

ABSTRACT
With next-generation sequencing technologies, it is now feasible to efficiently sequence patient-derived virus populations at a depth of coverage sufficient to detect rare variants. However, each sequencing platform has characteristic error profiles, and sample collection, target amplification, and library preparation are additional processes whereby errors are introduced and propagated. Many studies account for these errors by using ad hoc quality thresholds and/or previously published statistical algorithms. Despite common usage, the majority of these approaches have not been validated under conditions that characterize many studies of intrahost diversity. Here, we use defined populations of influenza virus to mimic the diversity and titer typically found in patient-derived samples. We identified single-nucleotide variants using two commonly employed variant callers, DeepSNV and LoFreq. We found that the accuracy of these variant callers was lower than expected and exquisitely sensitive to the input titer. Small reductions in specificity had a significant impact on the number of minority variants identified and subsequent measures of diversity. We were able to increase the specificity of DeepSNV to >99.95% by applying an empirically validated set of quality thresholds. When applied to a set of influenza virus samples from a household-based cohort study, these changes resulted in a 10-fold reduction in measurements of viral diversity. We have made our sequence data and analysis code available so that others may improve on our work and use our data set to benchmark their own bioinformatics pipelines. Our work demonstrates that inadequate quality control and validation can lead to significant overestimation of intrahost diversity.
IMPORTANCE Advances in sequencing technology have made it feasible to sequence patient-derived viral samples at a level sufficient for detection of rare mutations. These high-throughput, cost-effective methods are revolutionizing the study of within-host viral diversity. However, the techniques are error prone, and the methods commonly used to control for these errors have not been validated under the conditions that characterize patient-derived samples. Here, we show that these conditions affect measurements of viral diversity. We found that the accuracy of previously benchmarked analysis pipelines was greatly reduced under patient-derived conditions. By carefully validating our sequencing analysis using known control samples, we were able to identify biases in our method and to improve our accuracy to acceptable levels. Application of our modified pipeline to a set of influenza virus samples from a cohort study provided a realistic picture of intrahost diversity and suggested the need for rigorous quality control in such studies.
INTRODUCTION
Many viral pathogens are thought to exist as a cloud of closely related mutants within an infected individual (1). Until recently, our understanding of intrahost viral dynamics and the impact of viral diversity on evolution and pathogenesis has been limited by low-throughput sequencing methods. However, with the advent of next-generation sequencing (NGS), it is now feasible to sequence patient-derived samples at sufficient read depth to detect rare single-nucleotide variants (SNV). There has been an explosion of studies that employ NGS to quantify viral diversity within and between hosts (2–13). Although NGS produces the large quantities of sequence data needed to detect rare variants, the process is error prone (14–16), and many bioinformatics tools do not explicitly address the challenges inherent in studies of patient-derived viral populations.
A number of sample preparation protocols have been developed to control for the errors in NGS-based studies of virus populations, but each approach has its own caveats that ultimately limit its application. Cirseq is an ingenious technique in which template RNA is sheared and circularized prior to reverse transcription (RT) (17, 18). Subsequent rolling-circle cDNA synthesis produces tandem reads, generating a consensus sequence for each RNA fragment. While the method is likely to be highly sensitive for rare-variant detection and can control for reverse transcription, PCR, and sequencing errors, the requirement for a large and relatively pure population of viral RNA limits its applicability to patient-derived samples (17, 18). “Primer ID” methods require less input and target sequencing to the viral genome. This approach relies on barcoded primers to construct consensus sequences for each cDNA template and can control for PCR and sequencing errors (19, 20). Because Primer ID methods require that each bar code be physically attached to a PCR product, they are most easily applied to small, targeted regions of the genome. As such, they have limited application in whole-genome sequencing.
Sequence-independent single-primer amplification (SISPA) is an alternative approach that allows whole-genome sequencing and controls for errors propagated during library preparation (21). In this method, RT-PCR products are sheared and tagged with bar-coded random primers in a Klenow reaction prior to library preparation. SISPA controls for any errors that may arise during library amplification, including PCR biases. The method has been used in conjunction with statistical algorithms to control for accuracy in studies of intrahost influenza virus diversity (5, 22, 23). However, the bar-coding reaction used in SISPA can be biased in unpredictable ways, resulting in uneven coverage and sensitivity across the genome (24).
Statistical algorithms have also been developed to distinguish true variants from sequencing errors (25–34). These methods rely solely on sequencing data and are more easily applied to whole-genome sequencing. In general, variant-calling algorithms calculate base-specific error rates using various metrics, including mapping quality (MapQ), base quality (Phred), strand bias, and sequence context. True variants are identified as those with frequencies exceeding the expected error rate according to some predetermined statistical test. Despite being employed in many NGS-based studies of viral diversity (3, 4, 12, 35, 36), few of these algorithms have been benchmarked using defined viral populations. To our knowledge, none have been tested under conditions that mimic those found in patient-derived samples. The accuracy of such algorithms in the context of NGS studies of patient-derived viral populations is largely unknown.
Here, we use genetically defined populations of influenza A virus with variable input titers to determine the accuracy of rare-variant detection in patient-derived samples. We highlight the challenges that accompany NGS-based studies of viral diversity and include a means for improving accuracy. This work exemplifies the controls that should be run prior to any NGS-based study of viral populations and provides a comprehensive data set for benchmarking other pipelines.
MATERIALS AND METHODS
Viruses and cells.
Madin-Darby canine kidney cells were provided by Arnold S. Monto (University of Michigan School of Public Health) and were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) with 10% fetal bovine serum (Gibco and HyClone), 25 mM HEPES (Invitrogen), and 0.1875% bovine serum albumin (Life Technologies). Influenza A/WSN/33(H1N1) virus was rescued from transfected cells using an 8-plasmid reverse genetic system containing each genomic segment (pHW181 to -188), a kind gift from Robert Webster (St. Jude's Children's Research Hospital) (37, 38). A biological clone of influenza A/Puerto Rico/8/1934(H1N1) virus was obtained from the ATCC (VR-1469), and the genomic segments were cloned into the pHW2000 reverse-genetic system (38). The sequences of these clones were verified using Sanger sequencing.
Patient-derived samples of influenza A virus were collected as part of the Household Influenza Vaccine Effectiveness (HIVE) study (39, 40) and kindly provided by Arnold S. Monto and colleagues at the University of Michigan School of Public Health. The HIVE study was approved by the Institutional Review Board at the University of Michigan, and all subjects provided informed consent.
Viral populations.
We extracted viral RNA from infected supernatants using QIAamp viral RNA kits (Qiagen) and generated cDNA using Superscript III one step with HiFi platinum Taq (Invitrogen). PCR products were purified using the GeneJet PCR purification kit (ThermoFisher Scientific) according to the manufacturer's instructions.
PR8-WSN33 population.
For the experiment on the accuracy of DeepSNV (see Fig. 2), WSN33 and PR8 viruses were plaque purified and passaged three times in MDCK cells. We then verified the sequences of these viruses by Sanger sequencing. Two microliters of RNA template was used to generate cDNA in eight segment-specific one-step RT-PCRs with 0.2 μM the following primers: PB2-Forward-JT (5′-GCAGGTCAATTATATTCAATATGGAAA-3′), PB2-Reverse-JT (5′-CAAGGTCGTTTTTAAACTATTCGACAC-3′), PB1-Forward-JT (5′-GCAGGCAAACCATTTGAATGG-3′), PB1-Reverse-JT (5′-CAAGGCATTTTTTCATGAAGGACAAG-3′), PA-Forward-JT (5′-GCAGGTACTGATTCAAAATGGAAG-3′), PA-Reverse-JT (5′-CAAGGTACTTTTTTGGACAGTATGG-3′), NA-Forward-JT (5′-(GCAGGAGTTTAAATGAATCCAAACC-3′), NA-Reverse-JT (5′-CAATTG-3′), HA-Forward-JT (5′-GCAGGGGAAAATAAAAACAACCAAAAT-3′), HA-Reverse-JT (5′-CAAGGGTGTTTTTCCTTATATTTCTGAA-3′), NP-Forward-JT (5′-GCAGGGTAGATAATCACTCACAG-3′), NP-Reverse-JT (5′-CAAGGGTATTTTTCTTTAATTGTCGTACT-3′), M-Forward-JT (5′-GCAGGTAGATATTGAAAGATGAGTC-3′), M-Reverse-JT (5′-CAAGGTAGTTTTTTACTCCAGCTCT-3′), NS-Forward-JT (5′-GCAGGGTGACAAAGACATAATG-3′), and NS-Reverse-JT (5′-CAAAGGGTGTTTTTTATTATTAAATAAGCTG-3′). The reaction conditions were 50°C (60 min) and 94°C (2 min), followed by 30 cycles of 94°C (30 s), 54°C (30 s), and 68°C (3 min). Molar equivalents of each PCR product were pooled to generate reconstituted cDNA genomes of both WSN33 and PR8. The WSN33 cDNA pool was then serially diluted in the PR8 cDNA pool to yield WSN33-PR8 mixtures in which WSN33 made up 5, 2.5, 1.25, 0.63, and 0.16% of the population. Seven hundred and fifty nanograms of each mixture was sheared to an average size of 300 to 400 bp using a Covaris S220 focused ultrasonicator with the following settings: intensity, 4; duty cycle, 10%; bursts per second, 200; duration, 80 s. Sequencing libraries were prepared from these fragmented products using the NEBNext Ultra DNA library prep kit (NEB), Agencourt AMPure XP beads (Beckman Coulter), and NEBNext multiplex oligonucleotides for Illumina (NEB). The pooled libraries were sequenced on an Illumina MiSeq machine with 2 × 250 paired-end reads. A clonal plasmid control library was prepared from 8 plasmids containing PR8 genomic segments. The plasmids were mixed to equal molarity, and cDNA was generated using a multiplex one-step RT-PCR with the primers Uni12/Inf1 (5′-GGGGGGAGCAAAAGCAGG-3′), Uni12/Inf3 (5′-GGGGGAGCGAAAGCAGG-3′), and Uni13/Inf1 (5′-5CGGGTTATTAGTAGAAACAAGG-3′) as described previously (41, 42). The library was prepared in identical fashion to the experimental populations and was sequenced in the same MiSeq lane.
FIG 2.
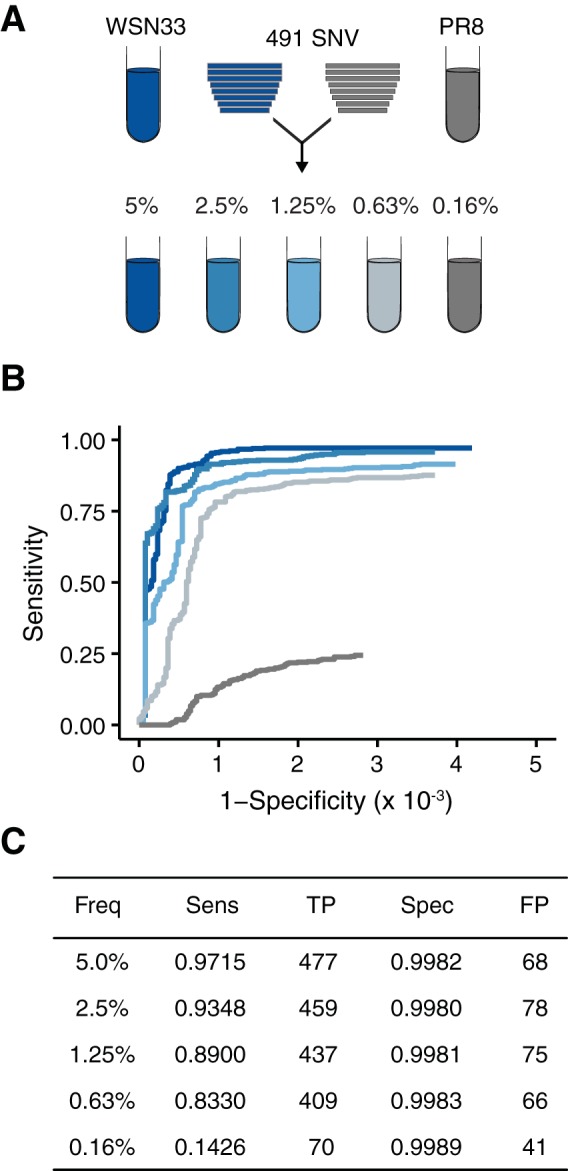
Accuracy of DeepSNV. (A) Reconstituted cDNA genomes of influenza virus strain WSN33 were diluted serially in reconstituted cDNA genomes of PR8, generating artificial populations with 491 single-nucleotide variants from WSN (relative to PR8) at the indicated frequencies. (B) ROC curve measuring the accuracy of DeepSNV in identifying WSN33 variants mixed with PR8 at the indicated (by colors matching those in panel A) frequencies. (C) Summary of the data in panel B at a P value threshold of 0.01. Freq, frequency; Sens, sensitivity; TP, true positives; FP, false positives.
Experimental intrahost population.
Twenty point mutants were generated in the WSN33 background using the pHW2000 reverse-genetics system (37; Elisa Visher, Shawn Whitefield, John T. McCrone, William Fitzsimmons, and Adam S. Lauring, unpublished data). In short, we used overlap PCR mutagenesis to introduce the following mutations: HA, T1583G; HA, G1006T; HA, G542T; M, T861G; M, A541C; NA, G1168T; NA, C454T; NP, A454C; NP, A1160T; NS, G227T; NS, A809G; PA, T964G; PA, T237A; PA, A1358T; PB1, G599A; PB1, G1764T; PB1, T1288A; PB2, A1854G; PB2, A440T; and PB2, A1167T. Viruses were rescued from transfected cells as described previously (38).
We passaged the 20 WSN33 point mutants and the WSN33 wild type (WT) once in MDCK cells and verified the identities of the mutants by sequencing each on an Illumina MiSeq as described above. We quantified the genome copy number of each supernatant using a SuperScript III Platinum One-Step RT-quantitative PCR (qPCR) kit (Invitrogen) and universal influenza A/B virus primer and probe sets (43). Equal genome equivalents of each infected supernatant were mixed and diluted to generate a population containing each of the 20 mutants present at 5% frequency and a total concentration of 105 genomes per microliter. We diluted this mixture in WT WSN33 supernatant to create populations in which each mutant was present at 2, 1, 0.5, and 0.2% frequency, all with a total concentration of 105 genomes per microliter. These 5 populations were diluted serially into basal medium to generate samples with total nucleic acid concentrations of 104 and 103 genomes per microliter. Viral RNA was extracted from these samples, and cDNA was generated in a one-step multiplex RT-PCR as described above. The WT WSN33 sample (105 genomes per microliter) was processed and sequenced in duplicate. We prepared libraries as before and used Quanti PicoGreen double-stranded DNA (dsDNA) quantification (ThermoFisher Scientific) to quantify the concentration of each indexed library. We pooled equal quantities (in nanograms) of each indexed library and removed adapter dimers by gel isolation with the GeneJet gel extraction kit (ThermoFisher Scientific) prior to sequencing on an Illumina HiSeq 2500 with 2 × 125 paired-end reads. A clonal control library was processed in an identical fashion starting from an equimolar mixture of 8 plasmids containing the WSN33 genomic segments.
For analysis (see Fig. 7), we isolated fresh RNA from the 5%, 2%, 1%, and 0.5% samples with 104 genomes per microliter. The samples were processed and sequenced in duplicate as described above.
FIG 7.
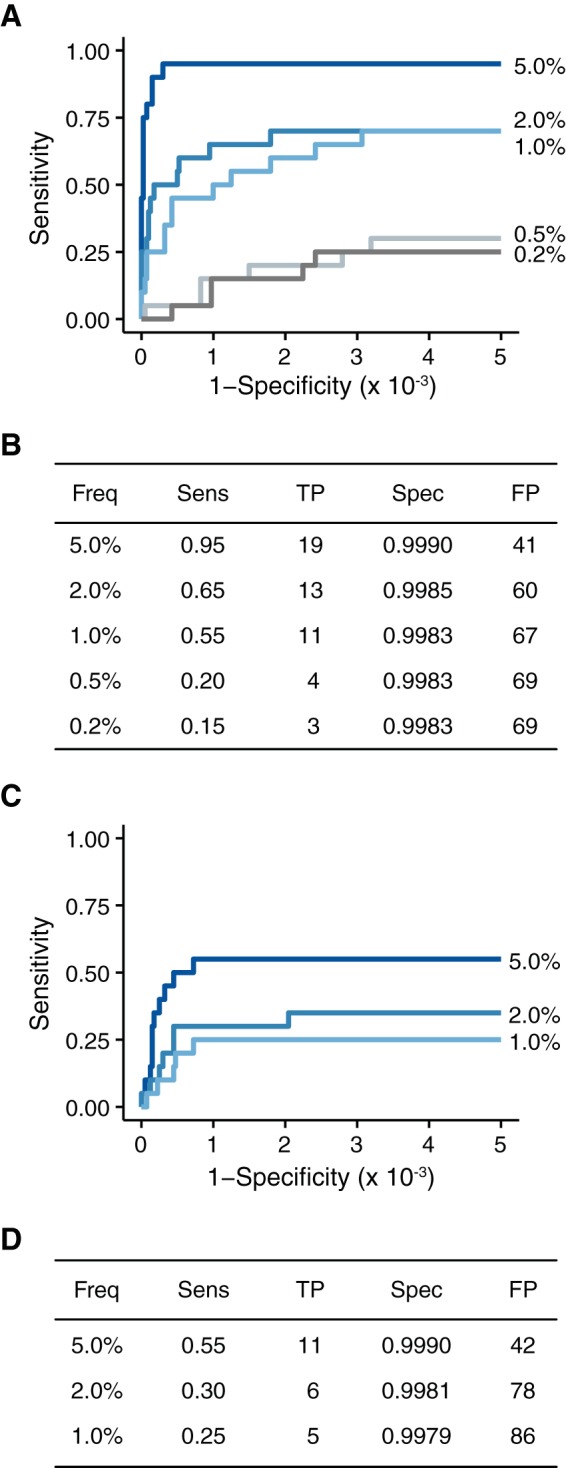
Accuracy of DeepSNV on populations with lower input nucleic acid levels. (A) ROC curve for the samples with 104 genomes/μl. (B) Summary of the data in panel A at a P value threshold of 0.01. (C) ROC curve for the samples with 103 genomes/μl. (D) Summary of the data in panel C at a P value threshold of 0.01.
Sequence analysis.
Reads were aligned to either a PR8 or a WSN33 reference sequence using Bowtie2 (44). The alignments were sorted, and PCR duplicates were removed using Picard (http://broadinstitute.github.io/picard/). Variants were called using either DeepSNV (26) or LoFreq (28) and filtered using the Pysam module in Python and custom R scripts available for download at https://github.com/lauringlab/Benchmarking_paper. Bases with a Phred score of <30 were masked in the DeepSNV analysis. We connected all of these steps into an analytical pipeline using bpipe (45), which is available for download at https://github.com/lauringlab/variant_pipeline. To save memory during SNV processing, only variants with P values of <0.9 were included in our receiver operating characteristic (ROC) curve analysis, as the vast majority of true negatives are trivial to identify and have a P value of 1. For ease of viewing, and to account for this analytical artifact, we extended the ROC curves horizontally from the last observed change in sensitivity. All the commands required to generate the figures are available for anonymous download at https://github.com/lauringlab/Benchmarking_paper. An interactive Shiny app of our benchmarking work can be downloaded at https://github.com/lauringlab/Benchmarking_shiny.
Diversity metrics.
The Shannon entropy (H) of each genomic position was calculated as follows: H = −Σi = 1nxiln(xi), where xi represents the frequency of the ith allele and n represents the number of alleles found at the given position. Since our data do not represent haplotypes, we report Shannon's entropy as the mean across all genomic positions.
The L1 norm (L) between 2 populations was calculated as follows: L = Σi = 1n|pi − qi|, where n represents the union of variants between the two samples and pi and qi represent the frequencies of the ith variant in each sample.
Data set accession number.
All raw fastq files have been submitted to the Sequence Read Archive (SRA) under BioProject accession number PRJNA317621.
RESULTS
The ability to reliably identify SNV is integral to accurate NGS-based studies of viral diversity. The accuracy of any SNV-calling pipeline can be described in terms of its sensitivity and specificity. Sensitivity is the proportion of true variants that are properly identified, and specificity is the proportion of true negatives that are properly identified. In other words, sensitivity measures an assay's ability to detect true variants present in a viral population, whereas specificity is determined by how many false variants (errors of some kind) are erroneously identified. An assay with perfect accuracy, in which all the true variants are found and only true variants are found, has a sensitivity and specificity of 1.
There is an obvious trade-off between sensitivity and specificity. Improved sensitivity often requires less stringent criteria in variant calling, which reduces specificity. Conversely, increased stringency can improve specificity but often reduces sensitivity. This relationship can be visualized using an ROC curve (Fig. 1). An ROC curve plots the sensitivity of an assay along the y axis and 1 minus specificity, or the false-positive rate, along the x axis. A variant-calling pipeline must be tested against known data in order to construct an ROC curve. The outcomes can then be stratified according to a metric that quantifies the probability that a given variant is real, often a P value or quality score. In a controlled benchmarking experiment, all true variants are known, and the sensitivity and specificity can be calculated at different cutoffs (Fig. 1A). These points are then used to construct the curve (Fig. 1B). A perfect ROC curve in which all the true positives can be separated from all the false positives is a right angle that follows the upper left perimeter of the plot.
FIG 1.
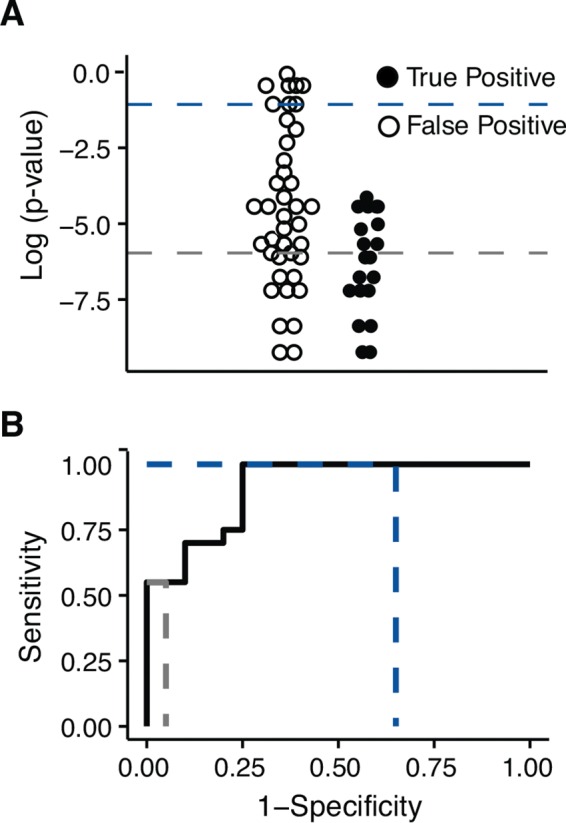
Example of an ROC curve. (A) Hypothetical variants are stratified by the log of the P value. P value thresholds are indicated as dashed colored lines. These “data” are intended to illustrate the concept and are not based on an actual experiment. (B) An ROC curve made from the hypothetical data shown in panel A. The dashed colored lines indicate the points on the curve corresponding to the thresholds in panel A.
Initial accuracy.
A comprehensive comparison of SNV-calling approaches is beyond the scope of this work. Instead, we robustly benchmark one variant caller, DeepSNV, and highlight approaches for improving its accurate application to patient-derived populations. In doing so, we demonstrate the importance of validating any variant-calling method under the experimental conditions to which it is applied. We chose DeepSNV as our starting point, because at the time, it was the only variant caller that had been benchmarked on a data set of known viral variants (26).
DeepSNV is a variant-calling algorithm that uses a clonal, plasmid-derived control to estimate local error rates across the genome (26). Because it is clonal, the sequence of the control is known with a high degree of confidence, and any nonconsensus base is indicative of an error in library preparation or sequencing. Additionally, the control and experimental samples are processed together and are assumed to have identical noise characteristics, thereby minimizing issues of “batch effect.” DeepSNV then applies a hierarchical binomial model at each genomic position and identifies true variants as those with frequencies significantly above the noise found in the plasmid control. Like many variant-calling algorithms, the accuracy of DeepSNV was initially determined using samples that required minimal PCR amplification. However, its accuracy has not been tested when applied to whole-genome sequencing of a viral population amplified by RT-PCR.
In our first benchmarking data set, we created defined mixtures of two plaque-purified and expanded influenza virus strains, WSN33 and PR8. cDNAs from both viruses were mixed serially so that WSN33 cDNA was present at frequencies of 5, 2.5, 1.25, 0.63, and 0.16% (Fig. 2A). The viruses differ at 491 positions (the primer sites used in RT-PCR were excluded from analysis), providing 491 true positives in each dilution. On plasmids subjected to limited PCR, DeepSNV identified known variants at 0.1% frequency with a sensitivity of 0.860 and a specificity of 1.0 (26). Under our experimental conditions, we found reductions in sensitivity (0.851 for variants at 0.63% and 0.173 for variants at 0.16%) and specificity (0.9980 and 0.9987 for variants at 0.63% and 0.16%, respectively). We were able to more closely approach the perfect specificity previously reported for DeepSNV by applying a more stringent P value of 0.01. A minor decrease in sensitivity accompanied this slightly more stringent P value (Fig. 2C). We used this P value cutoff in all subsequent experiments. The specificity was above 0.9980 at all dilutions. While the drop in specificity (from 1.0) appears small, it corresponds to 78 false positives when applied to the more than 39,000 potential variants in the 13,057-bp influenza virus genome.
An experimental intrahost population.
Although the initial benchmarking experiment validated our ability to accurately detect rare variants in influenza virus populations, the experiment was run under relatively artificial conditions. Patient-derived populations are typically less divergent than WSN33 and PR8 (4, 5, 23), and the number of viral genomes in patient samples is much lower than that found in cell culture. To mimic patient-specific conditions, we generated 20 viral clones, each with a single point mutation in the WSN33 background. We sequenced stocks of these mutants on an Illumina MiSeq instrument to account for any additional mutations that might have arisen between transfection and the passage 1 stock. Four additional mutations were found above 1% frequency (frequencies of 1.2% to 3.7%). We also determined the genome copy number of each stock using quantitative RT-PCR. We then mixed equal genome equivalents of these 20 viruses to generate a sample population with 105 copies per microliter, with each mutation present at 5% frequency. This population was serially diluted in a stock of wild-type WSN33, generating samples with each of the 20 mutations present at 2, 1, 0.5, 0.2, and 0.1% frequency (Fig. 3A). We then serially diluted these populations in basal medium to obtain mixtures with lower nucleic acid inputs. The range (103 to 105 copies per microliter) matches the inputs typically found in many patient-derived influenza virus samples (data not shown). We sequenced these populations on the Illumina HiSeq platform and called variants using DeepSNV. We also processed and sequenced the wild-type WSN33 stock in duplicate to control for any mutations in the viral diluent.
FIG 3.
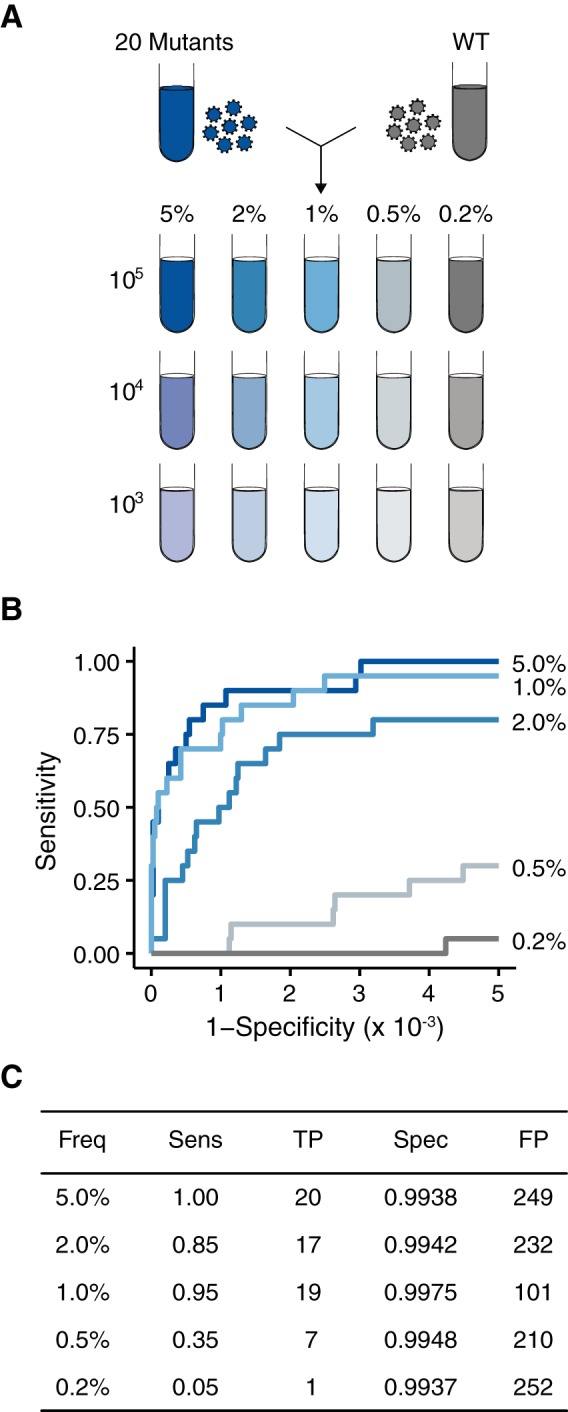
Accuracy of DeepSNV on populations approximating patient-derived samples. (A) Twenty viral supernatants, each with a single SNV, were diluted in a WSN33 viral supernatant to generate artificial viral populations with 20 mutations at the indicated frequencies. These populations were diluted further in basal medium to match the genome concentrations found in patient-derived samples (105 to 103 genomes/μl). (B) ROC curve measuring the accuracy of DeepSNV in identifying SNV at the indicated frequencies. (C) Summary of the data in panel B at a P value threshold of 0.01.
The 20 mutations present in our initial viral mixture were the only true positives considered in our analysis. Four SNV that were present at >1% frequency in either both duplicates of the wild-type stock or any one of the viral clones were masked, excluded from the analysis, and considered neither true positives nor true negatives. By applying these thresholds, we were able to validate our analysis using only variants identified a priori and to avoid the circular logic of validating a variant pipeline using SNV identified by the same pipeline.
In these populations with lower diversities and input titers, we maintained greater than 0.85 sensitivity for SNV at 1% frequency or higher. Despite a high depth of coverage (>10,000 reads per bp), our sensitivity was considerably lower for variants at or below 0.5% frequency (Fig. 3B and C). The drop in sensitivity, compared to the first data set (Fig. 2), was most likely due to the 1,000-fold decrease in the nucleic acid concentration and the fact that library preparation requires a number of sampling steps that may limit detection.
In our initial analysis of these data using the same default DeepSNV settings mentioned above, the specificity was significantly lower than what was observed in our PR8-WSN33 populations (a mean of 0.9812 with a minimum of 0.9598). These lower-input samples underwent more PCR cycles, which have been shown to skew the error distributions in the test libraries relative to the plasmid control (46). We were able to partially account for this variation by using an alternative beta binomial model available in DeepSNV, which more appropriately fits these conditions. With these settings, the specificity was greater than 0.9900 in all the samples with 105 genomes per microliter (Fig. 3C). As described above, while 0.9900 specificity appears adequate, it results in over 200 false-positive variants when applied to the over 39,000 potential variants in the influenza virus genome. The false positives outnumber the true positives by 10-fold in these populations, with realistic diversity and input. We were able to increase our specificity by applying a more stringent P value cutoff. However, as shown by the ROC curves in Fig. 3, this move toward the y axis markedly reduces sensitivity. Our data demonstrate that with moderate concentrations of input nucleic acid, even statistically significant P values from a robust variant caller are not sufficient to accurately separate true- from false-positive variants.
Additional filtering criteria.
Many next-generation sequencing studies utilize mapping quality (MapQ) and/or base quality (Phred) thresholds to ensure that only the highest-caliber sequencing data are used to call variants. Mapping quality measures the probability that a given read is mapped to the correct position in the genome, while base quality estimates the likelihood that the base call by the sequencer is correct. In the above analysis, we masked bases that had a Phred score of less than 30 (0.001 probability of being incorrect) and did not apply any MapQ cutoffs. In our next analysis, we applied seemingly stringent cutoffs, such as a MapQ score of 20 and a Phred score of 30, to our data (32, 47–49). These criteria were unable to distinguish true from false positives in our 105 samples (Fig. 4A) and indicate that many false positives occur on well-mapped reads with high-quality base calls.
FIG 4.
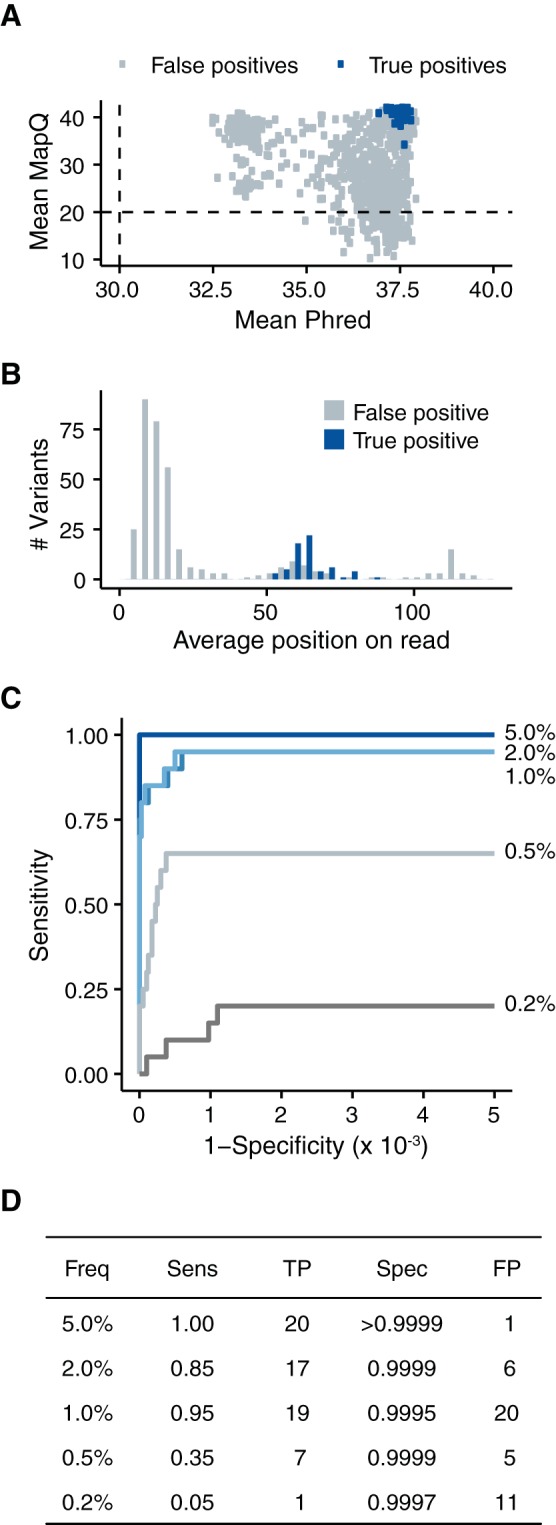
Accuracy can be improved through more stringent quality thresholds. (A) All called variants from the five samples with 105 genomes/μl and P values of <0.01 stratified by the mean mapping quality of the reads containing the variant and the mean Phred scores of the variant bases. The dashed lines indicate common cutoffs of 20 and 30 for mapping quality and Phred, respectively. (B) Histogram of average positions on a paired-end read of the variants that passed our mean MapQ threshold of 30 and mean Phred threshold of 35. (C) ROC curve measuring the accuracy of our analysis after applying the following quality cutoffs: mean MapQ score, >30; mean Phred score, >35; average read position, between 32 and 94 (the middle 50% of the read). (D) Summary of the data in panel C at a P value threshold of 0.01.
We further parsed our false variant calls by locating them within individual sequencing reads. It is well known that sequence quality drops near the end of a read (15, 50), and we found that our false positives clustered at the termini of our paired-end reads (Fig. 4B). The average Phred score of these false positives was 37.1, further demonstrating that filtering on the quality score alone is insufficient. In contrast, true positives were uniformly distributed across the reads, resulting in an average read position near the middle of the read.
Based on these results, we applied a number of empirically determined cutoffs, which markedly improved our specificity to >0.9990 without sacrificing sensitivity (Fig. 4C and D). For a variant to be considered in our analysis, we required a mean mapping quality of ≥30, a mean Phred score of ≥35, and an average read position within the middle 50% of the read. Under these conditions, we found 20 or fewer false positives in all 5 of the samples. Given this success, we applied a number of other strategies to further increase our accuracy, including Benjamini-Hochberg P value correction, more stringent P values (<0.01) or frequency cutoffs (>0.2%), retention of duplicate PCR reads, trimming the ends of the influenza virus genome, and employing alternative statistical distributions to estimate the error rate in the control sample. None of these approaches significantly improved our accuracy over the above-mentioned quality and read position criteria. The impacts of various filtering criteria on our data can be visualized in an interactive Shiny application available for download at https://github.com/lauringlab/benchmarking_shiny.git.
We also benchmarked the accuracy of our DeepSNV pipeline in estimating the frequency of the true-positive variants (Fig. 5). Although the medians of the measured frequencies match the expected values, we found substantial spread in each sample, and overall, the fit was modest (R2 = 0.65). The mean percent difference between the measured and expected frequencies was 41%. This error is likely due to amplification bias associated with RT-PCR (20) and library preparation and should be kept in mind when employing downstream analyses that depend on frequency measurements (e.g., variant fitness, haplotype reconstruction, Shannon's entropy, and other diversity metrics).
FIG 5.
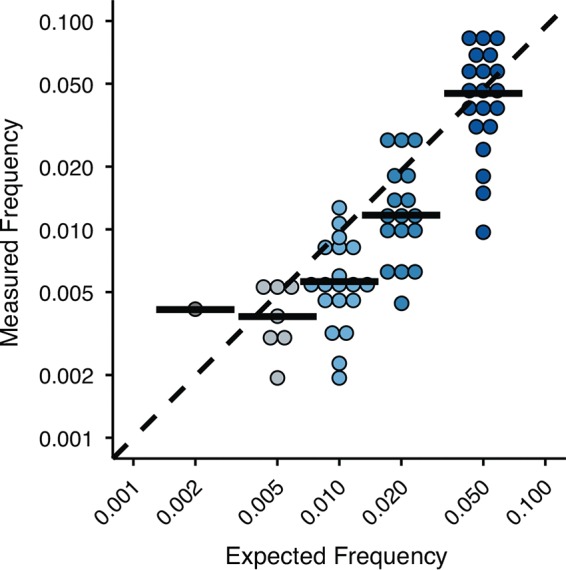
Accuracy of the frequency measurements for true-positive SNV in the samples with 105 genomes/μl. The black bars are the medians. The dashed line is where measured and expected frequencies are equal. Note that both axes are on a log scale.
Relatively low accuracy is not unique to DeepSNV.
DeepSNV is one of many variant callers that employ a combination of empirical and statistical approaches to model error rates. We asked whether the decreased accuracy observed in our data set was due simply to peculiarities specific to DeepSNV. We analyzed our 105 input populations using LoFreq, another variant caller commonly used in next-generation sequencing studies that has been reported to have perfect specificity (28). Under our experimental conditions, LoFreq had marginally reduced sensitivity compared to DeepSNV when applied to variant frequencies of ≥1.0% but marginally increased sensitivity when applied to variant frequencies of <1.0% (Fig. 6). The specificity of LoFreq was comparable to what we observed with DeepSNV in our high-input cell culture-derived populations (Fig. 2) and better than DeepSNV in our initial analysis of the 20 mutant populations (Fig. 3) prior to Phred, MapQ, and read position filtering. This increased specificity was most likely due to the fact that the LoFreq algorithm already takes MapQ and Phred scores into account when calling variants and has a stringent strand bias filter that removes many of the variants found only at one end of a paired-end read. Because it does not compare test samples to a plasmid control, it is also more robust in regard to issues of PCR skewing than DeepSNV. However, even with these additional characteristics, the specificity of LoFreq was lower than that of our improved DeepSNV pipeline (compare Fig. 4 and 6), with over 40 false positives per sample. It appears that higher than expected false-positive rates are not specific to DeepSNV and most likely plague many variant callers applied to patient-derived viral samples.
FIG 6.
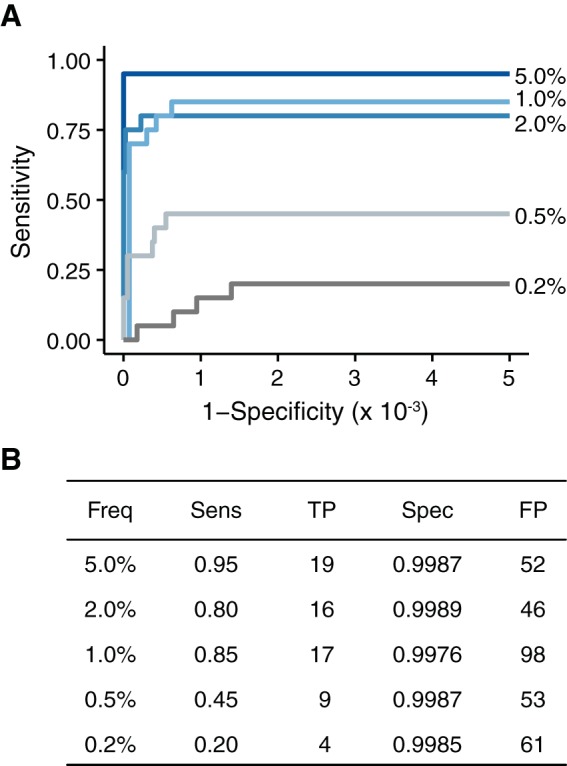
Accuracy of LoFreq on populations with 105 genomes/μl. (A) Accuracy of LoFreq using standard parameters. The specificity of LoFreq was scaled to account for the same number of tests as performed in DeepSNV. (B) Summary of the data in panel A at a P value threshold of 0.01.
Accuracy at lower input levels.
Host-derived viral populations vary in copy number and titer by several orders of magnitude (51–53). This variability can be attributed to a variety of factors, including the collection site, the ease of nucleic acid isolation, the presence of host nucleic acid, the efficiency of library preparation, and host and viral factors. To ensure accuracy across a range of input levels, we diluted our experimental populations serially in basal medium (Fig. 3A) and identified variants using our modified DeepSNV analysis pipeline (Fig. 7). As expected, our sensitivity was lower in populations with fewer genomes. For example, a variant at 0.5% frequency in a sample with 104 genomes per microliter is expected to be present on only 700 genomes in the initial RT-PCR. Many of these will be lost due to bottlenecks in the amplification and library preparation process. We also found reduced specificity in lower-input samples. The increase in false positives is presumably due to a greater dependence on RT-PCR amplification and consequent propagation of errors. These data highlight the importance of controlling for input levels when comparing diversity across experimental samples.
In most cases, RT-PCR errors should be sporadic and randomly distributed across the amplified region. If RT-PCR errors are responsible for the reduced specificity found at lower input levels, they should be easily identified as variants present in only one of two RT-PCRs performed on the same RNA (54). To test this hypothesis, we sequenced duplicate RT-PCRs of the 5, 2, 1, and 0.5% variant frequency samples from our collection of samples with 104 genomes per microliter. The duplicates were processed separately but sequenced on the same lane of an Illumina HiSeq. We applied the stringent quality cutoffs and required that a given variant be found in both duplicates. By analyzing samples in duplicate, we reduced the number of false positives in each sample to 10 or fewer, resulting in a specificity of >0.9998 (Fig. 8). This increased specificity was not accompanied by decreased sensitivity. In fact, we found a slight increase in sensitivity (compared to Fig. 4), most likely due to variability in library preparation. Thus, accurate analysis of low-input samples can be achieved through duplicate RT-PCRs and careful benchmarking experiments.
FIG 8.
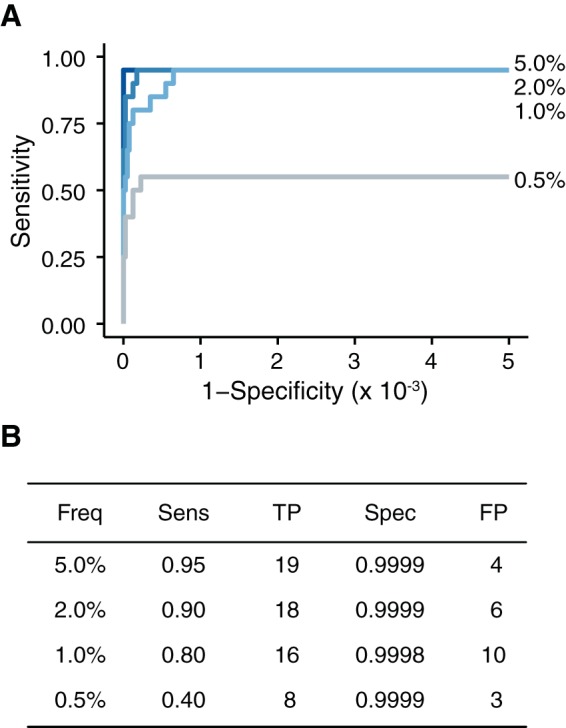
At lower inputs, duplicate samples improve accuracy. (A) ROC curve of the samples with 104 genomes/μl processed in duplicate. Only SNV present in both samples were considered. (B) Summary of the data in panel A at a P value threshold of 0.01.
Suboptimal SNV identification confounds diversity measurements.
NGS of intrahost populations is commonly used to determine the impact of host or environmental factors on viral diversity. Because measurements of viral diversity rely entirely on SNV identified in NGS data, they are very sensitive to the accuracy of these variant calls. To illustrate this problem, we calculated the diversity of our samples with 105 genomes per microliter at each step of our benchmarking process using three complementary metrics (Table 1). Richness is the count of nonconsensus variants present in a population (often referred to as intrahost SNV). Shannon's entropy is a diversity metric that accounts for both the number of variants present (richness) and their frequencies (evenness). Because our data are unphased (i.e., without haplotypes), we have reported the average entropy per nucleotide position (3). The last metric, L1 norm, is a distance measurement that describes how similar two populations are to one another based on the frequencies of variants present. Identical populations have an L1 norm of 0. To mimic experimental conditions, we included all the variants identified in each analysis regardless of whether subsequent benchmarking distinguished them as true or false positives. We found that the accuracy of the SNV-calling method had a profound effect on measurements of diversity. It is clear from the richness measurements in Table 1 that the number of false SNV (i.e., the specificity) largely determines the accuracy of the downstream analyses. Thus, our adapted DeepSNV protocol, which was able to distinguish between true and false SNV with the highest accuracy, gave the most accurate measures of diversity, followed by LoFreq and the default version of DeepSNV.
TABLE 1.
Diversity measurements in experimental populations
| Variant frequency (%)a | Diversity metric | Value |
|||
|---|---|---|---|---|---|
| Expected | LoFreq | DeepSNV | DeepSNV modified | ||
| 5.0 | Richness | 20 | 71 | 269 | 21 |
| Entropy | 2.97E−4 | 3.35E−4 | 1.60E−3 | 2.77E−4 | |
| L1 norm | 0 | 0.519 | 4.006 | 0.378 | |
| 1.0 | Richness | 20 | 115 | 120 | 39 |
| Entropy | 8.37E−5 | 2.78E−4 | 3.14E−4 | 7.30E−5 | |
| L1 norm | 0 | 2.702 | 0.704 | 0.133 | |
| 0.5 | Richness | 20 | 62 | 217 | 12 |
| Entropy | 4.71E−5 | 8.47E−5 | 1.12E−3 | 2.10E−5 | |
| L1 norm | 0 | 0.196 | 3.156 | 0.089 | |
The frequency of 20 true-positive variants. Only the input libraries with 105 genomes/μl were used.
To determine the impact of our improved variant-calling pipeline on actual host-derived populations, we applied our approach to 8 patient-derived samples collected as part of a household-based cohort study of influenza virus (39, 40) (Table 2). The samples were chosen from two influenza seasons and include H1N1 and H3N2 subtypes over a range of input titers and days of infection (measured as day post-symptom onset). As in our benchmarking data set, the estimated diversity of each sample was greatly reduced when we applied our empirically determined quality thresholds. The number of intrahost SNV and the Shannon entropy were reduced by up to 10-fold, suggesting the presence of a large number of false positives in our unmodified pipeline. These data show that validation is necessary to avoid overestimating the diversity present in patient-derived samples.
TABLE 2.
Diversity measurements in patient-derived samples
| Sample ID | DPSa | Strain | Seasonb | Log10 genomes/μl | Measurement |
|||
|---|---|---|---|---|---|---|---|---|
| DeepSNV |
DeepSNV modified |
|||||||
| Richness | Entropy | Richness | Entropy | |||||
| 1376 | 1 | A/H1N1 | 2013–2014 | 5.3 | 90 | 8.70E−4 | 5 | 6.04E−5 |
| 1401 | 2 | A/H1N1 | 2013–2014 | 5.0 | 110 | 1.04E−3 | 22 | 1.16E−4 |
| 1405 | 3 | A/H1N1 | 2013–2014 | 4.3 | 120 | 1.08E−3 | 30 | 1.55E−4 |
| 1374 | 4 | A/H1N1 | 2013–2014 | 4.6 | 185 | 1.25E−3 | 13 | 1.05E−4 |
| 1227 | 1 | A/H3N2 | 2012–2013 | 5.3 | 79 | 5.21E−4 | 8 | 4.43E−5 |
| 1321 | 1 | A/H3N2 | 2012–2013 | 4.3 | 20 | 2.35E−4 | 3 | 1.70E−5 |
| 1229 | 2 | A/H3N2 | 2012–2013 | 4.6 | 32 | 3.34E−4 | 8 | 7.07E−5 |
| 1245 | 3 | A/H3N2 | 2012–2013 | 4.4 | 197 | 1.38E−3 | 3 | 1.07E−5 |
DPS, days after symptom onset.
The influenza season was considered to run from September to May.
DISCUSSION
Robust validation is essential in NGS-based studies of viral diversity. Differences in experiment design and sample preparation can lead to wide variability in the accuracy of SNV identification. We found that the input nucleic acid concentration, which can vary greatly in patient-derived samples, had a large impact on both the sensitivity and specificity of rare-variant detection. At moderate levels of nucleic acid input, we could improve accuracy by filtering putative SNV based on quality metrics and read position. We further improved our accuracy at low input levels by processing the samples in duplicate. While our quality cutoffs may not be universally applicable to all samples and variant callers, our data suggest that experiment design is critical for accurate SNV detection. These findings are important, as few, if any, variant callers have been benchmarked under patient-derived conditions. Finally, we showed that inaccuracies in SNV calling drastically impact downstream analysis and lead to overestimations of intrahost diversity in patient-derived samples.
We initially chose DeepSNV for our studies, because it is one of the few variant callers that has been validated on viral sequencing reads in which all true-positive variants and their frequencies were known a priori and independent of NGS (26). A key strength of our study is that we applied DeepSNV to experimental populations that more closely mimic the diversity and levels of virus found in patient-derived samples of influenza virus. At a modest input of 105 genomes per microliter and default DeepSNV settings, false positives can outnumber true positives by a factor of 4. It should be noted that our specificity in all cases remained above 0.9900. When applied across an entire influenza virus genome, a specificity of >0.9995 is required to reduce false positives to low levels. As described above, the decreased accuracy of DeepSNV under these conditions is due to small but important differences in our experiment design compared to what has been previously reported, namely, the input nucleic acid concentration and RT-PCR amplification.
Because DeepSNV is somewhat agnostic toward the mapping quality and the base quality of a given variant, we sought to improve our accuracy by identifying thresholds that more effectively distinguished true-positive SNV. In our data sets, the distributions of average MapQ and Phred scores of putative SNV were bimodal, with true SNV found in the higher of the two distributions. In the data presented, these cutoffs include ≥98% of the true-positive variant calls. Our empirically determined thresholds were chosen to eliminate putative SNV found in the lower of the two distributions. These thresholds should be reproducible in our system, as we have observed consistent MapQ and Phred quality distributions over 300 influenza virus libraries and 5 HiSeq runs. We have also seen the same bimodal trend in libraries of poliovirus populations but have applied a lower empirically determined MapQ cutoff to these data. The shift in MapQ scores is most likely rooted in differences in genomic structure between the two viruses. While our MapQ and Phred thresholds are robust in our system, they may need to be adjusted for use in others.
Even in the face of stringent MapQ and Phred cutoffs, we found many high-quality false-positive SNV that were identified only at the termini of paired-end reads. We removed these by filtering putative SNV based on their average positions in a paired-end read. These false positives were found almost exclusively in regions of the genome that were enriched for read start sites. This enrichment may be a consequence of sequence context, the fragmentation process, or our size selection protocol. We suggest that there might also be a biological reason for this effect, as our PCR-amplified plasmid control samples did not exhibit this bias. For example, defective interfering particles, which commonly arise during cell passage, contain truncated genomic segments and would only be present in infected supernatants and not the plasmid control. We hypothesize that the large deletions in these segments increase the number of reads that start at certain genomic positions. As the beginning of reads can also be error prone, this enrichment would result in false-positive SNV. Our analysis was particularly vulnerable to this type of error, because we did not trim the ends of our reads, and DeepSNV, unlike other variant callers, does not directly test for strand bias or consider read position as a variable. While it is easy to diagnose these shortcomings in retrospect, such errors had not been previously reported for DeepSNV and were elucidated only through our extensive validation.
While read filtering and trimming are common in NGS data sets (55–57), we have taken a slightly different approach in our analysis. In the initial SNV identification step, we masked bases with Phred scores of <30 but made no additional restrictions on the raw data. We imposed additional quality restrictions only after putative SNV—those that exceeded the expected frequency given the plasmid control—were identified. While our approach treats variant nucleotides more stringently than consensus base calls, we do not think that this differential stringency introduces unnecessary bias. Because we identified specificity as the major problem in accurate SNV identification, stringent filtering of potential false positives seems appropriate. Furthermore, the vast majority of reads call a consensus base, and our mean quality score thresholds would therefore not be expected to remove many consensus base calls from the analysis.
Frequency thresholds of 0.1 to 1% represent an additional quality filter that is applied to SNV after identification (35). We did not apply direct frequency thresholds in our analysis, as we found that arbitrary cutoffs limited sensitivity without improving specificity. Read depth, or coverage, is another metric that can be used in conjunction with frequency to ensure accurate SNV identification. Although we did not apply a direct coverage cutoff, DeepSNV has been reported to require coverage of 10 times the reciprocal of frequency for sufficient power to call SNV. For example, a coverage of 1,000× is needed to detect a variant at 1% frequency. In our analysis, the lowest coverage for a true positive was 1,795 reads (4.8% frequency), while the lowest coverage for a false-positive variant was 966 reads (8.5% frequency). If a given data set has variability in read depth across the genome, SNV at identical frequencies may be detected with differing sensitivities. Under such conditions, the application of variant frequency or read depth thresholds would lead to severe ascertainment bias in subsequent analyses of diversity.
Few studies of intrahost diversity quantify or control for the number of genomes in a sample. This is important, because we found that the input copy number is a key factor in variant detection. Despite high accuracy at 105 genomes per microliter, we observed a decrease in sensitivity in our samples with 103 genomes per microliter. More importantly, this drop in sensitivity was accompanied by reduced specificity. At lower nucleic acid concentrations, NGS pipelines rely more heavily on RT-PCR amplification, which tends to propagate errors that are otherwise indistinguishable from true positives in sequence data. We were able to limit these sporadic and random errors by processing low-input samples in duplicate. Quantifying and controlling for RT-PCR errors in this way allows us to accurately compare patient-derived samples that vary over a range of inputs.
Many variant callers are benchmarked on simulated data sets, plasmids, or PCR products and may not have comparable sensitivities and specificities when applied to viral samples. Our goal was not to compare the strengths and weaknesses of a few algorithms, but rather, to highlight how accuracy can be experiment specific. We recognize that some variant callers may perform better than DeepSNV and that others may be better suited to other systems. However, our work with LoFreq suggests that all methods have inherent limitations and that understanding these limitations is essential. We have been able to greatly improve the accuracy of DeepSNV under our experimental conditions, and we are now equipped with an understanding of the limitations of our method.
Our study highlights previously underrecognized issues in variant calling and suggests factors that should be considered in future studies of viral diversity. The need for target amplification, the structure of the viral genome, and variation in input genome copy numbers may lead to errors specific to a given experiment. We have shown that these seemingly small differences in sensitivity and specificity (e.g., 0.9998 versus 0.9900) can have profound effects on measurements of viral diversity in experimental and patient-derived populations. These differences are especially important in comparative studies of intrahost diversity. We realize that there are many solutions to the problem of NGS accuracy and have made our data sets and code available to the community. We hope that this will allow others to benchmark their own pipelines or to improve on our work. This process should improve the reliability of NGS in studies of virus evolution and molecular epidemiology.
ACKNOWLEDGMENTS
We thank Pat Schloss, Mike Imperiale, and Robert Woods for helpful discussion and critical reading of the manuscript. We thank Chris Gates, and Peter Ulintz for help with bpipe and Arnold Monto, Suzanne Ohmit, Rachel Truscon, Emileigh Johnson, Ryan Malosh, and Josh Petrie for making the HIVE samples available to us.
This work was supported by a Clinician Scientist Development Award from the Doris Duke Charitable Foundation and grant R01 AI118886, both to A.S.L. J.T.M. was supported by the Michigan Predoctoral Training Program in Genetics (T32GM007544).
REFERENCES
- 1.Lauring AS, Frydman J, Andino R. 2013. The role of mutational robustness in RNA virus evolution. Nat Rev Microbiol 11:327–336. doi: 10.1038/nrmicro3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen KG, Shapiro BJ, Matranga CB, Sealfon R, Lin AE, Moses LM, Folarin OA, Goba A, Odia I, Ehiane PE, Momoh M, England EM, Winnicki S, Branco LM, Gire SK, Phelan E, Tariyal R, Tewhey R, Omoniwa O, Fullah M, Fonnie R, Fonnie M, Kanneh L, Jalloh S, Gbakie M, Saffa S, Karbo K, Gladden AD, Qu J, Stremlau M, Nekoui M, Finucane HK, Tabrizi S, Vitti JJ, Birren B, Fitzgerald M, McCowan C, Ireland A, Berlin AM, Bochicchio J, Tazon-Vega B, Lennon NJ, Ryan EM, Bjornson Z, Milner DA Jr, Lukens AK, Broodie N, Rowland M, Heinrich M, Akdag M, Schieffelin JS, Levy D, Akpan H, Bausch DG, Rubins K, McCormick JB, Lander ES, Günther S, Hensley L, Okogbenin S, Viral Hemorrhagic Fever Consortium, Schaffner SF, Okokhere PO, Khan SH, Grant DS, Akpede GO, Asogun DA, Gnirke A, Levin JZ, Happi CT, Garry RF, Sabeti PC. 2015. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 162:738–750. doi: 10.1016/j.cell.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grubaugh ND, Smith DR, Brackney DE, Bosco-Lauth AM, Fauver JR, Campbell CL, Felix TA, Romo H, Duggal NK, Dietrich EA, Eike T, Beane JE, Bowen RA, Black WC, Brault AC, Ebel GD. 2015. Experimental evolution of an RNA virus in wild birds: evidence for host-dependent impacts on population structure and competitive fitness. PLoS Pathog 11:e1004874. doi: 10.1371/journal.ppat.1004874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers MB, Song T, Sebra R, Greenbaum BD, Hamelin M-E, Fitch A, Twaddle A, Cui L, Holmes EC, Boivin G, Ghedin E. 2015. Intrahost dynamics of antiviral resistance in influenza A virus reflect complex patterns of segment linkage, reassortment, and natural selection. mBio 6:e02464–14. doi: 10.1128/mBio.02464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poon LLM, Song T, Rosenfeld R, Lin X, Rogers MB, Zhou B, Sebra R, Halpin RA, Guan Y, Twaddle A, DePasse JV, Stockwell TB, Wentworth DE, Holmes EC, Greenbaum B, Peiris JSM, Cowling BJ, Ghedin E. 2016. Quantifying influenza virus diversity and transmission in humans. Nat Genet 48:195–200. doi: 10.1038/ng.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olp LN, Jeanniard A, Marimo C, West JT, Wood C. 2015. Whole-genome sequencing of KSHV from Zambian Kaposi's sarcoma biopsies reveals unique viral diversity. J Virol 89:12299–12308. doi: 10.1128/JVI.01712-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kugelman JR, Kugelman-Tonos J, Ladner JT, Pettit J, Keeton CM, Nagle ER, Garcia KY, Froude JW, Kuehne AI, Kuhn JH, Bavari S, Zeitlin L, Dye JM, Olinger GG, Sanchez-Lockhart M, Palacios GF. 2015. Emergence of Ebola virus escape variants in infected nonhuman primates treated with the MB-003 antibody cocktail. Cell Rep 12:2111–2120. doi: 10.1016/j.celrep.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 8.Lakdawala SS, Jayaraman A, Halpin RA, Lamirande EW, Shih AR, Stockwell TB, Lin X, Simenauer A, Hanson CT, Vogel L, Paskel M, Minai M, Moore I, Orandle M, Das SR, Wentworth DE, Sasisekharan R, Subbarao K. 2015. The soft palate is an important site of adaptation for transmissible influenza viruses. Nature 526:122–125. doi: 10.1038/nature15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Slyke GA, Arnold JJ, Lugo AJ, Griesemer SB, Moustafa IM, Kramer LD, Cameron CE, Ciota AT. 2015. Sequence-specific fidelity alterations associated with West Nile virus attenuation in mosquitoes. PLoS Pathog 11:e1005009–21. doi: 10.1371/journal.ppat.1005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuevas JM, Willemsen A, Hillung J, Zwart MP, Elena SF. 2015. Temporal dynamics of intrahost molecular evolution for a plant RNA virus. Mol Biol Evol 32:1132–1147. doi: 10.1093/molbev/msv028. [DOI] [PubMed] [Google Scholar]
- 11.Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, Kanneh L, Jalloh S, Momoh M, Fullah M, Dudas G, Wohl S, Moses LM, Yozwiak NL, Winnicki S, Matranga CB, Malboeuf CM, Qu J, Gladden AD, Schaffner SF, Yang X, Jiang P-P, Nekoui M, Colubri A, Coomber MR, Fonnie M, Moigboi A, Gbakie M, Kamara FK, Tucker V, Konuwa E, Saffa S, Sellu J, Jalloh AA, Kovoma A, Koninga J, Mustapha I, Kargbo K, Foday M, Yillah M, Kanneh F, Robert W, Massally JLB, Chapman SB, Bochicchio J, Murphy C, Nusbaum C, Young S, Birren BW, Grant DS, Scheiffelin JS, Lander ES, Happi C, Gevao SM, Gnirke A, Rambaut A, Garry RF, Khan SH, Sabeti PC. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kundu S, Lockwood J, Depledge DP, Chaudhry Y, Aston A, Rao K, Hartley JC, Goodfellow I, Breuer J. 2013. Next-generation whole genome sequencing identifies the direction of norovirus transmission in linked patients. Clin Infect Dis 57:407–414. doi: 10.1093/cid/cit287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauck M, Alvarado-Mora MV, Becker EA, Bhattacharya D, Striker R, Hughes AL, Carrilho FJ, O'Connor DH, Pinho JRR. 2012. Analysis of hepatitis C virus intrahost diversity across the coding region by ultradeep pyrosequencing. J Virol 86:3952–3960. doi: 10.1128/JVI.06627-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam HYK, Clark MJ, Chen R, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, Butte AJ, Ji HP, Snyder M. 2012. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol 30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schirmer M, Ijaz UZ, D'Amore R, Hall N, Sloan WT, Quince C. 2015. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res 43:e37–e37. doi: 10.1093/nar/gku1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, Ishikawa S, Linak MC, Hirai A, Takahashi H, Altaf-Ul-Amin M, Ogasawara N, Kanaya S. 2011. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res 39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acevedo A, Brodsky L, Andino R. 2014. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acevedo A, Andino R. 2014. Library preparation for highly accurate population sequencing of RNA viruses. Nat Protoc 9:1760–1769. doi: 10.1038/nprot.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou S, Jones C, Mieczkowski P, Swanstrom R. 2015. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J Virol 89:8540–8555. doi: 10.1128/JVI.00522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djikeng A, Halpin R, Kuzmickas R, DePasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson NG, Ghedin E, Spiro DJ. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5–9. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson MI, Balmaseda A, Kuan G, Saborio S, Lin X, Halpin RA, Stockwell TB, Wentworth DE, Harris E, Gordon A. 2014. The evolutionary dynamics of influenza A and B viruses in the tropical city of Managua, Nicaragua. Virology 462-463:81–90. doi: 10.1016/j.virol.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghedin E, Laplante J, DePasse J, Wentworth DE, Santos RP, Lepow ML, Porter J, Stellrecht K, Lin X, Operario D, Griesemer S, Fitch A, Halpin RA, Stockwell TB, Spiro DJ, Holmes EC, George KS. 2011. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis 203:168–174. doi: 10.1093/infdis/jiq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosseel T, Van Borm S, Vandenbussche F, Hoffmann B, van den Berg T, Beer M, Höper D. 2013. The origin of biased sequence depth in sequence-independent nucleic acid amplification and optimization for efficient massive parallel sequencing. PLoS One 8:e76144–9. doi: 10.1371/journal.pone.0076144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flaherty P, Natsoulis G, Muralidharan O, Winters M, Buenrostro J, Bell J, Brown S, Holodniy M, Zhang N, Ji HP. 2012. Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res 40:e2. doi: 10.1093/nar/gkr861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, Beerenwinkel N. 2012. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 27.Isakov O, Bordería AV, Golan D, Hamenahem A, Celniker G, Yoffe L, Blanc H, Vignuzzi M, Shomron N. 2015. Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics 31:2141–2150. doi: 10.1093/bioinformatics/btv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macalalad AR, Zody MC, Charlebois P, Lennon NJ, Newman RM, Malboeuf CM, Ryan EM, Boutwell CL, Power KA, Brackney DE, Pesko KN, Levin JZ, Ebel GD, Allen TM, Birren BW, Henn MR. 2012. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol 8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. 2009. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson SJ, Welkers MRA, Depledge DP, Coulter E, Breuer JM, de Jong MD, Kellam P. 2013. Viral population analysis and minority-variant detection using short read next-generation sequencing. Phil Trans R Soc B 368:20120205. doi: 10.1098/rstb.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Charlebois P, Macalalad A, Henn MR, Zody MC. 2013. V-Phaser 2: variant inference for viral populations. BMC Genomics 14:674. doi: 10.1186/1471-2164-14-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerstung M, Papaemmanuil E, Campbell PJ. 2014. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 30:1198–1204. doi: 10.1093/bioinformatics/btt750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Combe M, Garijo R, Geller R, Cuevas JM, Sanjuán R. 2015. Single-cell analysis of RNA virus infection identifies multiple genetically diverse viral genomes within single infectious units. Cell Host Microbe 18:424–432. doi: 10.1016/j.chom.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck A, Tesh RB, Wood TG, Widen SG, Ryman KD, Barrett ADT. 2014. Comparison of the live attenuated yellow fever vaccine 17D-204 strain to its virulent parental strain Asibi by deep sequencing. J Infect Dis 209:334–344. doi: 10.1093/infdis/jit546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann E, Mahmood K, Yang C-F, Webster RG, Greenberg HB, Kemble G. 2002. Rescue of influenza B virus from eight plasmids. Proc Natl Acad Sci U S A 99:11411–11416. doi: 10.1073/pnas.172393399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pauly MD, Lauring AS. 2015. Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J Virol 89:3584–3597. doi: 10.1128/JVI.03483-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohmit SE, Petrie JG, Malosh RE, Fry AM, Thompson MG, Monto AS. 2015. Influenza vaccine effectiveness in households with children during the 2012-2013 season: assessments of prior vaccination and serologic susceptibility. J Infect Dis 211:1519–1528. doi: 10.1093/infdis/jiu650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monto AS, Malosh RE, Petrie JG, Thompson MG, Ohmit SE. 2014. Frequency of acute respiratory illnesses and circulation of respiratory viruses in households with children over 3 surveillance seasons. J Infect Dis 210:1792–1799. doi: 10.1093/infdis/jiu327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 42.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Centers for Disease Control and Prevention. 2009. CDC protocol of realtime RTPCR for influenza A(H1N1). Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 44.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sadedin SP, Pope B, Oshlack A. 2012. Bpipe: a tool for running and managing bioinformatics pipelines. Bioinformatics 28:1525–1526. doi: 10.1093/bioinformatics/bts167. [DOI] [PubMed] [Google Scholar]
- 46.Gerstung M, Beerenwinkel N. 2015. Calling subclonal mutations with deepSNV. https://www.bioconductor.org/packages/release/bioc/vignettes/deepSNV/inst/doc/deepSNV.pdf. [Google Scholar]
- 47.Koboldt DC, Larson DE, Wilson RK. 2013. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinformatics 44:15.4.1–15.4.17. doi: 10.1002/0471250953.bi1504s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dietz J, Schelhorn S-E, Fitting D, Mihm U, Susser S, Welker M-W, Füller C, Däumer M, Teuber G, Wedemeyer H, Berg T, Lengauer T, Zeuzem S, Herrmann E, Sarrazin C. 2013. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J Virol 87:6172–6181. doi: 10.1128/JVI.02778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. 2012. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A 109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang XV, Blades N, Ding J, Sultana R, Parmigiani G. 2012. Estimation of sequencing error rates in short reads. BMC Bioinformatics 13:185. doi: 10.1186/1471-2105-13-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau LLH, Ip DKM, Nishiura H, Fang VJ, Chan KH, Peiris JSM, Leung GM, Cowling BJ. 2013. Heterogeneity in viral shedding among individuals with medically attended influenza A virus infection. J Infect Dis 207:1281–1285. doi: 10.1093/infdis/jit034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teunis PFM, Sukhrie FHA, Vennema H, Bogerman J, Beersma MFC, Koopmans MPG. 2015. Shedding of norovirus in symptomatic and asymptomatic infections. Epidemiol Infect 143:1710–1717. doi: 10.1017/S095026881400274X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeyama A, Hashimoto K, Sato M, Kawashima R, Kawasaki Y, Hosoya M. 2016. Respiratory syncytial virus shedding by children hospitalized with lower respiratory tract infection. J Med Virol 88:938–946. doi: 10.1002/jmv.24434. [DOI] [PubMed] [Google Scholar]
- 54.Robasky K, Lewis NE, Church GM. 2014. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet 15:56–62. doi: 10.1038/nrg3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. 2013. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One 8:e85024–13. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nielsen R, Paul JS, Albrechtsen A, Song YS. 2011. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet 12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]