

ABSTRACT
Nonhuman primates (NHPs) are a historically important source of zoonotic viruses and are a gold-standard model for research on many human pathogens. However, with the exception of simian immunodeficiency virus (SIV) (family Retroviridae), the blood-borne viruses harbored by these animals in the wild remain incompletely characterized. Here, we report the discovery and characterization of two novel simian pegiviruses (family Flaviviridae) and two novel simian arteriviruses (family Arteriviridae) in wild African green monkeys from Zambia (malbroucks [Chlorocebus cynosuros]) and South Africa (vervet monkeys [Chlorocebus pygerythrus]). We examine several aspects of infection, including viral load, genetic diversity, evolution, and geographic distribution, as well as host factors such as age, sex, and plasma cytokines. In combination with previous efforts to characterize blood-borne RNA viruses in wild primates across sub-Saharan Africa, these discoveries demonstrate that in addition to SIV, simian pegiviruses and simian arteriviruses are widespread and prevalent among many African cercopithecoid (i.e., Old World) monkeys.
IMPORTANCE Primates are an important source of viruses that infect humans and serve as an important laboratory model of human virus infection. Here, we discover two new viruses in African green monkeys from Zambia and South Africa. In combination with previous virus discovery efforts, this finding suggests that these virus types are widespread among African monkeys. Our analysis suggests that one of these virus types, the simian arteriviruses, may have the potential to jump between different primate species and cause disease. In contrast, the other virus type, the pegiviruses, are thought to reduce the disease caused by human immunodeficiency virus (HIV) in humans. However, we did not observe a similar protective effect in SIV-infected African monkeys coinfected with pegiviruses, possibly because SIV causes little to no disease in these hosts.
INTRODUCTION
Viruses that naturally infect wild nonhuman primates (NHPs) are of considerable interest because of their zoonotic potential (1, 2). For example, the study of naturally occurring simian immunodeficiency virus (SIV) infections in African monkeys (i.e., “natural hosts” for SIV) has provided invaluable insight into the origins and pathogenesis of human immunodeficiency virus (HIV) infection in humans and SIV infection in endangered great apes (see references 3–5 for reviews). However, with the exception of SIV, which infects monkeys from over 40 species throughout Africa, the RNA viruses naturally infecting wild NHPs remain largely uncharacterized. This lack of characterization is due in part to limitations in technology and the technical and ethical challenges inherent in invasive sampling of wild NHPs (6–8).
Previously, we used unbiased deep sequencing, a technique that utilizes random hexamers to prime cDNA synthesis from RNA in combination with next-generation sequencing, to discover and characterize blood-borne RNA viruses in wild monkeys from several cercopithecoid (i.e., Old World monkey [OWM]) species in Uganda, Tanzania, and Zambia (9–16). We consistently detected viruses from three genera: lentiviruses (i.e., SIV, Retroviridae family), pegiviruses (i.e., simian pegivirus [SPgV], Flaviviridae family), and simian arteriviruses (i.e., viruses distantly related to simian hemorrhagic fever virus [SHFV], Arteriviridae family). Viruses from these genera are relevant to NHP and human health: human pegivirus (HPgV) is associated with a reduction in pathological immune activation and mortality in HIV-infected people (17–19), while simian arteriviruses are prototypical preemergent zoonotic pathogens that have caused numerous outbreaks of viral hemorrhagic fever in captive Asian-origin macaque monkeys (20, 21).
We hypothesized that in addition to lentiviruses, simian pegiviruses and simian arteriviruses are common RNA viruses found in plasma (i.e., the “plasma RNA virome”) of monkeys throughout sub-Saharan Africa. To test this hypothesis, we sought to expand our knowledge of the host and geographic ranges of these viruses by applying unbiased deep sequencing to plasma samples collected from wild African green monkeys (AGMs) in Zambia (malbroucks [Chlorocebus cynosuros]) and South Africa (vervet monkeys [Chlorocebus pygerythrus]). Although AGMs in Gambia (sabaeus monkeys [Chlorocebus sabaeus]) are known to harbor simian pegiviruses (22), the AGM species examined in this study are not known to harbor simian pegiviruses or simian arteriviruses and are separated by as much as 2,000 km from the closest known natural host of these viruses (baboons [Papio spp.] in Zambia [21]). Additionally, extensive population and cytokine data were already available for a large subset of the South African vervet population included in this study (23). Here, we demonstrate prevalent infection of these AGMs with simian lentiviruses, simian pegiviruses, and simian arteriviruses (but not other RNA viruses), supporting our idea that viruses from these genera are major constituents of the plasma RNA virome in monkeys from a diversity of cercopithecoid species living across a large part of sub-Saharan Africa.
MATERIALS AND METHODS
Ethics statement.
All the animals sampled in this study were used according to regulations set forth by the Animal Welfare Act. Sampling of vervet monkeys in South Africa was approved by the Interfaculty Animal Ethics Committee (project no. 13/2010) at the University of Free State. Sampling of vervet monkeys in South Africa and malbrouck monkeys in Zambia was approved by the University of Wisconsin—Milwaukee Animal Care and Use Committee (protocol 07-08 32).
Sample collection.
Vervet monkeys were individually trapped by using established methods (24), the details of which were described previously (23). Briefly, each animal was trapped, sedated, and bled via venous puncture. During sampling, a detailed physical examination was performed, clinical signs were assessed, and the approximate age and sex of each individual were noted. Each vervet monkey sampled had a microchip implanted for further identification and for the prevention of duplicate sampling. Upon blood collection, plasma was purified and immediately stored at −80°C.
Unbiased deep sequencing.
Samples were processed for sequencing in a biosafety level 3 laboratory as described previously (14), with slight modifications. Briefly, for each sample, RNA was isolated from ∼200 μl of plasma by using the Qiagen QIAamp MinElute virus spin kit (Qiagen, Hilden, Germany), omitting carrier RNA. Samples were then treated with DNase, and cDNA synthesis was primed by using random hexamers from a double-stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). Samples were fragmented, and sequencing adaptors were ligated by using the Nextera DNA sample preparation kit (Illumina, San Diego, CA, USA). Deep sequencing was performed on the Illumina MiSeq instrument. Sequence data were processed by using CLC Genomics Workbench 6.5 (CLC Bio, Aarhus, Denmark) and Geneious R5 (Biomatters, Auckland, New Zealand). Low-quality (<Q30, Phred quality score) and short (<100-bp) reads were removed, and coding-complete genome sequences for each virus were acquired by using a combination of mapping and the de novo assembly algorithm in CLC Genomics Workbench version 6.5. Viral genomes were annotated in CLC Genomics Workbench version 6.5, and open reading frames (ORFs) were confirmed by querying the NCBI GenBank database (25).
Phylogenetic analysis.
Sequences were aligned by using a codon-based version of the open-source software Multiple Alignment Using Fast Fourier Transform (MAFFT), implemented in TranslatorX, without GBLOCKS gene fragment cleaning (26). Phylogenetic history was inferred from aligned nucleotide sequences by using the maximum likelihood method (1,000 bootstrap replicates) via Molecular Evolutionary Genetics Analysis version 6 (MEGA6) open-source software (27). The best nucleotide substitution model, a general time-reversible model coupled with a Γ distribution for rate variation (GTR+Γ), with five rate categories and a +Γ parameter of 1.1092, was estimated by using MEGA6. All positions containing gaps and missing data were eliminated (complete deletion), resulting in a final data set of 4,380 positions. The initial tree for the heuristic search was obtained by applying the neighbor-joining method to a matrix of pairwise distances, estimated by using the maximum composite likelihood approach.
qRT-PCR.
Vervet monkey SIV (SIVver)-specific TaqMan quantitative reverse transcriptase PCR (qRT-PCR) was performed on plasma samples (23). TaqMan qRT-PCR assays were then developed to quantify concentrations of plasma viral RNA for both SPgVver and DMVV-1 (Drakensberg Mountain vervet virus). First, alignments of the GBV-Cver and DMVV-1 genomes obtained by unbiased deep sequencing were constructed in CLC Genomics Workbench version 6.5 to identify highly conserved regions. Primer3 (28) was then used to design primers and probes specific for these regions [i.e., SPgVver forward primer 5′-CAGC-(deoxy-inosine)-GACATCGGAGAAGC-3′, SPgVver reverse primer 5′-CTAACACTTCCCGGCACATT-3′, SPgVver probe 5′-FAM (6-carboxyfluorescein)-CGGCTGTAACTGGCCTTTAC-BHQ1 (black hole quencher 1)-3′, DMVV-1 forward primer 5′-GTCAGGGCTTCACCCTAGC-3′, DMVV-1 reverse primer 5′-GCCATACCTCCGAAGGGTTA-3′, and DMVV-1 probe 5′-Quasar670-CTTGGTCCCTGACGTGAAAA-BHQ3-3′]. PCR amplicons flanking the primer binding sites for each virus were generated, cloned into the Zero Blunt PCR vector (Invitrogen), and linearized (HindIII; New England BioLabs, Ipswich, MA). Transcription was performed in vitro for 6 h (MEGAscript T7 transcription kit; Invitrogen), followed by RNA transcript purification (MEGAclear transcription cleanup kit; Invitrogen), quantification (Qubit RNA high-sensitivity assay kit; Invitrogen), and dilution to a concentration of 1 × 1010 transcript copies/μl. Tenfold dilutions of this transcript were used as a standard curve.
Viral RNA was extracted from 100 μl of plasma with the Viral Total Nucleic Acid Purification kit (Promega, Madison, WI) on the Maxwell 16 MDx instrument (Promega). RNA was reverse transcribed and amplified by using the SuperScript III One-Step qRT-PCR system (Invitrogen) on a LightCycler 480 instrument (Roche, Indianapolis, IN). Reverse transcription was carried out at 37°C for 15 min and then at 50°C for 30 min, followed by 2 min at 95°C. Amplification was accomplished over 50 cycles as follows: 95°C for 15 s and 60°C for 1 min. The reaction mixture contained MgSO4 at a final concentration of 3.0 mM, two amplification primers at a concentration of 500 nM, and a probe at a concentration of 100 nM. The standard curve was linear over 8 orders of magnitude and was sensitive down to 10 copies of RNA transcript per reaction.
Evolutionary analyses.
For within-host nucleotide diversity analyses, single-nucleotide polymorphism (SNP) reports were generated by mapping pooled sequencing reads to their corresponding consensus sequence. Next, SNPGenie (29) was used to estimate synonymous nucleotide diversity (πS) and nonsynonymous nucleotide diversity (πN) for each ORF by using a new method for pooled-sequencing analyses (30) based on that of Nei and Gojobori (31) (accessible at https://github.com/hugheslab/snpgenie). Briefly, ORF sequences were translated, aligned at the amino acid level, and untranslated by using the ClustalW algorithm in MEGA6 (default settings) (27). These alignments were used to pool nucleotide diversity data for all within-host viral populations. Population-level πN and πS values were then calculated in sliding windows of 9 codons. Data were analyzed with R version 3.0.2 (32).
Cytokine and chemokine testing.
Cytokine testing in plasma was done by using a sandwich immunoassay-based protein array system, the Cytokine Monkey Magnetic 28-Plex panel (Invitrogen), according to the manufacturer's instructions. Results were read by using the Bio-Plex array reader (Bio-Rad Laboratories, Hercules, CA), which uses Luminex fluorescent-bead-based technology (Luminex Corporation, Austin, TX).
Statistical analyses of host data.
All analyses were conducted by using the MASS library (33) with the statistical programming language R (32). For demographic data, linear models were used to determine the relationship between viral infection (response variable) and host age, sex, and viral coinfection (predictor variables). For cytokine data, following outlier removal, the relationship between log cytokine concentration and age, sex, virus binary infection status (infected/uninfected for SIV, simian pegivirus, and simian arterivirus), and coinfection between SIV and simian pegivirus was assessed by using Gaussian-distributed linear models. For each cytokine, final models were selected by using stepwise backward elimination. Mixed-effect models that included sampling location as a random effect did not outperform the null expectation (models were compared using the Akaike information criterion) (results not shown) and were therefore not considered further.
Nucleotide sequence accession numbers.
Coding-complete genome sequences for SPgVver, DMVV-1, and SIVver have been made available in GenBank under accession numbers KR611946 to KR611983 and KR862293 to KR862363. Sequences for malbrouck SPgV (SPgVmal) and Zambian malbrouck virus 1 (ZMbV-1) can be found in GenBank under accession numbers KT166442 and KT166441, respectively.
RESULTS
Virus discovery.
We performed unbiased deep sequencing of RNA extracted from plasma collected from 12 malbroucks sampled at 3 locations in Zambia and 50 vervets sampled at 9 locations in South Africa. To demonstrate the sensitivity of this method for detecting RNA viruses in these samples, we first assessed our ability to detect SIVver, an SIV type previously detected in these samples by RT-PCR (23). We found 100% congruence between detection of SIVver by deep sequencing and detection by PCR (see Table S1 in the supplemental material for sequencing statistics and coverage details).
In addition to SIVver, deep sequencing revealed two novel pegiviruses, SPgVmal and SPgVver, infecting Zambian malbrouck monkeys and South African vervet monkeys, respectively. Compared to other pegiviruses, SPgVmal and SPgVver were most closely related to one another (Fig. 1A). These viruses were more closely related to simian pegiviruses from baboons (genus Papio) sampled in Zambia and Uganda than to simian pegiviruses from the more closely related but geographically distant sabaeus monkeys (Chlorocebus sabaeus) sampled in Gambia (9, 16, 22).
FIG 1.

Species-level phylogenetic relationships of all known pegiviruses (A) and arteriviruses (B). Viruses are shown adjacent to the silhouette of their respective host, with host common names in italics. Primate hosts are shown in black, and nonprimate hosts are shown in gray. Viruses discovered in this study are depicted by host silhouettes with solid coloring: green for AGM pegiviruses and blue for AGM arteriviruses. Host silhouettes with a colored outline draw attention to viruses of importance. Sabaeus SPgV (SPgVsab) has a green outline because this virus does not group with the AGM pegiviruses presented here. SHEV has a blue outline because of its close relationship to the AGM arteriviruses presented here. White host silhouettes symbolize arteriviruses that have caused outbreaks of viral hemorrhagic fever in captive macaques. Question marks emphasize that the natural host(s) of these viruses remains unknown. Shown is a maximum likelihood tree with 1,000 bootstrap replicates. Black dots indicate splits that are supported by 100% of bootstrap replicates. Bootstrap values below 70 are not shown. The bar shows the calculated genetic distance. SPgVkrc, Kibale red colobus; SPgVkrtg, Kibale red-tailed guenon; SPgVob, SPgV from olive baboon; SPgVmyb, Mikumi yellow baboon; BPgV, bat pegivirus; SPgVtri, owl monkey; SPgVcal-mx, marmoset-mystax; SPgVlab, tamarin; EqPgV, equine pegivirus; TDAV, Theiler's disease-associated virus; RPgV, rat PgV; KRTGV-1, Kibale red-tailed guenon virus; DeMBV-1, Debrazza's monkey virus; PBJV, Peter B. Jahrling virus; KKCBV-1, Kafue kinda-chacma baboon virus; MYBV-1, Mikumi yellow baboon virus; SWBV-1, Southwest baboon virus; KRCV-1, Kibale red colobus virus; LaDV-1, lactate dehydrogenase elevating virus; PRRSV-1, porcine reproductive and respiratory syndrome virus; APRAV-1, African pouched rat virus; EAV, equine arteritis virus; WPDV, wobbly possum disease virus.
We also discovered two novel simian arteriviruses, which we named Drakensberg Mountain vervet virus (DMVV-1) and Zambian malbrouck virus (ZMbV-1). These viruses contained the additional ORFs characteristic of all simian arteriviruses discovered to date (34, 35) and phylogenetically clustered with other simian arteriviruses (Fig. 1B). Of all known simian arteriviruses, DMVV-1 and ZMbV-1 were most closely related to one another. However, DMVV-1 and ZMbV-1 also fell within the same lineage as simian hemorrhagic encephalitis virus (SHEV). SHEV is the simian arterivirus responsible for the first recognized outbreak of simian hemorrhagic fever (SHF) in captive macaques in Sukhumi, former Soviet Union, in 1964 (20, 36), making DMVV-1 and ZMbV-1 the closest relatives of an SHF-causing virus identified to date.
Geographic distribution and prevalence.
To examine the prevalence of these viruses in a larger group of AGMs, we tested RNA extracted from the plasma samples of 161 South African vervet monkeys for the presence of SPgVver and DMVV-1 using highly sensitive qRT-PCR assays designed to amplify conserved regions of the SPgVver and DMVV-1 genomes. We also used deep-sequencing data, RT-PCR, and serological assays (described previously in reference 23) to screen for SIVver infection in these animals (see Table S1 in the supplemental material). Taken together with deep-sequencing data from Zambian malbroucks, SIVver (or SIVmal) and SPgVver (or SPgVmal) were present in monkeys from each sampling site, with 27 to 100% and 6 to 48% of individuals testing positive for lentiviral and pegiviral RNAs, respectively (Fig. 2). In contrast, DMVV-1 was found only in wild AGM populations west of the Drakensberg Mountains in South Africa (sites A, B, and C, i.e., Free State province), with 4 to 47% of individuals from these sites testing positive for viral RNA. ZMbV-1 was found in 25% of malbrouck monkeys in Zambia. All three virus types (lentiviruses, pegiviruses, and simian arteriviruses) were detected in the semi-free-living vervet monkeys of the Riverside Wildlife Rehabilitation Center (sites I and J), which serves as a semicaptive sanctuary for AGMs from across South Africa.
FIG 2.

Geographic distribution and prevalence of AGM plasma viruses. Shown are prevalences of SIV, SPgV, and simian arteriviruses in wild AGMs sampled from Zambia (malbrouck monkey [Chlorocebus cynosuros]) and South Africa (vervet monkey [Chlorocebus pygerythrus]). Venn diagrams of each monkey population show the percentages of uninfected, monoinfected, coinfected, and triple-infected monkeys for each of these three viruses. Gray circles represent the total numbers of monkeys sampled and are proportional to the sample size from each location. Numbers within the gray circles but outside the colored circles are the percentages of each population that are triple negative for virus (white). Colored circles within gray circles show the percentages of each population infected with SIV, SPgV, or DMVV-1/ZMbV-1 that are monoinfected, coinfected, or triple infected. Adjacent colored numbers outside the circles show the percentages of each population infected with the respective virus. Sites I and J are from the Riverside Rehabilitation Center, which houses vervet monkeys from across the region. The prevalence of each virus was determined by using a combination of deep sequencing, qRT-PCR, and RT-PCR.
Evolutionary relationships among viruses.
We amplified regions of the SPgVver and DMVV-1 genomes corresponding to the highly conserved nonstructural protein 3 (NS3) and ORF1b, respectively, from each monkey that tested positive for viral RNA by qRT-PCR that was not subjected to unbiased deep sequencing. Phylogenetic analysis of SPgVver and SPgVmal NS3 sequences revealed general clusters of sequence similarity reflective of the various sampling locations, although admixture among locations was observed (Fig. 3). In particular, SPgVver sequences from monkeys sampled west of the Drakensberg Mountains formed a monophyletic group representing a subclade of viral sequences sampled east of the mountains, which were more diverse and paraphyletic. Two SPgVver sequences (animals VSAM0017 and VSAM0018) from the southernmost sampling site (site M) were relatively divergent: one grouped with SPgVver in the western clade, and the other shared the greatest similarity with SPgVmal. The phylogeny of DMVV-1 and ZMbV-1 in wild AGMs found to the west of the Drakensberg Mountains revealed a pattern similar to that of SPgVver in animals west of the Drakensberg Mountains (Fig. 4). In both phylogenies, viruses from the Zambian malbroucks (SPgVmal and ZMbV-1) formed outgroups.
FIG 3.

Phylogenetic relationships of all SPgV variants discovered in this study. A PCR amplicon spanning the putative NS3 coding region of the SPgVver genome was sequenced from samples that tested positive for SPgV RNA by qRT-PCR that were not subjected to unbiased deep sequencing. Shown is a maximum likelihood tree, not rooted, with 1,000 bootstrap replicates. Black dots indicate splits that are supported by 100% of bootstrap replicates. Bootstrap values below 70 are not shown. The bar shows the calculated genetic distance.
FIG 4.
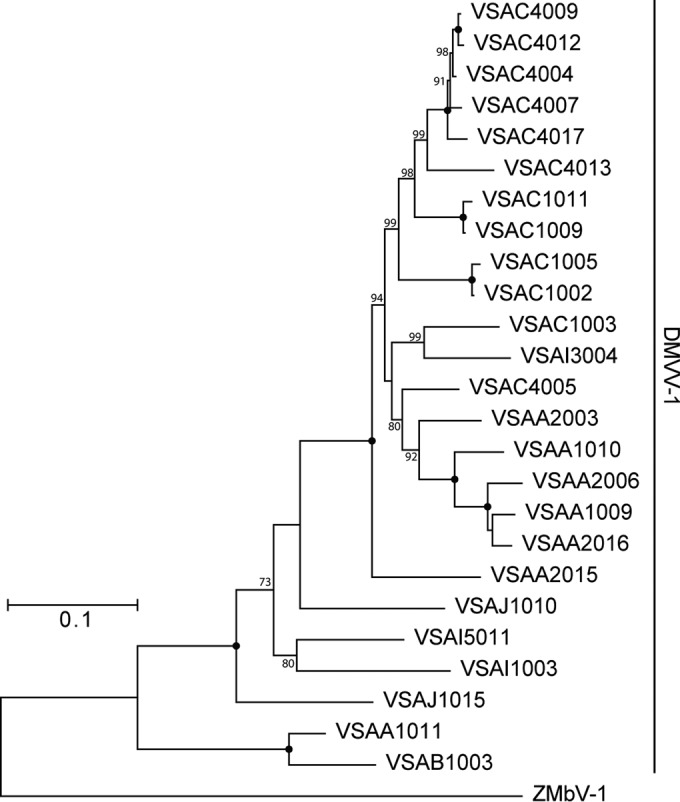
Phylogenetic relationships of all AGM simian arteriviruses discovered in this study. A PCR amplicon spanning ORF1b of the DMVV-1 genome was sequenced from samples that tested positive for DMVV-1 RNA by qRT-PCR that were not subjected to unbiased deep sequencing. Shown is a maximum likelihood tree, not rooted, with 1,000 bootstrap replicates. Black dots indicate splits that are supported by 100% of bootstrap replicates. Bootstrap values below 70 are not shown. The bar shows the calculated genetic distance.
Viremia.
To quantify viremia in infected vervet monkeys, we performed virus-specific qRT-PCR on RNA extracted from plasma collected from each vervet (additional analyses were not performed on samples from Zambian malbrouck monkeys due to very limited sample volumes). Virus titers of SPgVver and DMVV-1, averaging 1.98 × 107 and 1.89 × 107 viral genome copies per ml of plasma, respectively, were significantly higher than those detected for SIVver, which averaged 1.29 × 106 genome copies per ml of plasma in infected individuals (P < 0.001 and P < 0.001, respectively, by a two-tailed unpaired t test on log-transformed values) (Fig. 5A). Coinfection with either of the two other viruses did not have a significant effect on plasma titers of SIVver, SPgVver, or DMVV-1 (Fig. 5B and C).
FIG 5.

Viremia of the AGM plasma viruses. (A) SPgVver and DMVV-1 RNA concentrations were quantified from plasma samples of 161 South African vervet monkeys by using highly sensitive virus-specific qRT-PCR assays designed from deep-sequencing data. Only positive results are shown. SIVver loads were determined previously (23) by the same method. Significance was assessed by using a two-tailed unpaired t test on log-transformed values, with error bars showing the standard errors of the means. (B) South African vervets were stratified by coinfection status, and plasma viral load values for the virus in question were plotted. Significance was assessed by using a two-tailed unpaired t test on log-transformed values, with error bars showing the standard errors of the means. (C) Linear regression correlating the viral load of each virus in coinfected individuals. Data points with a colored halo indicate triple-infected individuals.
Viral genetic diversity.
Vast genetic diversity is a hallmark of primate lentiviruses and plays an integral role in the immune evasion (i.e., persistence) strategy employed by SIV/HIV (3, 37, 38). The genetic diversity of simian pegiviruses and simian arteriviruses is less well defined. To examine the genetic diversity of these viruses at the population level, we aligned consensus sequences spanning the entire protein-coding region of each viral genome obtained from South African vervet monkeys by deep sequencing. We then created a pairwise matrix to compare each viral sequence to all other sequences in the alignment. SIVver sequences shared the lowest percent identity on average (81.48% ± 0.23% [mean ± standard error of the mean {SEM}]) (Fig. 6A). However, when SIVver analysis was restricted to sampling sites A, B, and C, the percent identity among SIVver sequences was higher (86.35% ± 0.18% [mean ± SEM]) (Fig. 6A) and comparable to the percent identity observed for DMVV-1 sequences from these same locations (85.00% ± 0.38% [mean ± SEM]) (Fig. 6A). Although SPgVver was found to infect monkeys over a much larger geographical region than DMVV-1, the percent identity among SPgVver sequences was appreciably higher than that observed for DMVV-1 or SIVver sequences (95.65% ± 0.20% [mean ± SEM]) (Fig. 6A).
FIG 6.

Genetic diversity of AGM plasma viruses. (A) Complete consensus sequences spanning the entire coding region of each virus were aligned, and a pairwise comparison between each aligned sequence was performed. Percent identity values from each comparison were plotted and compared by using a two-tailed unpaired t test. Boxes show the middle two quartiles, and whiskers show the minimum and maximum percent identities observed. (B) Synonymous (πS) and nonsynonymous (πN) viral nucleotide diversities within each infected monkey, determined by calculating πS and πN values across the entire viral genome. Only samples that yielded virus sequences with >100× coverage for >99% of the protein-coding region of the genome were used for this analysis. Significance was assessed by using a two-tailed unpaired t test, with error bars showing the standard errors of the means. (C) Linear regression correlating within-host nucleotide diversity and plasma viral load for each virus.
To examine whether this pattern of genetic diversity was also observed at the level of the individual host, we determined the within-host synonymous (πS) and nonsynonymous (πN) nucleotide diversities of viral populations for infected vervet monkeys. For this analysis, we considered only samples that yielded sequences with >100× coverage for >99% of the protein-coding regions of the respective viral genome. There were highly significant differences among viruses with respect to both πS and πN (determined by a two-tailed unpaired t test) (Fig. 6B). Similar to our population-level analysis, we found that SIVver had the greatest within-host nucleotide diversity (mean πS, 1.82 × 10−2 differences/site; mean πN, 3.69 × 10−3 differences/site) (Fig. 6B). DMVV-1 displayed less within-host diversity than did SIVver (mean πS, 1.13 × 10−2; mean πN, 1.58 × 10−3), with an ∼2-fold lower πN value averaged across the genome. However, DMVV-1 diversity was still significantly greater than that of SPgVver (mean πS, 2.08 × 10−3; mean πN, 2.16 × 10−4), which had πS and πN values that were a full order of magnitude lower than those observed for SIVver (Fig. 6.B). Viral loads did not correlate significantly with πS or πN values obtained for SIVver, SPgVver, or DMVV-1 (Fig. 6C). For each virus studied, πS was significantly higher than πN (P < 0.001 for DMVV-1 and P < 0.01 for SPgVver and SIVver, determined by a paired t test), which is indicative of purifying selection acting across the viral genome (overall πN/πS ratios of 0.23 for SIVver, 0.19 for DMVV-1, and 0.21 for SPgVver).
To examine synonymous and nonsynonymous nucleotide diversities across the genome of each virus, we examined πS and πN in a sliding window of 9 codons across each ORF for each virus. For 19 of 23 total ORFs tested, πS significantly exceeded πN (determined by a paired t test), which is indicative of the widespread action of purifying selection (Table 1). Of the remaining ORFs, πN exceeded πS only in tat of SIV, and this difference was not significant. Regions experiencing peaks of nonsynonymous nucleotide diversity (πN > πS) indicative of overdominant positive selection were limited to the E1-, E2-, P7-, NS2-, NS5a-, and NS5b-encoding regions of SPgVver; the env and tat ORFs of SIVver; and ORFs 1a, TF, 2b, 3, 3′, 4, and 5 of DMVV (Table 2). The proportion of variants with nonsynonymous polymorphisms in these regions ranged from 12 to 47% in SPgVver, 31 to 92% in SIV, and 21 to 100% in DMVV. All 14 variants of DMVV-1 displayed nonsynonymous polymorphisms in a 22-codon region ranging from nucleotides 13922 to 13987 in the viral genome, a region that corresponds to the primary neutralizing antibody epitope that has been mapped in other arteriviruses (39–41), making this an extremely likely target of positive selection in most hosts.
TABLE 1.
Nonsynonymous and synonymous nucleotide diversities for all open reading frames of SPgVver, SIVver, and DMVV-1 in African green monkeysa
| Virus | ORF | Mean πN ± SE | Mean πS ± SE | P valueb |
|---|---|---|---|---|
| SIVver | gag*** | 0.002387 ± 0.00029 | 0.02153 ± 0.00185 | 1.24 × 10−35 |
| pol*** | 0.001595 ± 0.00014 | 0.01808 ± 0.00101 | 8.59 × 10−70 | |
| vif*** | 0.002705 ± 0.00038 | 0.00939 ± 0.00216 | 5.05 × 10−6 | |
| vpx*** | 0.001078 ± 0.00033 | 0.01158 ± 0.00319 | 1.62 × 10−6 | |
| tat | 0.006165 ± 0.00118 | 0.00351 ± 0.00137 | 0.189 | |
| rev*** | 0.005369 ± 0.00117 | 0.02000 ± 0.00324 | 9.87 × 10−6 | |
| env*** | 0.006657 ± 0.00057 | 0.02102 ± 0.00138 | 1.20 × 10−30 | |
| nef*** | 0.004809 ± 0.00069 | 0.01738 ± 0.00218 | 1.19 × 10−10 | |
| SPgVver | SPgV*** | 0.000214 ± 0.00003 | 0.00208 ± 0.00019 | 7.00 × 10−33 |
| DMVV-1 | 1a*** | 0.000954 ± 0.00008 | 0.01208 ± 0.00078 | 9.02 × 10−65 |
| TF | 0.001643 ± 0.00035 | 0.00287 ± 0.00088 | 0.0251 | |
| 1b*** | 0.000385 ± 0.00006 | 0.01289 ± 0.00071 | 1.70 × 10−82 | |
| 2a′*** | 0.001221 ± 0.00027 | 0.01175 ± 0.00158 | 5.73 × 10−13 | |
| 3′*** | 0.002715 ± 0.00052 | 0.01108 ± 0.00203 | 1.96 × 10−6 | |
| 4′*** | 0.001358 ± 0.00036 | 0.01023 ± 0.00195 | 1.96 × 10−8 | |
| 2a*** | 0.001255 ± 0.00044 | 0.01212 ± 0.00262 | 5.41 × 10−5 | |
| 2b** | 0.001889 ± 0.00032 | 0.00551 ± 0.00109 | 0.000391 | |
| 3*** | 0.003706 ± 0.00064 | 0.01117 ± 0.00179 | 3.34 × 10−5 | |
| 4 | 0.005481 ± 0.00082 | 0.00730 ± 0.00124 | 0.0433 | |
| 5a* | 0.007072 ± 0.00153 | 0.02214 ± 0.00511 | 0.000655 | |
| 5 | 0.009080 ± 0.00181 | 0.01100 ± 0.00195 | 0.156 | |
| 6*** | 0.000371 ± 0.00014 | 0.00705 ± 0.00180 | 4.45 × 10−6 | |
| 7*** | 0.001382 ± 0.00033 | 0.00887 ± 0.00197 | 2.68 × 10−5 |
Bonferroni significance levels were used to account for the use of 23 tests, one for each open reading frame, as follows: *, α value of <0.05 if the P value was <0.00217; **, α value of <0.01 if the P value was <0.000435; ***, α value of <0.001 if the P value was <4.35 × 10−5. Significance levels refer to a paired t test where πN equals πS.
Determined by a paired t test.
TABLE 2.
Peaks of nonsynonymous nucleotide diversity for all open reading frames of SPgVver, SIVver, and DMVV-1 in South African vervet monkeysa
| Virus | ORF | Start position | Stop position | Length (no. of codons) | % of variants with nonsynonymous polymorphism |
|---|---|---|---|---|---|
| SIVver (n = 13) | gag | — | — | — | — |
| pol | — | — | — | — | |
| vif | — | — | — | — | |
| vpx | — | — | — | — | |
| tat | 5685 | 5735 | 17 | 31 | |
| 7980 | 8120 | 47 | 85 | ||
| rev | — | — | — | — | |
| env | 6107 | 6226 | 40 | 92 | |
| 6335 | 6376 | 14 | 85 | ||
| 7013 | 7078 | 22 | 62 | ||
| nef | — | — | — | — | |
| SPgVver (n = 17) | 1053 | 1082 | 10 | 24 | |
| 1326 | 1376 | 17 | 29 | ||
| 2244 | 2282 | 13 | 24 | ||
| 2403 | 2438 | 12 | 12 | ||
| 2604 | 2630 | 9 | 29 | ||
| 6696 | 6746 | 17 | 12 | ||
| 8922 | 8966 | 15 | 47 | ||
| DMVV-1 (n = 14) | 1a | 733 | 768 | 12 | 21 |
| TF | 2924 | 3064 | 47 | 71 | |
| 3152 | 3232 | 27 | 50 | ||
| 3512 | 3541 | 10 | 21 | ||
| 1b | — | — | — | — | |
| 2a′ | — | — | — | — | |
| 3′ | 11389 | 11442 | 18 | 64 | |
| 11527 | 11565 | 13 | 57 | ||
| 4′ | — | — | — | — | |
| 2a | — | — | — | — | |
| 2b | 12470 | 12526 | 19 | 79 | |
| 3 | 13201 | 13230 | 10 | 64 | |
| 4 | 13227 | 13307 | 27 | 86 | |
| 13512 | 13571 | 20 | 71 | ||
| 5a | — | — | — | — | |
| 5 | 13748 | 13849 | 34 | 79 | |
| 13922 | 13987 | 22 | 100 | ||
| 6 | — | — | — | — | |
| 7 | — | — | — | — |
Peaks were identified conservatively as 9-codon sliding windows in which πN exceeded both the respective window's πS and the mean value of πS for the ORF. Start and stop sites refer to approximate nucleotide coordinates in the genome sequence. Dashes indicate the absence of a nonsynonymous peak in that ORF.
Individual-level correlates of infection.
Several host variables associated with natural SIV infection have been described (3, 23). However, host parameters associated with simian pegivirus and simian arterivirus infections have not been evaluated in wild NHPs, including coinfection with other viruses. Consistent with data from previous studies, we found that increased age and female sex were associated with SIV infection in South African vervets (Table 3). In contrast, neither age nor sex was significantly associated with SPgVver or DMVV-1 infection. We found a strong association between SIVver and SPgVver coinfections, similar to the association observed between HIV-1 and HPgV infections in people (42). However, DMVV-1 infection was not significantly associated with either SPgVver or SIVver coinfection.
TABLE 3.
Relationship between viral infection status and demographic features of South African vervet monkeys
| Response variable | Predictor variablea | df | t | P |
|---|---|---|---|---|
| SIVver | Age* | 112 | 6.208 | <0.001 |
| Male sex* | −2.287 | 0.02 | ||
| SPgV infection* | 4.402 | <0.001 | ||
| DMVV-1 infection | 0.585 | 0.559 | ||
| SPgVver | Age | 112 | −0.938 | 0.350 |
| Male sex | 1.241 | 0.217 | ||
| SIV infection* | 4.402 | <0.001 | ||
| DMVV-1 infection | 1.420 | 0.158 | ||
| DMVV-1 | Age | 112 | −0.361 | 0.719 |
| Male sex | −0.675 | 0.501 | ||
| SIV infection | 0.585 | 0.560 | ||
| SPgV infection | 1.420 | 0.158 |
Asterisks signify predictor variables with a statistically significant relationship.
Cytokines associated with viral infection and coinfection.
We obtained data for 28 different plasma cytokines evaluated in 118 vervet monkeys (reported previously in reference 23) and used multivariate linear models to assess the effect of SIVver infection, SPgVver infection, SIVver/SPgVver coinfection, and DMVV-1 infection on plasma cytokine concentrations. We found that plasma concentrations of several cytokines were negatively correlated with age and female sex (Table 4).
TABLE 4.
Relationship between viral infection status and plasma cytokine concentrations in South African vervet monkeysa
| Virus | Cytokine | t value for cytokine | P value (uncorrected) for cytokine | P (corrected) for cytokine | Model covariate | t value for model covariate | P value (uncorrected) for model covariate | P (corrected) for model covariate |
|---|---|---|---|---|---|---|---|---|
| DMVV-1 | EGF | 2.27 | 0.040 | >1.0 | Age | −2.07 | 0.040 | >1.0 |
| GM-CSF | 3.19 | 0.002 | 0.05 | Age | −0.27 | 0.788 | >1.0 | |
| Sex (male) | 2.22 | 0.029 | 0.81 | |||||
| IL-1β | 2.29 | 0.024 | 0.67 | Age | −2.42 | 0.017 | 0.48 | |
| Sex (male) | 3.19 | 0.002 | 0.05 | |||||
| IL-10 | 2.36 | 0.020 | 0.56 | Sex (male) | 1.71 | 0.089 | >1.0 | |
| MCP-1 | 1.95 | 0.049 | >1.0 | Age | −2.05 | 0.043 | >1.0 | |
| Sex (male) | 3.83 | <0.01 | <0.01 | |||||
| MIP-1β | 3.09 | 0.003 | 0.07 | Age | −2.53 | 0.012 | 0.36 | |
| TNF-α | 2.10 | 0.038 | >1.0 | Sex (male) | 2.10 | 0.038 | >1.0 | |
| SPgVver | MIG | −2.24 | 0.027 | 0.76 | Sex (male) | 4.06 | <0.01 | <0.01 |
| SIV | −0.84 | 0.404 | >1.0 | |||||
| SIV*SPgV | 2.46 | 0.016 | 0.43 | |||||
| SIVver | IFN-γ | −2.79 | 0.006 | 0.18 | ||||
| IL-6 | 1.79 | 0.043 | >1.0 | SPgV | −1.19 | 0.236 | >1.0 | |
| SIV*SPgV | 1.52 | 0.131 | >1.0 | |||||
| I-TAC | 2.20 | 0.030 | 0.84 | |||||
| MIF | 2.15 | 0.034 | 0.95 | Age | −2.14 | 0.034 | 0.95 |
A multivariate linear model was developed, and model covariates were identified by using stepwise backward elimination. Uncorrected P values are those inferred by the model. Corrected P values represent Bonferroni-corrected P values for a total of 28 hypotheses tested (one for each cytokine). For a full list of cytokines tested, see Table S1 in the supplemental material.
With respect to viral infection, we found a positive association between the presence of SIVver and plasma concentrations of interleukin-6 (IL-6), as reported previously (23). We also found positive associations between the presence of SIVver and plasma concentrations of interferon-inducible T cell alpha chemoattractant (I-TAC) (i.e., CXCL11) and macrophage inhibition factor (MIF) and a negative association between interferon gamma (IFN-γ) and the presence of SIVver. SPgVver infection was not associated with changes in any plasma cytokine concentrations, with the exception of monokine induced by interferon gamma (MIG) (i.e., CXCL9), for which we found a negative association. When we examined cytokine concentrations in SIVver-infected animals, SPgVver coinfection did not significantly impact cytokine levels. Additionally, there was a suggestion that the plasma concentrations of several cytokines were increased in association with DMVV-1 infection, including several cytokines and chemokines produced by activated macrophages (IL-1β, granulocyte macrophage colony-stimulating factor [GM-CSF], macrophage inflammatory protein 1β [MIP-1β], epidermal growth factor [EGF], monocyte chemoattractant protein 1[MCP-1], and tumor necrosis factor alpha [TNF-α]) and the anti-inflammatory cytokine IL-10. Importantly, none of the associations that we found between viral infection and plasma cytokine concentrations reached statistical significance after correction for multiple comparisons (Bonferroni correction).
DISCUSSION
Wild AGMs have long been known to harbor species-specific variants of SIV (3) and have played an important role in SIV/HIV pathogenesis research as a “natural host” (5). However, other coinfecting viruses of AGMs have not been similarly well characterized. Here, we searched for blood-borne RNA viruses in AGMs from Zambia and several locations in South Africa using molecular techniques. In addition to SIVver, we found new viruses from two other genera, pegivirus and simian arterivirus, providing the first comparative analysis of viral infections with these viruses, in their natural state.
Defining the OWM plasma virome: variations on a theme.
Our discovery of novel pegiviruses and simian arteriviruses in AGMs in Zambia and South Africa adds to an emerging theme: with few exceptions (43), the plasma RNA virome of African monkeys is a distinct and definable entity consisting of various combinations of simian immunodeficiency viruses, simian pegiviruses, and simian arteriviruses. To date, species-specific simian pegiviruses and simian arteriviruses have been found in cercopithecoid monkeys belonging to both major African subfamilies (i.e., Cercopithecinae and Colobinae) and both cercopithecine tribes (i.e., Cercopithecini and Papionini). This finding suggests that like SIV, simian pegiviruses and simian arteriviruses are widely distributed among African monkeys of different species, with many more variants likely remaining to be discovered. Our discovery of these viruses in monkeys from South Africa also greatly expands the known geographic range of these virus groups in African monkeys (Fig. 7).
FIG 7.

Known geographic and host ranges of African monkey plasma viruses. (A) Map of Africa showing the sampling locations of monkeys in which simian pegivirus or simian arterivirus infection was identified. In the table, a colored dot indicates that SIV (red), SPgV (green), or a simian arterivirus (blue) infection was detected in that particular primate from that particular location (61–64). NYP, not yet published. (B) Genus-level phylogenetic tree of African OWMs and great apes. Colored dots indicate that SIV (red), GBV-C (green), or SHFV (blue) infection was detected in a primate from that genus. Names in boldface type indicate genera from which we have sampled more than 10 wild primates by unbiased deep sequencing (for a comprehensive list of species naturally infected with SIV, see reference 65). (Adapted from PLoS Genetics [66].)
The widespread distribution of pegiviruses in African monkeys is not entirely surprising because pegiviruses are already known to infect a diversity of primates, including humans (HPgV), chimpanzees (SPgVcpz), and New World monkeys (SPgV-A). This wide species range suggests that pegiviruses may have infected an ancient ancestor common to Old World and New World monkeys and cospeciated with these hosts, a hypothesis generally supported by the phylogenetic relationships among pegiviruses from these different hosts. However, a more detailed examination of the simian pegivirus phylogeny suggests a more nuanced history, with evidence for cross-species transmission of simian pegiviruses among OWMs and a viral phylogeny seemingly better predicted by host geographical location than by host phylogeny. More extensive sampling and more detailed analyses would be required to differentiate cospeciation from geographic spread of simian pegiviruses, as has been done for SIV (44).
Locally within South Africa, geography seems to be important for the distribution of simian arteriviruses, as evidenced by the lack of detection of simian arteriviruses in AGMs east of the Drakensberg Mountains. The absence of simian arteriviruses in AGMs from Gambia (Chlorocebus sabaeus) despite the detection of simian pegiviruses and SIV in these monkeys by unbiased deep sequencing suggests that simian arteriviruses generally have a patchy distribution across African OWM populations (22). One possible explanation for this observation is that simian arteriviruses are susceptible to “bottleneck” effects and local extinction, as was recently shown in the case of SIV and SPgV in the AGMs that now populate the Caribbean island of St. Kitts (22). Alternatively, it is possible that simian arteriviruses entered African OWMs relatively recently and have not yet spread beyond certain geographical confines. Sampling of wild primates from additional geographic locations, including locations that allow geography-based dating of simian pegiviruses and simian arteriviruses, similar to what has been done for SIV (45), will hopefully provide additional data points and add clarity to the natural histories of these viruses.
Genetic and genomic features of RNA plasma viruses in African monkeys.
Lifelong viremia is a hallmark of SIV infection and explains why the prevalence of SIV infection in AGMs increases with age. Persistent/prolonged infection also appears to be a feature of both simian pegivirus and simian arterivirus infections, which may explain the relatively high prevalence of these viruses in this study (9, 11). However, we did not find any correlation between the prevalence of simian pegivirus or simian arterivirus and age in AGMs, suggesting that these infections are not lifelong but are cleared after some undetermined period of time.
A key component of SIV's persistence strategy is the integration of proviral DNA into the host's genome, a persistence mechanism that, to our knowledge, is not utilized by simian pegiviruses or simian arteriviruses. However, SIV also relies upon several additional methods to maintain high levels of viral replication. The rapid accumulation of mutations that alter the amino acid sequence of viral proteins targeted by host antibodies and T cells decreases the effectiveness of these responses and enables viral persistence, a mechanism known as “immune escape,” which leaves a signature of nonsynonymous nucleotide diversity in the viral genome (37, 38). To gain insight into the persistence mechanisms used by simian pegiviruses and simian arteriviruses, we searched for signatures of nonsynonymous nucleotide diversity in the genomes of SIVver, SPgVver, and DMVV-1. As expected, we found hotspots of nonsynonymous variation in the regions of the SIVver envelope gene that correspond to the “highly variable loops” that are preferentially targeted by host antibodies (23, 46). We found a similar pattern of adaptive changes in the DMVV-1 genome, with signatures of nonsynonymous diversity found primarily in genes that code for envelope glycoproteins and/or regions corresponding to antibody epitopes that have been extensively mapped in other arteriviruses (47–51). This observation suggests that vervet monkeys infected with DMVV-1 produce antibodies that target DMVV-1 glycoproteins but that these antibodies may not be effective in clearing DMVV-1 infection, a scenario that may be akin to immune escape observed during SIV infection. Longitudinal studies of monkeys infected with simian arteriviruses in captivity would be required to more thoroughly evaluate the relationship between host immune responses and viral sequence changes.
Low levels of nonsynonymous diversity were found in the SPgVver genome despite the high levels of viremia found within infected individuals. This finding is consistent with data from previous studies of pegiviruses in captive NHPs (9, 52, 53) and suggests that the persistence mechanism(s) used by pegiviruses is fundamentally different from that of DMVV-1 or SIV, that pegivirus genomes are relatively constrained by purifying selection, or that pegivirus genomes are subject to a much lower mutation rate, as also suggested by their low levels of synonymous diversity (54). Whether the low level of nonsynonymous genetic diversity observed in simian pegiviruses is due to an increase in selective constraint, an inherently low mutation rate, or both remains an important question that future in vivo and in vitro studies need to address.
Pathogenesis and coinfection.
The study of SIV infection in wild and captive African monkeys has been invaluable for understanding the pathogenesis of HIV, because unlike HIV in humans, SIV infection in these natural hosts does not lead to the development of AIDS (5). The leading explanation for the apparently low pathogenicity of SIV in African monkeys is that SIV has coevolved with these hosts over thousands of years, and this host-virus relationship has now reached a state of near commensalism. However, the impact of simian pegivirus infection on SIV pathogenesis in African monkeys has never been assessed, as pegiviruses in African monkeys have been described only recently. Given that HPgV infection attenuates HIV pathogenesis and improves mortality in HIV-infected humans (17–19, 55), we examined the impact of SPgVver coinfection on cytokines influenced by SIVver using a multivariate linear model. We did not find a significant difference in cytokine concentrations in SIVver/SPgVver-coinfected monkeys compared to SIVver-monoinfected monkeys, nor did we find a difference in SIV loads between SPgVver-positive and SPgVver-negative AGMs. These findings suggest that simian pegiviruses do not significantly impact SIV pathogenesis (as measured by cytokine levels) in African OWMs, possibly because SIV in these natural hosts (unlike SIV in macaque monkeys and HIV in humans) causes little to no change in these parameters.
Similarly, we used a multivariate linear model to identify cytokines associated with DMVV-1 infection in vervet monkeys. In DMVV-1-infected animals, we found mildly elevated concentrations of several proinflammatory cytokines that are produced by activated macrophages, including IL-1β, GM-CSF, MIP-1β, EGF, MCP-1, and TNF-α. Although these associations did not reach statistical significance, this pattern of cytokine associations is biologically intriguing: macrophages support the replication of simian hemorrhagic fever virus (SHFV) (the simian arterivirus type strain) in vitro and are thought to be the primary cell type infected by simian arteriviruses in vivo, as has been shown for other nonsimian arteriviruses (34, 56). Additionally, increased expression of IL-10, an anti-inflammatory cytokine which we also found to have elevated levels in DMVV-1-infected vervets (albeit to nonsignificant levels), is associated with a reduction in virulence for both simian and nonsimian arteriviruses (56–58). Macaques infected with SHFV have increased plasma concentrations of proinflammatory cytokines but not increased levels of IL-10, possibly suggesting that IL-10 may be a host factor that protects African monkeys from simian arterivirus pathogenesis and SHF (59, 60).
Taken together, these data suggest that the simian lentiviruses, simian pegiviruses, and simian arteriviruses have a small to negligible effect on blood cytokine profiles in South African vervet monkeys. Extrapolating from these findings, we predict that these viral infections result in little to no overt disease in African OWMs, although the examination of other disease parameters, with greater statistical power, is needed to further clarify the long-term effect of these viral infections on NHP health.
Conclusion.
It is reasonable to expect that viruses from additional families will be discovered in the coming years as the sensitivity and throughput of virus discovery technology improve. Indeed, the prevalence of simian immunodeficiency viruses, simian pegiviruses, and simian arteriviruses in many OWM populations and the high levels of viremia that these viruses cause in infected individuals have simplified their discovery and characterization. Viruses that cause acute infections, replicate to low titers, or are primarily cell associated are undoubtedly more difficult to identify with current sequencing-based methods. While efforts to discover novel animal viruses will continue to be of high yield, further characterization of newly discovered viruses in controlled laboratory settings will also be important, especially for select viruses with the potential to impact human and wildlife health. Such studies are already under way for simian pegiviruses and simian arteriviruses (9, 21). Considering that NHP models faithfully recapitulate many important features of viral diseases in humans, future research to discover novel primate viruses has the potential to be particularly fruitful.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University of Wisconsin Department of Pathology and Laboratory Medicine and the Wisconsin National Primate Research Center (WNPRC) for the use of their facilities and services. We thank Erin Bailey, Thomas Friedrich, and Jeffrey Rogers for helpful discussion. We thank Jiro Wada (IRF-Frederick) for artistic rendering of animal silhouettes and Laura Bollinger (IRF-Frederick) for critically editing the manuscript. We thank the Department of Environmental Affairs, South Africa; Department of Tourism, Environmental and Economic Affairs, Free State Province; Ezemvelo KZN Wildlife in KwaZulu Natal Province; the Department of Economic Development and Environmental Affairs, Eastern Cape; and the Zambia Wildlife Authority.
This publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of the Office of Research Infrastructure Programs, NIH, U.S. Department of Health and Human Services, or of the institutions and companies affiliated with the authors. The funders of this research had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
This work is dedicated to the memory of Austin L. Hughes, who died unexpectedly during the preparation of the manuscript. He was our collaborator, mentor, and friend, and we are deeply indebted to him for the great contributions he made, both to our lives and to this work.
Funding Statement
This work was funded by the National Institutes of Health (NIH) (grants R01AI077376-01, R01AI084787, R01RR025781, P01AI088564, and R01AI116382-01), the National Science Foundation (NSF) (grants NSF1029302, NSF1029323, NSF1029451, and DGE0929297), the joint NIH-NSF Ecology of Infectious Diseases program and the United Kingdom Economic and Social Research Council (grant TW009237), and the Wisconsin Partnership Program through the Wisconsin Center for Infectious Diseases. This publication was made possible in part by grants from the National Institutes of Health National Center for Research Resources (grant P51RR000167 to the WNPRC, University of Wisconsin—Madison, and grant R01OD010980, formerly R01RR016300, to the University of California, Los Angeles) and grant OD011104 to the Tulane National Primate Research Center from the Office of Research Infrastructure Programs (ORIP). This research was conducted in part at a facility constructed with support from Research Facilities Improvement Program grants RR15459-01 and RR020141-01. A.L.B. performed this work with support from the University of Wisconsin Medical Scientist Training Program (MSTP) (grant T32 GM008692) and a National Research Service Award (NRSA) through the Microbes in Health and Disease (MHD) training program at the University of Wisconsin (grant T32 AI55397). C.W.N. performed this work with support from NSF Graduate Research Fellowship DGE-0929297, the University of South Carolina (USC) Presidential Fellowship, and the USC Department of Biological Sciences Kathryn Hinnant-Johnson, M.D., Memorial Fellowship. This work was also funded in part through Battelle Memorial Institute's prime contract with the U.S. National Institute of Allergy and Infectious Diseases (NIAID) under contract no. HHSN272200700016I. A subcontractor to Battelle Memorial Institute who performed this work is J.H.K., an employee of Tunnell Government Services, Inc.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00573-16.
REFERENCES
- 1.Wolfe ND, Dunavan CP, Diamond J. 2007. Origins of major human infectious diseases. Nature 447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies TJ, Pedersen AB. 2008. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc Biol Sci 275:1695–1701. doi: 10.1098/rspb.2008.0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apetrei C, Robertson DL, Marx PA. 2004. The history of SIVS and AIDS: epidemiology, phylogeny and biology of isolates from naturally SIV infected non-human primates (NHP) in Africa. Front Biosci 9:225–254. doi: 10.2741/1154. [DOI] [PubMed] [Google Scholar]
- 4.Sharp PM, Hahn BH. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1:a006841. doi: 10.1101/cshperspect.a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. 2012. Natural SIV hosts: showing AIDS the door. Science 335:1188–1193. doi: 10.1126/science.1217550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper N, Nunn CL. 2013. Identifying future zoonotic disease threats: where are the gaps in our understanding of primate infectious diseases? Evol Med Public Health 2013:27–36. doi: 10.1093/emph/eot001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Ali Khan S, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JA, Daszak P, Lipkin WI. 2013. A strategy to estimate unknown viral diversity in mammals. mBio 4:e00598-13. doi: 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones RC, Greek R. 2014. A review of the Institute of Medicine's analysis of using chimpanzees in biomedical research. Sci Eng Ethics 20:481–504. doi: 10.1007/s11948-013-9442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey AL, Lauck M, Mohns M, Peterson EJ, Beheler K, Brunner KG, Crosno K, Mejia A, Mutschler J, Gehrke M, Greene J, Ericsen AJ, Weiler A, Lehrer-Brey G, Friedrich TC, Sibley SD, Kallas EG, Capuano S, Rogers J, Goldberg TL, Simmons HA, O'Connor DH. 2015. Durable sequence stability and bone marrow tropism in a macaque model of human pegivirus infection. Sci Transl Med 7:305ra144. doi: 10.1126/scitranslmed.aab3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey AL, Lauck M, Weiler A, Sibley SD, Dinis JM, Bergman Z, Nelson CW, Correll M, Gleicher M, Hyeroba D, Tumukunde A, Weny G, Chapman C, Kuhn JH, Hughes AL, Friedrich TC, Goldberg TL, O'Connor DH. 2014. High genetic diversity and adaptive potential of two simian hemorrhagic fever viruses in a wild primate population. PLoS One 9:e90714. doi: 10.1371/journal.pone.0090714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey AL, Lauck M, Sibley SD, Pecotte J, Rice K, Weny G, Tumukunde A, Hyeroba D, Greene J, Correll M, Gleicher M, Friedrich TC, Jahrling PB, Kuhn JH, Goldberg TL, Rogers J, O'Connor DH. 2014. Two novel simian arteriviruses in captive and wild baboons (Papio spp.). J Virol 88:13231–13239. doi: 10.1128/JVI.02203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauck M, Switzer WM, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Taylor B, Shankar A, Ting N, Chapman CA, Friedrich TC, Goldberg TL, O'Connor DH. 2013. Discovery and full genome characterization of two highly divergent simian immunodeficiency viruses infecting black-and-white colobus monkeys (Colobus guereza) in Kibale National Park, Uganda. Retrovirology 10:107. doi: 10.1186/1742-4690-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauck M, Bailey AL, Andersen KG, Goldberg TL, Sabeti PC, O'Connor DH. 2015. GB virus C coinfections in West African Ebola patients. J Virol 89:2425–2429. doi: 10.1128/JVI.02752-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauck M, Hyeroba D, Tumukunde A, Weny G, Lank SM, Chapman CA, O'Connor DH, Friedrich TC, Goldberg TL. 2011. Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS One 6:e19056. doi: 10.1371/journal.pone.0019056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauck M, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Chapman CA, Ting N, Switzer WM, Kuhn JH, Friedrich TC, O'Connor DH, Goldberg TL. 2013. Exceptional simian hemorrhagic fever virus diversity in a wild African primate community. J Virol 87:688–691. doi: 10.1128/JVI.02433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sibley SD, Lauck M, Bailey AL, Hyeroba D, Tumukunde A, Weny G, Chapman CA, O'Connor DH, Goldberg TL, Friedrich TC. 2014. Discovery and characterization of distinct simian pegiviruses in three wild African Old World monkey species. PLoS One 9:e98569. doi: 10.1371/journal.pone.0098569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanteri MC, Vahidnia F, Tan S, Stapleton JT, Norris PJ, Heitman J, Deng X, Keating S, Brambilla D, Busch MP, Custer B. 2015. Downregulation of cytokines and chemokines by GB virus C after transmission via blood fusion in HIV-positive blood recipients. J Infect Dis 211:1585–1596. doi: 10.1093/infdis/jiu660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, Chaloner K, Tillmann HL, Williams CF, Stapleton JT. 2006. Effect of early and late GB virus C viraemia on survival of HIV-infected individuals: a meta-analysis. HIV Med 7:173–180. doi: 10.1111/j.1468-1293.2006.00366.x. [DOI] [PubMed] [Google Scholar]
- 19.Bhattarai N, Stapleton JT. 2012. GB virus C: the good boy virus? Trends Microbiol 20:124–130. doi: 10.1016/j.tim.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauck M, Alkhovsky SV, Bào Y, Bailey AL, Shevtsova ZV, Shchetinin AM, Vishnevskaya TV, Lackemeyer MG, Postnikova E, Mazur S. 2015. Historical outbreaks of simian hemorrhagic fever in captive macaques were caused by distinct arteriviruses. J Virol 89:8082–8087. doi: 10.1128/JVI.01046-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey AL, Lauck M, Sibley SD, Friedrich TC, Kuhn JH, Freimer NB, Jasinska AJ, Phillips-Conroy JE, Jolly CJ, Marx PA, Apetrei C, Rogers J, Goldberg TL, O'Connor DH. 2016. Zoonotic potential of simian arteriviruses. J Virol 90:630–635. doi: 10.1128/JVI.01433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapusinszky B, Mulvaney U, Jasinska AJ, Deng X, Freimer N, Delwart E. 2015. Local virus extinctions following a host population bottleneck. J Virol 89:8152–8161. doi: 10.1128/JVI.00671-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma D, Jasinska A, Kristoff J, Grobler JP, Turner T, Jung Y, Schmitt C, Raehtz K, Feyertag F, Martinez Sosa N, Wijewardana V, Burke DS, Robertson DL, Tracy R, Pandrea I, Freimer N, Apetrei C, International Vervet Research Consortium. 2013. SIVagm infection in wild African green monkeys from South Africa: epidemiology, natural history, and evolutionary considerations. PLoS Pathog 9:e1003011. doi: 10.1371/journal.ppat.1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grobler JP, Turner TR. 2010. A novel trap design for the capture and sedation of vervet monkeys (Chlorocebus aethiops). S Afr J Wildl Res 40:163–168. doi: 10.3957/056.040.0208. [DOI] [Google Scholar]
- 25.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 29.Nelson CW, Moncla LH, Hughes AL. 2015. SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 31:3709–3711. doi: 10.1093/bioinformatics/btv449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson CW, Hughes AL. 2015. Within-host nucleotide diversity of virus populations: insights from next-generation sequencing. Infect Genet Evol 30:1–7. doi: 10.1016/j.meegid.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. [DOI] [PubMed] [Google Scholar]
- 32.R Core Team. 2015. R: a language and environment for statistical computing. R Foundation For Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 33.Venables WN, Ripley BD. 2002. Modern applied statistics with S. Springer-Verlag, New York, NY. [Google Scholar]
- 34.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bào YM, Ng TFF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo Jle D, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O'Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lapin BA, Pekerman SM, Iakovleva LA, Dzhikidze EK, Shevtsova ZV, Kuksova MI, Dan'ko LV, Krylova RI, Akbroit EI, Agrba VZ. 1967. Hemorrhagic fever in monkeys. Vopr Virusol 12:168–173. (In Russian.) [PubMed] [Google Scholar]
- 37.da Silva J. 2003. The evolutionary adaptation of HIV-1 to specific immunity. Curr HIV Res 1:363–371. doi: 10.2174/1570162033485249. [DOI] [PubMed] [Google Scholar]
- 38.Snoeck J, Fellay J, Bartha I, Douek DC, Telenti A. 2011. Mapping of positive selection sites in the HIV-1 genome in the context of RNA and protein structural constraints. Retrovirology 8:87. doi: 10.1186/1742-4690-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang Y, Kim DY, Ropp S, Steen P, Christopher-Hennings J, Nelson EA, Rowland RR. 2004. Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Res 100:229–235. doi: 10.1016/j.virusres.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 40.Oleksiewicz MB, Bøtner A, Toft P, Normann P, Storgaard T. 2001. Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: the nsp2 fragment of the replicase polyprotein contains a cluster of B-cell epitopes. J Virol 75:3277–3290. doi: 10.1128/JVI.75.7.3277-3290.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang FX, Song N, Chen LZ, Cheng SP, Wu H, Wen YJ. 2013. Non-structural protein 2 of the porcine reproductive and respiratory syndrome (PRRS) virus: a crucial protein in viral pathogenesis, immunity and diagnosis. Res Vet Sci 95:1–7. doi: 10.1016/j.rvsc.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Compston LI, Li C, Sarkodie F, Owusu-Ofori S, Opare-Sem O, Allain JP. 2009. Prevalence of persistent and latent viruses in untreated patients infected with HIV-1 from Ghana, West Africa. J Med Virol 81:1860–1868. doi: 10.1002/jmv.21614. [DOI] [PubMed] [Google Scholar]
- 43.Lauck M, Sibley SD, Lara J, Purdy MA, Khudyakov Y, Hyeroba D, Tumukunde A, Weny G, Switzer WM, Chapman CA, Hughes AL, Friedrich TC, O'Connor DH, Goldberg TL. 2013. A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild Old World primate. J Virol 87:8971–8981. doi: 10.1128/JVI.00888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charleston MA, Robertson DL. 2002. Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst Biol 51:528–535. doi: 10.1080/10635150290069940. [DOI] [PubMed] [Google Scholar]
- 45.Worobey M, Telfer P, Souquière S, Hunter M, Coleman CA, Metzger MJ, Reed P, Makuwa M, Hearn G, Honarvar S, Roques P, Apetrei C, Kazanji M, Marx PA. 2010. Island biogeography reveals the deep history of SIV. Science 329:1487. doi: 10.1126/science.1193550. [DOI] [PubMed] [Google Scholar]
- 46.Mayr LM, Zolla-Pazner S. 2015. Antibodies targeting the envelope of HIV-1. Microbiol Spectr 3:AID-0025-2014. doi: 10.1128/microbiolspec.AID-0025-2014. [DOI] [PubMed] [Google Scholar]
- 47.Balasuriya UB, Patton JF, Rossitto PV, Timoney PJ, McCollum WH, MacLachlan NJ. 1997. Neutralization determinants of laboratory strains and field isolates of equine arteritis virus: identification of four neutralization sites in the amino-terminal ectodomain of the G(L) envelope glycoprotein. Virology 232:114–128. doi: 10.1006/viro.1997.8551. [DOI] [PubMed] [Google Scholar]
- 48.Plagemann PG. 2001. Complexity of the single linear neutralization epitope of the mouse arterivirus lactate dehydrogenase-elevating virus. Virology 290:11–20. doi: 10.1006/viro.2001.1139. [DOI] [PubMed] [Google Scholar]
- 49.Plagemann PGW, Rowland RRR, Faaberg KS. 2002. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Arch Virol 147:2327–2347. doi: 10.1007/s00705-002-0887-2. [DOI] [PubMed] [Google Scholar]
- 50.Plagemann PGW. 2004. The primary GP5 neutralization epitope of North American isolates of porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol 102:263–275. doi: 10.1016/j.vetimm.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Ostrowski M, Galeota JA, Jar AM, Platt KB, Osorio FA, Lopez OJ. 2002. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol 76:4241–4250. doi: 10.1128/JVI.76.9.4241-4250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bukh J, Apgar CL. 1997. Five new or recently discovered (GBV-A) virus species are indigenous to New World monkeys and may constitute a separate genus of the Flaviviridae. Virology 229:429–436. doi: 10.1006/viro.1997.8461. [DOI] [PubMed] [Google Scholar]
- 53.Bukh J, Kim JP, Govindarajan S, Apgar CL, Foung SK, Wages J, Yun AJ, Shapiro M, Emerson SU, Purcell RH. 1998. Experimental infection of chimpanzees with hepatitis G virus and genetic analysis of the virus. J Infect Dis 177:855–862. doi: 10.1086/515255. [DOI] [PubMed] [Google Scholar]
- 54.Stapleton JT, Xiang J, McLinden JH, Bhattarai N, Chivero ET, Klinzman D, Kaufman TM, Chang Q. 2014. A novel T cell evasion mechanism in persistent RNA virus infection. Trans Am Clin Climatol Assoc 125:14–24; discussion 24–26. [PMC free article] [PubMed] [Google Scholar]
- 55.Xiang J, Wünschmann S, Diekema DJ, Klinzman D, Patrick KD, George SL, Stapleton JT. 2001. Effect of coinfection with GB virus C on survival among patients with HIV infection. N Engl J Med 345:707–714. doi: 10.1056/NEJMoa003364. [DOI] [PubMed] [Google Scholar]
- 56.Vatter HA, Brinton MA. 2014. Differential responses of disease-resistant and disease-susceptible primate macrophages and myeloid dendritic cells to simian hemorrhagic fever virus infection. J Virol 88:2095–2106. doi: 10.1128/JVI.02633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weesendorp E, Stockhofe-Zurwieden N, Popma-De Graaf DJ, Fijten H, Rebel JM. 2013. Phenotypic modulation and cytokine profiles of antigen presenting cells by European subtype 1 and 3 porcine reproductive and respiratory syndrome virus strains in vitro and in vivo. Vet Microbiol 167:638–650. doi: 10.1016/j.vetmic.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 58.Amarilla SP, Gómez-Laguna J, Carrasco L, Rodríguez-Gómez IM, Caridad y Ocerín JM, Morgan SB, Graham SP, Frossard JP, Drew TW, Salguero FJ. 2015. A comparative study of the local cytokine response in the lungs of pigs experimentally infected with different PRRSV-1 strains: upregulation of IL-1α in highly pathogenic strain induced lesions. Vet Immunol Immunopathol 164:137–147. doi: 10.1016/j.vetimm.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Johnson RF, Dodd LE, Yellayi S, Gu W, Cann JA, Jett C, Bernbaum JG, Ragland DR, St Claire M, Byrum R, Paragas J, Blaney JE, Jahrling PB. 2011. Simian hemorrhagic fever virus infection of rhesus macaques as a model of viral hemorrhagic fever: clinical characterization and risk factors for severe disease. Virology 421:129–140. doi: 10.1016/j.virol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vatter HA, Donaldson EF, Huynh J, Rawlings S, Manoharan M, Legasse A, Planer S, Dickerson MF, Lewis AD, Colgin LM, Axthelm MK, Pecotte JK, Baric RS, Wong SW, Brinton MA. 2015. A simian hemorrhagic fever virus isolate from persistently infected baboons efficiently induces hemorrhagic fever disease in Japanese macaques. Virology 474:186–198. doi: 10.1016/j.virol.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma D, Jasinska AJ, Feyertag F, Wijewardana V, Kristoff J, He T, Raehtz K, Schmitt CA, Jung Y, Cramer JD. 2014. Factors associated with simian immunodeficiency virus transmission in a natural African nonhuman primate host in the wild. J Virol 88:5687–5705. doi: 10.1128/JVI.03606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bibollet-Ruche F, Bailes E, Gao F, Pourrut X, Barlow KL, Clewley JP, Mwenda JM, Langat DK, Chege GK, McClure HM. 2004. New simian immunodeficiency virus infecting De Brazza's monkeys (Cercopithecus neglectus): evidence for a cercopithecus monkey virus clade. J Virol 78:7748–7762. doi: 10.1128/JVI.78.14.7748-7762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldberg TL, Sintasath DM, Chapman CA, Cameron KM, Karesh WB, Tang S, Wolfe ND, Rwego IB, Ting N, Switzer WM. 2009. Coinfection of Ugandan red colobus (Procolobus [Piliocolobus] rufomitratus tephrosceles) with novel, divergent delta-, lenti-, and spumaretroviruses. J Virol 83:11318–11329. doi: 10.1128/JVI.02616-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lauck M, Switzer WM, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Shankar A, Greene JM, Ericsen AJ, Zheng H. 2014. Discovery and full genome characterization of a new SIV lineage infecting red-tailed guenons (Cercopithecus ascanius schmidti) in Kibale National Park, Uganda. Retrovirology 11:1–8. doi: 10.1186/1742-4690-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Locatelli S, Peeters M. 2012. Cross-species transmission of simian retroviruses: how and why they could lead to the emergence of new diseases in the human population. AIDS 26:659–673. doi: 10.1097/QAD.0b013e328350fb68. [DOI] [PubMed] [Google Scholar]
- 66.Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MAM, Kessing B, Pontius J, Roelke M, Rumpler Y, Schneider MPC, Silva A, O'Brien SJ, Pecon-Slattery J. 2011. A molecular phylogeny of living primates. PLoS Genet 7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.