene lineages among H1N1 and H2N2 viruses across 1918 to 1967. A single lineage of M1 and NP protein genes has continuously circulated in humans over the 50 years period since 1918 (Fig. 3B and C), whereas the PB1 protein genes were introduced in 1957, along with the surface glycoproteins (Fig. 2B and 3D). The most immunogenic protein, NP (34, 36), drifted gradually since 1957 reflecting their evolution in recent years (Fig. 3C). A similar but less pronounced drift was observed for the M1 and PB1 genes (Fig. 3BD). These phylogenetic relationships partly explain the gain of epitopes in children observed in the 1941-1956 period (Table 2 and Fig. 3A). In comparison to the drift rate of surface HA and NA proteins (37), the internal NP, M1, and PB1 that represent important CTL targets drifted at a lower rate (Fig. 3B to D). Remarkably, even when the viral PB1 was replaced in pH2N2-1957 as a result of the emergence of novel genes or reassortment (37), our data showed that the majority of experimentally tested CTL peptides remained conserved.
Variations within CTL antigenic regions and immune escape.
Immune escape can impair CTL-mediated viral control (38), as shown for HLA-B*3501-restricted NP418–426 or HLA-B*2705-restricted NP383–391 influenza epitopes (39–42). Furthermore, CTL-mediated immune pressure can drive influenza virus evolution, as shown by selection of NP variants with variation within CTL antigenic regions (43, 44). Given the importance of CTL variants in escaping protective CTL-mediated immunity, we performed a comprehensive analysis (across 142 experimentally validated CTL Ag-p presented by 51 HLA-A an HLA-B molecules) of mutations in pH2N2-1957 antigenic regions that may have potentially escaped recognition from established CTL memory prior to 1957. Our study identified two overlapping immunogenic peptides within the viral NP (HLA-A*03:01-restricted-NP407–416 and HLA-B*15:01-restricted-NP404–414), which resembled pH1N1/1918 virus but not the H1N1 strains isolated after the pandemic period (1935 to 1956) (data not shown). We also found that the emergent PB1 protein gene of pH2N2-1957 contained three novel mutations affecting two epitopes (M→I at position 6 [p6] within the HLA-A2-restricted PB1166–174 and L→V at p3 and N→I at p6 within the A*03:01-restricted PB1471–480 [referred to here as PB1471-L473V/N476I]) (data not shown).
To assess the impact of these variations on CTL recognition, we probed their ability to recall memory CD8+ T cells from PBMCs isolated from healthy volunteers (Table 3) using CTL peptide stimulation, followed by IFN-γ/TNF-α ICS (29). The novel PB1471-L473V/N476I variant emerging with pH2N2-1957 failed to recall preestablished memory CTLs (Fig. 4), suggesting that HLA-A*03:01+ individuals of all ages had a diminished CTL memory response to pH2N2-1957. Unexpectedly, no responses were detected toward any HLA-A2-restricted PB1166–174 orHLA-A3-restricted NP407–416 variants in our donors (data not shown).
TABLE 3.
Ages and HLA types of donors
| Donor | Age (yr)a | HLA type(s) |
|
|---|---|---|---|
| HLA-A | HLA-B | ||
| D-3A | 57 | 0301, 3201 | 4402 |
| D-3B | 58 | 0301, 1101 | 0702, 4403 |
| D-3C | 43 | 0301, 1101 | 3501, 4402 |
| D-15A | 61 | 0201, 3101 | 1501, 4402 |
| D-15B | 40 | 0301, 2601 | 0702, 1501 |
| D-15C | 39 | 0101, 0301 | 0801, 1501 |
| D-15D | NA | 0201 | 1501 |
| D-3D | NA | 0301 | 0702, 1501 |
| D-2A | 71 | 0201, 2402 | 3906, 4002 |
| D-2B | 25 | 0201, 6801 | 4402, 5101 |
| D-2C | 33 | 0201, 0301 | 1402, 5801 |
| D-2D | 69 | 0201, 3201 | 0702, 4403 |
NA, not available.
FIG 4.

Loss of CTL immunity toward A*03:01+-PB1471–480 L473V/N476I variant with the emergence of the pH2N2-1957. CTL responses to PB1471–480 in HLA-A*03:01+ healthy donors (D-3A, D-3B, and D-3C) were assessed. Polyclonal CTL lines were generated by pulsing with wild-type PB1471 or variant PB1471L473V/N476I peptides, followed by 10 days of in vitro expansion. On day 10, the cells were stimulated with cognate peptide, followed by a 5-h ICS assay. Reactivation was measured on live CD3+ CD4− CD8+ T cells by TNF-α/IFN-γ ICS. The results for no-peptide controls are shown in parentheses. The presence of preexisting influenza-specific CTL memory was verified by assessing the immunogenic A*03:01-restricted NP265–273 response as a positive control (left column). FACS plots (A) and graphed data with background subtracted (B) are shown. The data are representative of three independent experiments.
Furthermore, we found that the HLA-B*15:01-restricted NP404 V408I variant, present in pH2N2-1957 isolates (data not shown) failed to reactivate preexisting CTL memory pools in donor D-15A (Fig. 5). In contrast, donor D-15B displayed reactivity to both H1N1 wild-type (wt) and H2N2 (V408I) variants of B*15:01-restricted-NP404–414 (Fig. 5). To determine whether this was due to the interepitope cross-reactivity or resulted from infection with distinct influenza strains, we analyzed TCRαβ signatures toward the NP404-wt and NP404-V408I antigenic peptides. Epitope-specific CTLs were single cell sorted for paired TCRαβ analysis (Fig. 6A). Although there was a high degree of shared TRAV and TRBV usage (Fig. 6B), distinct TCRαβ profiles (defined by the hypervariable complementary determining region 3 [CDR3] sequence) responded to each B*1501-restrcited NP404 variant, with only one TCRαβ clonotype (Vα12.2-DGGNKL-Jα47/Vβ19-SIIGWDYEQ-Jβ2.7) shared at low frequency (∼5%) between B*15:01-NP404-wt and B*15:01-NP404-V408I (Fig. 6C). These data indicate that donor 15B was infected with at least two different influenza strains eliciting distinct TCRαβ repertoire pools and thus point to a non-cross-reactive nature of memory CTL pools toward the pH2N2-1957 variant in B*15:01+ individuals. Given that the NP404 variant was shared between pH1N1-1918 and pH2N2-1957, but not with the intervening strains (data not shown), our data suggest that only the adults were likely protected from viruses capable of generating NP404-specific memory toward pH2N2-1957.
FIG 5.
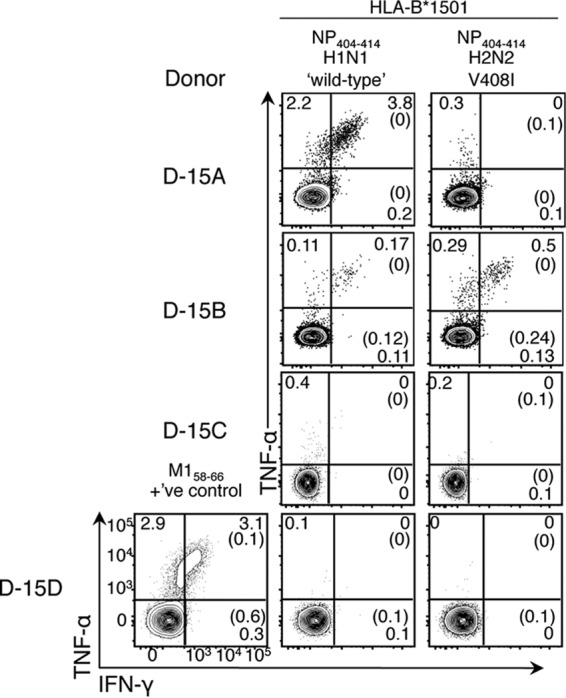
Mutations in the subdominant HLA-B*15:01-restricted NP404–414 during the 1933-1957 period compromised CTL recognition of pH2N2-1957. Influenza-specific CD8+ T-cell responses in HLA-B*15:01+ healthy donors (D-15A, D-15B, D-15C, and D-15D) were assessed by IFN-γ/TNF-α ICS as described in Fig. 4. Reactivity to NP404 was confirmed in two of four donors (D-15A and D-15B) despite prominent influenza-specific CTL reactivation toward A2+-M158–66 found in D-15D. Cells were gated on live CD3+ CD4− CD8+ T cells. Percentages of background staining of no-peptide controls are shown in parentheses. Data for D-15A and D-15B are representative of three independent experiments, those for D-15C are representative of two independent experiments, and those for D-15D are representative of one experiment due to limited PBMC samples.
FIG 6.

Minimal TCRαβ cross-reactivity between HLA-B*15:01-restricted NP404–414-H1N1 and NP404–414-H2N2-V408I variants. (A) Specific cell lines from donor 15B were generated by stimulation with NP404–414-H1N1(‘wt')- or NP404–414-H2N2-V408I peptides, followed by 10 days of culture in the presence of human plasma serum and subsequent restimulation with cognate peptide for detection of epitope-specific CD8+ T cells by an IFN-γ surface capture assay. Live CD3+ CD4− CD8+ IFN-γ+ T cells were single cell sorted, and TCRαβ profiles were determined by single-cell nested multiplex reverse transcription-PCR using TRAV and TRBV primers (31). (B) TRAV and TRBV usage. (C) CDR3 clonotypes.
Taken together, our functional analysis of CTL variants revealed potential CTL immune evasion toward pH2N2-1957 within HLA-A*03:01+ individuals for PB1471-L473V/N476I variant and HLA-B*15:01+ population for NP404–414-V408I epitope. The wt and likely escape variants are roughly evenly distributed among contemporary H1N1 and H3N2 seasonal vaccine strains (data not shown). Although unlikely, it cannot be excluded that the donors analyzed here were exposed to viruses containing only wt CTL peptides and, in turn, masked the absence of CTL responses toward the likely escape variants identified. Nonetheless, the loss of immunogenicity in these epitopes may have affected the CTL response capacity against pH2N2 in HLA-A*0301+ and HLA-B*1501+ individuals, which, for instance, among Caucasians comprise ∼14.4 and ∼6% of the population, respectively (45). However, these escape variants could not explain heterogeneous demographic susceptibility to pH2N2-1957. Conversely, higher conservation of CTL epitopes during the 1941-1957 period suggest that children would have generated CD8+ T cell memory to the vast majority of epitopes present in pH2N2-1957. Thus, it is highly unlikely that higher pH2N2-1957 influenza susceptibility in children was due to CTL immune escape.
Repeated influenza exposures lead to increased protection irrespective of B cell immunity.
A possible explanation for differential susceptibility to pH2N2-1957 between children and adults is that adults had “accumulated” heterosubtypic immunity as a result of multiple influenza exposures over decades (Fig. 1B). Circulation of antigenically distinct H1N1 viruses (22) associated with influenza outbreaks prior to pH2N2-1957 (20, 21) further reinforces this possibility and raises a question of how repeated influenza infections impact the protective capacity of CD8+ T cell memory (Fig. 1B). To investigate whether reduced influenza disease morbidity relates to numerical and/or phenotypic attributes of primary versus “boosted” CTL memory, we set up a triple infection experimental regimen in B-cell-deficient μMT mice (32). Naive μMT C57BL/6 mice were sequentially infected at ≥30-day intervals one, two, or three times with HKx31-H3N2 virus (HKx31) i.n. (Fig. 7A). Body weight (BW) loss in mice undergoing primary (1°), secondary (2°), or tertiary (3°) infection was recorded, and CTL memory was analyzed at day 51 after the last infection (Fig. 7).
FIG 7.

Repeated influenza infections result in reduced morbidity and increased numbers of lung memory CD8+ TRM cells. (A) Triple-boost regimen of μMT mice by sequential i.n. infection (at ≥30-day intervals) one, two, or three times with H3N2-x31 virus (103 PFU). (B) Weight loss from mice undergoing primary (1°), secondary (2°), or tertiary (3°) infection is shown for individual or grouped mice. (C) Representative FACS plots and total numbers of bulk CD8+ TRM cells at day 51 postinfection. (D and E) Total numbers of lung bulk CD8+ TRM cells were calculated according to the percentages of CD8+, CD62Llo, CD103+, and total cell counts in the lung. Data in panel B are means of n = 14 mice in the 1° infection, n = 9 mice in the 2° infection, and n = 5 mice in the 3° infection; error bars indicate the standard errors of the mean (SEM; unpaired t test; *, P < 0.05; **, P < 0.01). Upper asterisks indicate P values for the 3° infection with respect to 1° infection; lower asterisks indicate P values of 3° with respect to the 1° infection. The data in panels D and E are means of five mice for the 1° infection, four mice for the 2° infection, and five mice for the 3° infection (error bars indicate the SEM).
Our data showed decreased morbidity and enhanced recovery in mice that received two influenza infections prior to the 3° challenge (Fig. 7B). In marked contrast to primary or secondary influenza infections in which μMT mice showed significant BW loss (1° infection = 92.6% ± 10.8% [P < 0.05]; 2° infection = 89.6% ± 9% [P < 0.05] at day 11) and did not recover for >20 days, mice undergoing tertiary challenge did display any influenza-induced morbidity and had no BW loss (mean BW = 100.5% ± 5.1% at day 11) (Fig. 7B). Although recovery from primary influenza challenge in intact C57BL/6 mice takes ∼10 days (46), influenza disease recovery was significantly delayed by an average of 30days in the primary but not the tertiary mice (Fig. 7B), highlighting CTL protection offered during the tertiary challenge.
Furthermore, as demonstrated in several recent reports (47–49), CD8+ TRM cells have an important contribution to protection from viral challenge and are necessary for inducing protective heterosubtypic immunity against influenza (50). Notably, multiple sequential skin infections with vaccinia virus (VACV) results in an accumulation at CD8+ TRM cells that can populate the entire skin surface and mediate enhanced viral protection (51).
Therefore, we tested the correlation of CD8+ TRM compartmentalization with repeated antigen experience and the presence of association with a superior influenza control. In particular, we enumerated lung, “bulk” CD8+ TRM cells in mice receiving consecutive infections at resting memory phase (day 51 after infection) (Fig. 7A). CD8+ TRM cells were characterized by the expression of the early activation marker (CD69), either alone or in conjunction with the α-chain of the αEβ7 integrin (CD103), with both phenotypes defining CD8+ TRM cells (52–54). Remarkably, we found that the frequency of CD8+ TRM cells increased with repetitive antigen experience (Fig. 7C). The mean numbers of CD8+ CD62Llo CD69+ CD103+ T cells during the 1° memory were 1.39 × 104 ± 1.27 × 104, increased by 2-fold to 2.73 × 104 ± 0.96 × 104 during the 2° memory, and then further increased by 3-fold during the 3° memory, with a mean of 7.5 × 104 ± 5.1 × 104 (Fig. 7D). With mounting evidence suggesting that CD69 is a requirement for tissue retention (54), it is notable that a large proportion of CD8+ CD62Llo T cells in the lung were CD69+ CD103− and those cells increased with the sequential boosting (mean total numbers in 1° memory of 3.6 × 105 ± 0.8 × 105 versus 2° memory of 4.2 × 105 ± 1 × 105 versus 3° memory of 4.6 × 105 ± 5.1 × 104; Fig. 7E). Together, these data are indicative of numeric differences in CD8+ TRM cells generated by multiple infections and add to the notion that the infection history (the number of influenza exposures) results in superior protective capacity of CTL memory against influenza viruses. In respiratory infections such as influenza viruses, this may be associated with the enrichment of lung CD8+ TRM cells.
DISCUSSION
The main findings of our study are as follows: (i) the majority of the known CTL antigenic regions in immunodominant influenza proteins remained largely conserved across 1918 to 1957, including the two major pH1N1-1918 and pH2N2-1957 pandemics; (ii) conserved CTL Ag-p with pH2N2-1957 were substantially higher for influenza viruses circulating immediately prior 1957, indicating that differential susceptibility is not due to age-specific exposure to influenza viruses with distinct CTL peptides; and (iii) demographic susceptibility might be more closely linked to the number of influenza exposures. Our data show that repeated infections with influenza viruses increased numbers of protective CD8+ TRM cells in the lung, which in turn could have influenced pH2N2-1957 demographic susceptibility, with adults having higher numbers of infections than children.
Although CTL escape variants are a hallmark of chronic viral infections (e.g., chronic HIV or hepatitis) (55–57), we find that the CTL antigenic regions within influenza viruses causing acute respiratory disease remain largely conserved across a 40-year period. Our findings support previous models of emergence/fixation of influenza escape variants during the conditions of selective advantage (longer persistence), as well as specific environmental (long virus incubation periods between influenza seasons) and stochastic dynamics (58). This conservation might indicate that influenza-specific CTLs target internal segments necessary for viral replication/function and thus limit viral fitness, which is the case for the highly conserved and prominent HLA-A*0201-restricted M158 epitope (18, 29, 59). The seasonality of influenza epidemics and reduced duration of illness and of the contagious period would decrease the opportunity for selection and transmission of replicatively fit escape variants capable of persisting in an enormously diverse HLA population.
Nonetheless, we found viral variants within A*03:01-restricted PB1471 and B*15:01-restricted NP404 epitopes associated with immune evasion, a finding consistent with previous observations of drift and fixation of CTL escape in influenza-specific B35-NP418 and B27-NP383 epitopes (39–42). The extent of potential CTL escape is in line with a recent study comparing the rate of evolution in the NP proteins across human and swine influenza viruses (43). That study demonstrated that CTL-mediated selective pressure drives the emergence of mutations in CTL Ag-p. Interestingly, the authors reported that variants emerged with selective advantage and thus could fixate at the population level (43). Importantly, some of the identified mutations were previously shown to negatively impact CTL-mediated immunity (43).
One limitation in linking CTL immunity to past pandemics is the fact that global influenza surveillance only became intensive in recent years. A further complication in the influenza field is the lack of understanding of CTL immunodominance, the breadth of the response, and the extent of epitope discovery, which have been limited to a small number of HLA types. Therefore, it remains possible that our results on conservation and extent of immune escape may only partially represent the full extent of immune evasion that could have played a role in susceptibility to pH2N2-1957.
Our results on the role of CTL immunity in determining pandemic susceptibility most closely circumscribe in the context H2N2 pandemic of 1957 and in the unique household study, with laboratory-confirmed influenza before and after the pandemic, demonstrating heterogeneous demographic influenza susceptibility (13). Influenza activity the 1918-1956 period spiked in several outbreaks, i.e., the 1928-1929, 1932-1933, 1936-1937, 1943-1944, 1947, and 1950-1951 outbreaks (60, 61), with the latter two outbreaks being remarkably severe. Early evidence suggests homogenous demographic attack rates during the 1968 H3N2 “Hong Kong” pandemic (pH3N2-1968) (62); however, wide geographical differences in pH3N2 severity have been reported (63). Thus, the epidemiology of pH3N2-1968 virus remains to some extent controversial (64). In contrast, low susceptibility was observed in the elderly during the 2009 H1N1 pandemic (65), a finding partially explained by the presence of preexisting cross-reactive CD8+ T cells (66) and nAbs (65), the latter elicited by prior exposure to seasonal H1N1 virus. Furthermore, it has been observed that infection history with exposure to seasonal H1N1 strains circulating during the 1983-1996 period, but not in other years, prompted antibody responses against a homologous HA epitope present in pH1N1-2009 (67). Thus, it is possible that the adult population might have acquired more broadly reactive antibodies than children, which could have contributed to protective immunity against H2N2 pandemic strain.
Historically, susceptibility to influenza has been disproportionate in children (68). Although naïveté of the adaptive immune component or failure to mount an appropriately coordinated T cell response (69) are possible explanations, our data suggest the importance of the size and quality of the preestablished CTL memory.
Early evidence from the murine models suggested that increasing the magnitude of CTL memory recall responses by double priming with antigenically distinct influenza viruses resulted in unprecedented protection from lethal H7N7 challenge (70). Our observation that experienced CD8+ T cell memory is more protective than 1° memory is consistent with Listeria monocytogenes, lymphocytic choriomeningitis virus (LCMV-Armstrong), and VACV models (71, 72). The protective capacity of 2° and 3° CD8+ T cell memory is a result of both increased CTL numbers (72) and their qualitative characteristics (71, 73). The qualitative protective features of secondary CD8+ T cell memory include retention of effector functions such as potent cytokine production and cytolytic capacity characterized by a distinct transcriptional signature (72, 74), enhanced migratory potential and localization to extralymphoid tissue such as the lungs (71, 72), or enhanced ability to rapidly migrate into the site of pathogen penetration (73). Furthermore, CD8+ TRM cells have been implicated as a major component mediating cross-strain protective immunity against influenza (50).
In the context of VACV infection of the skin, sequential infections resulted in an accumulation of CD8+ TRM cells that could populate the entire surface of the skin, resulting in enhanced protection upon VACV reinfection (51). This prompted us to investigate how sequential influenza infections, in the absence of any B-cell-mediated immunity, shape the CD8+ TRM compartment. To the best of our knowledge, these are the first data showing a positive association between repeated prior influenza infections, an increase in the number of CD8+ TRM cells, and reduced influenza-induced morbidity upon new challenge in the absence of antibodies in μMT mice.
Unexpectedly, we found no significant difference in the body weight loss between the mice undergoing primary and secondary infections. The reasons for this are unknown; however, a possible explanation could involve T-cell-mediated immunopathology (75). It is still possible that the secondarily challenged μMT mice had accelerated viral clearance. To the best of our knowledge, this is the first time the memory CD8+ T cell responses and body weight loss have been assessed following secondary and tertiary H3N2-HKx31 influenza virus infection using μMT mice.
Our results add to the notion that infection history appears to have a major impact in the quantity and quality of preestablished CTL memory, with multiple infections driving the accumulation of protective CD8+ TRM cells that could mediate protection during influenza rechallenge. Therefore, our results indicate that it might be highly feasible to recall heterologous CTL memory in the population by a CTL vaccine or a vaccine component. The optimal regimen for such a vaccine should include both priming and sequential booster phases to ensure establishment of CD8+ TRM cells and protective efficacy. In line with this idea, recent studies demonstrated that mucosal vaccination induces protective TRM cells that are highly efficient in controlling Chlamydia trachomatis infection (76). Further studies on mucosal influenza vaccine delivery systems are warranted.
ACKNOWLEDGMENTS
We acknowledge the animal house staff from the WEHI as well as Azad Rihampour for technical assistance and Linda Wakim for helpful discussions.
E. Bridie Clemens is an Australian National Health and Medical Research Council (NHMRC) Peter Doherty Fellow. Katherine Kedzierska and Jodie McVernon are NHMRC Career Development Fellows, level 2.
Funding Statement
This work was supported by Australian National Health and Medical Research Council (NHMRC) project grant GNT1008854 and program grant GNT1071916 to Katherine Kedzierska. Sergio M. Quiñones-Parra was supported by a University of Melbourne International Research Scholarship and was a CONACyT scholar. Dhanasekaran Vijaykrishna is supported by the Duke-NUS Signature Research Program funded by the Agency of Science, Technology and Research, Singapore, and the Ministry of Health, Singapore, and by contract HHSN272201400006C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, USA.
REFERENCES
- 1.World Health Organization. 2011. Global health observatory data repository. World Health Organization, Geneva, Switzerland: http://apps.who.int/ghodata/?vid=10012. [Google Scholar]
- 2.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the United States: measuring disease burden and costs. Vaccine 25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 3.Uyeki TM, Cox NJ. 2013. Global concerns regarding novel influenza A (H7N9) virus infections. N Engl J Med 368:1862–1864. doi: 10.1056/NEJMp1304661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kilbourne ED, Smith C, Brett I, Pokorny BA, Johansson B, Cox N. 2002. The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post-World War II epidemic. Proc Natl Acad Sci U S A 99:10748–10752. doi: 10.1073/pnas.162366899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rimmelzwaan GF, Kreijtz JH, Bodewes R, Fouchier RA, Osterhaus AD. 2009. Influenza virus CTL epitopes, remarkably conserved and remarkably variable. Vaccine 27:6363–6365. doi: 10.1016/j.vaccine.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Assarsson E, Bui HH, Sidney J, Zhang Q, Glenn J, Oseroff C, Mbawuike IN, Alexander J, Newman MJ, Grey H, Sette A. 2008. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J Virol 82:12241–12251. doi: 10.1128/JVI.01563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boon AC, de Mutsert G, van Baarle D, Smith DJ, Lapedes AS, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF. 2004. Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J Immunol 172:2453–2460. doi: 10.4049/jimmunol.172.4.2453. [DOI] [PubMed] [Google Scholar]
- 8.Lee LY, Ha do, Simmons LAC, de Jong MD, Chau NV, Schumacher R, Peng YC, McMichael AJ, Farrar JJ, Smith GL, Townsend AR, Askonas BA, Rowland-Jones S, Dong T. 2008. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Invest 118:3478–3490. doi: 10.1172/JCI32460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreijtz JH, de Mutsert G, van Baalen CA, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2008. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T lymphocyte populations directed to human influenza A virus. J Virol 82:5161–5166. doi: 10.1128/JVI.02694-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Sandt CE, Kreijtz JH, de Mutsert G, Geelhoed-Mieras MM, Hillaire ML, Vogelzang-van Trierum SE, Osterhaus AD, Fouchier RA, Rimmelzwaan GF. 2014. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. J Virol 88:1684–1693. doi: 10.1128/JVI.02843-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap KL, Ada GL, McKenzie IFC. 1978. Transfer of specific cytotoxic T lymphocytes protects mice inoculated with influenza virus. Nature 273:238–239. doi: 10.1038/273238a0. [DOI] [PubMed] [Google Scholar]
- 12.Bender BS, Croghan T, Zhang L, Small PA Jr. 1992. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 175:1143–1145. doi: 10.1084/jem.175.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Epstein SL, Lo CY, Misplon JA, Bennink JR. 1998. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol 160:322–327. [PubMed] [Google Scholar]
- 14.McMichael AJ, Gotch FM, Noble GR, Beare PA. 1983. Cytotoxic T-cell immunity to influenza. N Engl J Med 309:13–17. doi: 10.1056/NEJM198307073090103. [DOI] [PubMed] [Google Scholar]
- 15.Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, Bean T, Barclay W, Deeks JJ, Lalvani A. 2013. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 19:1305–1312. doi: 10.1038/nm.3350. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Wan Y, Qiu C, Quinones-Parra S, Zhu Z, Loh L, Tian D, Ren Y, Hu Y, Zhang X, Thomas PG, Inouye M, Doherty PC, Kedzierska K, Xu J. 2015. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat Commun 6:6833. doi: 10.1038/ncomms7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayward AC, Wang L, Goonetilleke N, Fragaszy EB, Bermingham A, Copas A, Dukes O, Millett ER, Nazareth I, Nguyen-Van-Tam JS, Watson JM, Zambon M, Johnson AM, McMichael AJ, Flu Watch G. 2015. Natural T cell-mediated protection against seasonal and pandemic influenza: results of the Flu Watch Cohort Study. Am J Respir Crit Care Med 191:1422–1431. doi: 10.1164/rccm.201411-1988OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Sandt CE, Kreijtz JH, Geelhoed-Mieras MM, Nieuwkoop NJ, Spronken MI, van de Vijver DA, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2016. Differential recognition of influenza A viruses by M158-66 epitope-specific CD8+ T cells is determined by extraepitopic amino acid residues. J Virol 90:1009–1022. doi: 10.1128/JVI.02439-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epstein SL. 2006. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: an experiment of nature. J Infect Dis 193:49–53. doi: 10.1086/498980. [DOI] [PubMed] [Google Scholar]
- 20.Rasmussen AF Jr, Stokes JC, Smadel JE. 1948. The Army experience with influenza, 1946-1947: laboratory aspects. Am J Hyg 47:142–149. [DOI] [PubMed] [Google Scholar]
- 21.Isaacs A, Gledhill AW, Andrewes CH. 1952. Influenza A viruses; laboratory studies, with special reference to European outbreak of 1950-1. Bull World Health Organ 6:287–315. [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson MI, Viboud C, Simonsen L, Bennett RT, Griesemer SB, St George K, Taylor J, Spiro DJ, Sengamalay NA, Ghedin E, Taubenberger JK, Holmes EC. 2008. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 4:e1000012. doi: 10.1371/journal.ppat.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joseph U, Linster M, Suzuki Y, Krauss S, Halpin RA, Vijaykrishna D, Fabrizio TP, Bestebroer TM, Maurer-Stroh S, Webby RJ, Wentworth DE, Fouchier RA, Bahl J. 2015. Adaptation of pandemic H2N2 influenza A viruses in humans. J Virol 89:2442–2447. doi: 10.1128/JVI.02590-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webster RG. 1997. Predictions for future human influenza pandemics. J Infect Dis 176(Suppl 1):S14–S19. doi: 10.1086/514168. [DOI] [PubMed] [Google Scholar]
- 25.Schafer J, Khristova ML, Busse TL, Sinnecker R, Kharitonenkov IG, Schrader C, Suss J, Bucher D. 1992. Analysis of internal proteins of influenza A (H2N2) viruses isolated from birds in East Germany in 1983. Acta Virol 36:113–120. [PubMed] [Google Scholar]
- 26.Makarova NV, Kaverin NV, Krauss S, Senne D, Webster RG. 1999. Transmission of Eurasian avian H2 influenza virus to shorebirds in North America. J Gen Virol 80(Pt 12):3167–3171. doi: 10.1099/0022-1317-80-12-3167. [DOI] [PubMed] [Google Scholar]
- 27.Jones JC, Baranovich T, Marathe BM, Danner AF, Seiler JP, Franks J, Govorkova EA, Krauss S, Webster RG. 2014. Risk assessment of H2N2 influenza viruses from the avian reservoir. J Virol 88:1175–1188. doi: 10.1128/JVI.02526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinones-Parra S, Loh L, Brown LE, Kedzierska K, Valkenburg SA. 2014. Universal immunity to influenza must outwit immune evasion. Front Microbiol 5:285. doi: 10.3389/fmicb.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, Miller A, Doherty PC, Vijaykrishna D, Rossjohn J, Gras S, Kedzierska K. 2014. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci U S A 111:1049–1054. doi: 10.1073/pnas.1322229111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen TH, Rowntree LC, Pellicci DG, Bird NL, Handel A, Kjer-Nielsen L, Kedzierska K, Kotsimbos TC, Mifsud NA. 2014. Recognition of distinct cross-reactive virus-specific CD8+ T cells reveals a unique TCR signature in a clinical setting. J Immunol 192:5039–5049. doi: 10.4049/jimmunol.1303147. [DOI] [PubMed] [Google Scholar]
- 31.Wang GC, Dash P, McCullers JA, Doherty PC, Thomas PG. 2012. T cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci Transl Med 4:128ra142. doi: 10.1126/scitranslmed.3003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitamura D, Roes J, Kuhn R, Rajewsky K. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 33.Wakim LM, Gupta N, Mintern JD, Villadangos JA. 2013. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol 14:238–245. doi: 10.1038/ni.2525. [DOI] [PubMed] [Google Scholar]
- 34.Wu C, Zanker D, Valkenburg S, Tan B, Kedzierska K, Zou QM, Doherty PC, Chen W. 2011. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc Natl Acad Sci U S A 108:9178–9183. doi: 10.1073/pnas.1105624108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan Y, Vijaykrishna D, Bahl J, Zhu H, Wang J, Smith GJ. 2010. The emergence of pandemic influenza viruses. Protein Cell 1:9–13. doi: 10.1007/s13238-010-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant E, Wu C, Chan KF, Eckle S, Bharadwaj M, Zou QM, Kedzierska K, Chen W. 2013. Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol Cell Biol 91:184–194. doi: 10.1038/icb.2012.78. [DOI] [PubMed] [Google Scholar]
- 37.Smith GJ, Bahl J, Vijaykrishna D, Zhang J, Poon LL, Chen H, Webster RG, Peiris JS, Guan Y. 2009. Dating the emergence of pandemic influenza viruses. Proc Natl Acad Sci U S A 106:11709–11712. doi: 10.1073/pnas.0904991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlson JM, Le AQ, Shahid A, Brumme ZL. 2015. HIV-1 adaptation to HLA: a window into virus-host immune interactions. Trends Microbiol 23:212–224. doi: 10.1016/j.tim.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2000. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J Virol 74:6800–6807. doi: 10.1128/JVI.74.15.6800-6807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boon AC, de Mutsert G, Graus YM, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF. 2002. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J Virol 76:2567–2572. doi: 10.1128/jvi.76.5.2567-2572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berkhoff EG, Geelhoed-Mieras MM, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2007. Assessment of the extent of variation in influenza A virus cytotoxic T-lymphocyte epitopes by using virus-specific CD8+ T-cell clones. J Gen Virol 88:530–535. doi: 10.1099/vir.0.82120-0. [DOI] [PubMed] [Google Scholar]
- 42.Rimmelzwaan GF, Boon AC, Voeten JT, Berkhoff EG, Fouchier RA, Osterhaus AD. 2004. Sequence variation in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. Virus Res 103:97–100. doi: 10.1016/j.virusres.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 43.Machkovech HM, Bedford T, Suchard MA, Bloom JD. 2015. Positive selection in CD8+ T-cell epitopes of influenza virus nucleoprotein revealed by a comparative analysis of human and swine viral lineages. J Virol 89:11275–11283. doi: 10.1128/JVI.01571-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valkenburg SA, Quinones-Parra S, Gras S, Komadina N, McVernon J, Wang Z, Halim H, Iannello P, Cole C, Laurie K, Kelso A, Rossjohn J, Doherty PC, Turner SJ, Kedzierska K. 2013. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat Commun 4:2663. doi: 10.1038/ncomms3663. [DOI] [PubMed] [Google Scholar]
- 45.National Center for Biotechnology Information. 2016. dbMHC: diversity anthropology component. National Center for Biotechnology Information, Bethesda, MD: http://www.ncbi.nlm.nih.gov/projects/gv/mhc/main.fcgi?cmd=init. [Google Scholar]
- 46.Bird NL, Olson MR, Hurt AC, Oshansky CM, Oh DY, Reading PC, Chua BY, Sun Y, Tang L, Handel A, Jackson DC, Turner SJ, Thomas PG, Kedzierska K. 2015. Oseltamivir prophylaxis reduces inflammation and facilitates establishment of cross-strain protective T cell memory to influenza viruses. PLoS One 10:e0129768. doi: 10.1371/journal.pone.0129768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iijima N, Iwasaki A. 2014. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346:93–98. doi: 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN. 2014. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346:101–105. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- 49.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. 2014. T cell memory: resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, Cauley LS. 2014. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol 95:215–224. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. 2012. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T. 2013. The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat Immunol 14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 53.Bergsbaken T, Bevan MJ. 2015. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol 16:406–414. doi: 10.1038/ni.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park CO, Kupper TS. 2015. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel R. 1990. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature 346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 56.Moore C, John M, James I, Christiansen F, Witt C, Mallal S. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 57.Fernandez C, Stratov I, De Rose R, Walsh K, Dale C, Smith M, Agy M, Hu S, Krebs K, Watkins D, O'Connor D, Davenport M, Kent S. 2005. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J Virol 79:5721–5731. doi: 10.1128/JVI.79.9.5721-5731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gog J, Rimmelzwaan G, Osterhaus A, Grenfell B. 2003. Population dynamics of rapid fixation in cytotoxic T lymphocyte escape mutants of influenza A. Proc Natl Acad Sci U S A 100:11143–11147. doi: 10.1073/pnas.1830296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terajima M, Ennis FA. 2012. Nuclear export signal and immunodominant CD8+ T cell epitope in influenza A virus matrix protein 1. J Virol 86:10258–10260. doi: 10.1128/JVI.00894-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Logan WP, Mac KD. 1951. Development of influenza epidemics. Lancet i:264–265. [PubMed] [Google Scholar]
- 61.Viboud C, Tam T, Fleming D, Miller MA, Simonsen L. 2006. 1951 influenza epidemic, England and Wales, Canada, and the United States. Emerg Infect Dis 12:661–668. doi: 10.3201/eid1204.050695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glezen WP. 1996. Emerging infections: pandemic influenza. Epidemiol Rev 18:64–76. doi: 10.1093/oxfordjournals.epirev.a017917. [DOI] [PubMed] [Google Scholar]
- 63.Viboud C, Grais RF, Lafont BA, Miller MA, Simonsen L. 2005. Multinational impact of the 1968 Hong Kong influenza pandemic: evidence for a smoldering pandemic. J Infect Dis 192:233–248. doi: 10.1086/431150. [DOI] [PubMed] [Google Scholar]
- 64.Reichert TA, Christensen RA. 2005. It's not about smoldering or neuraminidase: there were 2 variants of the A(H3N2) pandemic virus differing in internal genes. J Infect Dis 192:1858–1862. doi: 10.1086/497153. [DOI] [PubMed] [Google Scholar]
- 65.Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H, Liu F, Dong L, DeVos JR, Gargiullo PM, Brammer TL, Cox NJ, Tumpey TM, Katz JM. 2009. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 361:1945–1952. doi: 10.1056/NEJMoa0906453. [DOI] [PubMed] [Google Scholar]
- 66.Gras S, Kedzierski L, Valkenburg SA, Laurie K, Liu YC, Denholm JT, Richards MJ, Rimmelzwaan GF, Kelso A, Doherty PC, Turner SJ, Rossjohn J, Kedzierska K. 2010. Cross-reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc Natl Acad Sci U S A 107:12599–12604. doi: 10.1073/pnas.1007270107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Myers JL, Bostick DL, Sullivan CB, Madara J, Linderman SL, Liu Q, Carter DM, Wrammert J, Esposito S, Principi N, Plotkin JB, Ross TM, Ahmed R, Wilson PC, Hensley SE. 2013. Immune history shapes specificity of pandemic H1N1 influenza antibody responses. J Exp Med 210:1493–1500. doi: 10.1084/jem.20130212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poehling KA, Edwards KM, Weinberg GA, Szilagyi P, Staat MA, Iwane MK, Bridges CB, Grijalva CG, Zhu Y, Bernstein DI, Herrera G, Erdman D, Hall CB, Seither R, Griffin MR. 2006. The underrecognized burden of influenza in young children. N Engl J Med 355:31–40. doi: 10.1056/NEJMoa054869. [DOI] [PubMed] [Google Scholar]
- 69.You D, Ripple M, Balakrishna S, Troxclair D, Sandquist D, Ding L, Ahlert TA, Cormier SA. 2008. Inchoate CD8+ T cell responses in neonatal mice permit influenza-induced persistent pulmonary dysfunction. J Immunol 181:3486–3494. doi: 10.4049/jimmunol.181.5.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Christensen JP, Doherty PC, Branum KC, Riberdy JM. 2000. Profound protection against respiratory challenge with a lethal H7N7 influenza A virus by increasing the magnitude of CD8+ T-cell memory. J Virol 74:11690–11696. doi: 10.1128/JVI.74.24.11690-11696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nolz JC, Harty JT. 2011. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity 34:781–793. doi: 10.1016/j.immuni.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Masopust D, Ha SJ, Vezys V, Ahmed R. 2006. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol 177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 73.Slutter B, Pewe LL, Kaech SM, Harty JT. 2013. Lung airway-surveilling CXCR3hi memory CD8+ T cells are critical for protection against influenza A virus. Immunity 39:939–948. doi: 10.1016/j.immuni.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. 2010. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity 33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.La Gruta NL, Kedzierska K, Stambas J, Doherty PC. 2007. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85:85–92. doi: 10.1038/sj.icb.7100026. [DOI] [PubMed] [Google Scholar]
- 76.Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi J, Yethon JA, Farokhzad OC, Langer R, Starnbach MN, von Andrian UH. 2015. Vaccines: a mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]