

ABSTRACT
The liver constitutes a prime site of cytomegalovirus (CMV) replication and latency. Hepatocytes produce, secrete, and recycle a chemically diverse set of bile acids, with the result that interactions between bile acids and cytomegalovirus inevitably occur. Here we determined the impact of naturally occurring bile acids on mouse CMV (MCMV) replication. In primary mouse hepatocytes, physiological concentrations of taurochenodeoxycholic acid (TCDC), glycochenodeoxycholic acid, and to a lesser extent taurocholic acid significantly reduced MCMV-induced gene expression and diminished the generation of virus progeny, while several other bile acids did not exert antiviral effects. The anticytomegalovirus activity required active import of bile acids via the sodium-taurocholate-cotransporting polypeptide (NTCP) and was consistently observed in hepatocytes but not in fibroblasts. Under conditions in which alpha interferon (IFN-α) lacks antiviral activity, physiological TCDC concentrations were similarly effective as IFN-γ. A detailed investigation of distinct steps of the viral life cycle revealed that TCDC deregulates viral transcription and diminishes global translation in infected cells.
IMPORTANCE Cytomegaloviruses are members of the Betaherpesvirinae subfamily. Primary infection leads to latency, from which cytomegaloviruses can reactivate under immunocompromised conditions and cause severe disease manifestations, including hepatitis. The present study describes an unanticipated antiviral activity of conjugated bile acids on MCMV replication in hepatocytes. Bile acids negatively influence viral transcription and exhibit a global effect on translation. Our data identify bile acids as site-specific soluble host restriction factors against MCMV, which may allow rational design of anticytomegalovirus drugs using bile acids as lead compounds.
INTRODUCTION
Cytomegaloviruses (CMV) are members of the Betaherpesvirinae subfamily and persist for life in infected individuals during alternating phases of latency and productive reactivation. Seroprevalence studies indicate that the large majority of the global human population is currently infected with human cytomegalovirus (HCMV) (human herpesvirus 5 [HHV-5]; taxonomy ID 10359). Although HCMV-related fatalities in apparently healthy individuals sporadically occur (1, 2), HCMV replication usually remains subclinical. This drastically changes under immunocompromising conditions, where untreated CMV infections often cause overt disease, including hepatitis (3) and dysfunction, bleeding, ulceration, and perforation of organs of the upper as well as lower gastrointestinal tract (4). HCMV hepatitis is particularly common in liver transplant recipients and is associated with organ dysfunction and graft failure (5–9).
The species specificity of CMV precludes meaningful in vivo experimentation with HCMV in small animal models. Therefore, the homologous mouse CMV (MCMV) (Murid herpesvirus 1, taxonomy ID 10366), which infects Mus musculus (taxonomy ID 10090) as its natural host, has been established as a relevant small-animal model to study the pathobiology of CMV infections. The two viruses share colinear and partially homologous genomes and cause analogous organ manifestations and disease. Hepatocytes, liver sinusoidal endothelial cells (LSECs), biliary epithelial cells, and Kupffer cells are prime targets for CMV infection (10–12). Under intravenous infection conditions, viral particles reach the liver via the bloodstream. However, LSECs and the space of Disse separate hepatocytes from direct contact with the blood. The fact that virus replication has been initially detected in hepatocytes without prior replication in LSECs (13) suggests that MCMV can bypass the sinusoid endothelium, presumably through pores (fenestrae) in the LSEC layer. In the first infection round, infection of LSECs is five times more common than that of hepatocytes or Kupffer cells (14). While hepatocytes constitute a major site for the generation of cytomegalovirus progeny, LSECs were identified as an important niche for MCMV latency and reactivation (15). Remarkably, MCMV reactivation upon immunoablative treatment was first detectable in the liver (16).
Hepatocytes are polarized epithelial cells that synthesize primary bile acids from cholesterol. Primary bile acids are further conjugated with taurine or glycine and secreted into the canaliculi by the bile salt export pump (BSEP), which is located on canalicular membranes of hepatocytes (17, 18). Together with other bile contents, bile acids are stored in the gallbladder until food uptake stimulates their secretion into the intestine. There, bile acids undergo further chemical modifications, resulting in a variety of structurally related biomolecules that facilitate absorption of dietary lipids but also act as signaling molecules. Ninety-five percent of the bile acids get reabsorbed in the terminal ileum through the apical sodium-dependent bile acid transporter (ASBT) (19). After passing through enterocytes, they are secreted into the bloodstream. Within the blood, bile acids reach the portal liver vein. In the liver, bile acids pass the space of Disse through endothelial fenestrae (20) and are reabsorbed by hepatocytes through the sodium-taurocholate-cotransporting polypeptide (NTCP) and to a lesser extent by other bile acid transporters in the basolateral membrane (20). NTCP is expressed exclusively on hepatocytes and is responsible for ∼80% of the bile acid uptake (21). During this enterohepatic circulation, bile acids affect gene expression in multiple tissues, triggering alterations of bile acid metabolism, glucose homeostasis, lipid and lipoprotein metabolism, energy expenditure, inflammation, liver regeneration, and hepatocarcinogenesis (22). Bile acids regulate a broad spectrum of cellular signaling pathways, e.g., p38MAPK, Jun N-terminal kinase, and phosphatidylinositol 3-kinase (PI3-kinase) (22–24), and activate nuclear receptors such as the farnesoid X receptor (FXR), vitamin D receptor, pregnane X receptor, and constitutive androstane receptor (25). During continuous cell culture, primary hepatocytes rapidly lose their bile acid transporter surface polarity (26). The mRNA expression levels for bile acid transporters are comparable to those in liver samples only for the first 24 h (27), whereas longer cultivation leads to a decline in mRNA expression of bile acid importers (28). Consistently, concentrations of corresponding proteins also vanish in a time-dependent manner (27). Therefore, meaningful experiments with isolated hepatocytes and bile acids have to be performed within the first few days after isolation. Since established and immortalized hepatocyte cell lines do not express bile acid transporters and it is impossible to determine the biological impact of individual bile acids in in vivo settings, freshly isolated primary hepatocytes represent the best available model system to examine the potential impact of bile acids on MCMV replication.
Gastrointestinal and hepatotrophic viruses replicate in the presence of bile acids that influence cellular signaling, innate and adaptive immune responses (22, 29, 30), and interferon (IFN) signaling (31). Hepatitis C virus (HCV) and hepatitis B virus (HBV) benefit from bile acid-dependent FXR activation (32, 33), while for rotaviruses a significantly reduced fecal shedding was shown at 1 and 3 days postinfection in chenodeoxycholic acid-fed mice (34).
In this study, we took advantage of MCMV-permissive primary murine hepatocytes and recombinant MCMV mutants to analyze the antiviral effects exerted by distinct bile acids on virus replication at the cellular and molecular levels. We found that physiological concentrations of bile acids such as taurochenodeoxycholic acid (TCDC), glycochenodeoxycholic acid (GCDC), and to a lesser extent taurocholic acid (TC) possess potent antiviral activity against a ubiquitous hepatotropic herpesvirus, MCMV. The antiviral effect depends on bile acid import by NTCP, is FXR independent, and targets viral mRNA expression and global translation in infected cells.
MATERIALS AND METHODS
Antibodies and chemicals.
Bile acids, cycloheximide, irbesartan, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT), and the hemagglutinin (HA)-specific antibody were obtained from Sigma-Aldrich (St. Louis, USA). The following reagents were purchased from the indicated manufacturers: Z-VAD-FMK (R&D, Wiesbaden, Germany); mouse IFN-α, IFN-γ, and tumor necrosis factor alpha (TNF-α) (PBL, Piscataway, NJ, USA); GW4064 (Tocris, Bristol, United Kingdom); GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (Biodesign, Asbach, Germany); and secondary antibodies (DakoCytomation, Glostrup, Denmark). The biological activities of the cytokines (IFN-α, IFN-γ, and TNF-α) were routinely confirmed using IFN-stimulating response element (ISRE), IFN-γ activation site (GAS), and NF-κB reporter constructs (data not shown). All other chemicals and inhibitors were purchased from either Calbiochem, Merck (Darmstadt, Germany), or Sigma-Aldrich. The specific antibodies used against pp89-IE1 (CROMA 101), early 1 (E1), (M112/M113, CROMA 103), M45 (4D4-A3), and glycoprotein B (gB/M55, MCMV1.01) were kindly provided by Stipan Jonjić, Rijeka, Croatia.
Isolation of primary hepatocytes and cell culture.
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL/6, FXR-deficient (35), or IFN receptor (IFNAR)-deficient (36) mice by a collagenase perfusion technique as described previously (37). NIH 3T3 cells were grown in Dulbecco's modified Eagle medium containing 10% (vol/vol) fetal calf serum, streptomycin, penicillin, and glutamine. Mouse embryonic fibroblasts (MEF) were isolated from embryos of BALB/c or C57BL/6 mice as described previously (38). Animal care and experiments were performed according to the German law for animal protection.
Viruses and infection conditions.
For the generation of Δm157-MCMV:hMIEP-gfp, an FLP recombination target (FRT) site-flanked fragment encompassing the HCMV-derived major immediate early (IE) promoter/enhancer (MIEP) in front of the enhanced green fluorescent protein (eGFP) gene was introduced into a recombinant MCMV bacterial artificial chromosome (which already harbored an FRT site instead of the m157 coding sequence [CDS]) by FLP-mediated recombination. The generation of IE3-HA-MCMV was described elsewhere (39). Δm157-MCMV:luciferase has been described previously (40). Δie1-deficient MCMV was kindly provided by M. Messerle (41). Infections were amplified by centrifugal enhancement (800 × g, 30 min, room temperature). Virus titers were determined after 48 h of Δm157-MCMV:hMIEP-gfp infection of primary hepatocytes by standard plaque titration on MEF.
Immunoblotting.
Immunoblotting was performed following standard procedures. Protein concentrations were normalized according to Bradford staining and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to nitrocellulose membranes (Whatman, Dassel, Germany). Proteins were detected with specific primary and peroxidase-coupled secondary antibodies and visualized with the ECL chemiluminescence reagent (Western Lightning-ECL; PerkinElmer, USA).
Luciferase assay.
Phosphate-buffered saline (PBS)-washed cells were lysed in luciferase lysis buffer, a component of the luciferase reporter gene assay (Roche, Mannheim, Germany). Luciferase activity was quantified according to the manufacturer's instructions using a microplate luminometer (Mithras LB940; Berthold, Bad Wildbad, Germany).
TaqMan PCR.
Primary hepatocytes were infected with Δm157-MCMV:luciferase (0.1 PFU/cell) for 2 h before being washed with trypsin-EDTA (0.05%–0.02% [wt/vol]) (Biochrom, Berlin, Germany) to remove viral particles that had attached but not entered. DNA was isolated with a QIAamp DNA blood minikit (Qiagen, Hilden, Germany) and subjected to quantification by a TaqMan PCR assay (TaqMan universal PCR master mix; Applied Biosystems, Carlsbad, CA, USA) using the following specific primers: M92_forw (5′-CAACCGCGTCTTCAAGCA-3′), M92_rev (5′-AACAGCTGACCTATGACGCTGAT-3′), M92_probe (5′-CATAACGCGCTGCAGAAGGTGCC-3′), Actin_forw (5′-GTATCCCGGGTAACCCTTCTCT-3′), Actin_rev (5′-AACGGCAGAAGAAAGACAATTGA-3′), and Actin_probe (5′-CCAGCTTCTCAGCGCCCTTT-3′).
Reverse transcription-PCR (RT-PCR).
RNA for quantitative PCR (qPCR) was isolated with the ReliaPrep RNA cell miniprep system (Promega, Mannheim, Germany). For the reverse transcription reaction, a mix of oligo(dT) and random primers and the QuantiTect reverse transcription kit (Qiagen, Hilden, Germany) were used. Semiquantitative real-time PCR (GoTaq qPCR master mix; Promega) was performed using the following primers: IE1_forw (5′-TGACCACGTGGGGAATGATA-3′), IE1_rev (5′-GGTGTGCAATCTTACAGGACA-3′), IE3_forw (5′-ATCCTCATGGACCGCATCG-3′), IE3_rev (5′-TATGTTCATCTCGGGTCCTGC-3′), E1_forw (5′-CTGGAGCGGTCTCCTTTTTG-3′), E1_rev (5′-AAAACGCGGGTTGTTGTCC-3′), M45_forw (5′-CTTCGACGCCGTTTCTTACC-3′), M45_rev (5′-GTCACCATGTCGCAGATGTC-3′), M55_forw (5′-GGCAGGGATTGAAAGCGAAA-3′), M55_rev (5′-AGGAGGGCTAACAGTCCCTA-3′), SDHA_forw (5′-TGGGGAGTGCCGTGGTGTCA-3′), and SDHA_rev (5′-GTGCCGTCCCCTGTGCTGGT-3′). Note that the primers for ie1, ie3, e1, and the cellular housekeeping gene sdha have been designed to amplify spliced mRNAs. To our knowledge, splicing has not been described for M45 and M55. Therefore, design of exon-spanning primer sets was not possible. To exclude amplification of genomic DNA contaminants, control experiments without the reverse transcription reaction were performed in parallel.
RNA for other PCRs was isolated with the RNeasy minikit (Qiagen, Hilden, Germany), and PCR was performed using a one-step RT-PCR kit (Qiagen, Hilden, Germany) and the primers IE1_forw (5′-GAATAAAAGAGGGGGTGTGGTGTTA-3′), IE1_rev (5′-CAGCAACTCATCCTATCCAGACCT-3′), IE3_forw (5′-CAGAGCCTCATGCACACCTGCT-3′), IE3_rev (5′-GGAGGAGACCGTGCATGACAAG-3′), GAPDH_forw (5′-ACCACAGTCCATGCCATCAC-3′), and GAPDH_rev (5′-TCCACCACCCTGTTGCTGTA-3′).
[35S]methionine biosynthetic labeling of proteins.
Cells were incubated for 6 h with 27 μCi 35S-labeled l-Met/l-Cys (Easy Taq Express protein labeling mix [35S]; PerkinElmer, Waltham, MA, USA) before lysis. Proteins were separated by SDS-PAGE and (after gel fixation) visualized by autoradiography. Protein lane intensities were quantified using Fujifilm imaging plates and a Fujifilm FLA3000 image analyzer (Fujifilm, Tokyo, Japan).
Statistics.
Statistical significance was calculated for raw data using Wilcoxon tests for all experiments expect those with the results shown in Fig. 1E and 5D, for which a Mann-Whitney U test was used. (*/#, P < 0.05; **, P < 0.01; ***, P > 0.001) To present data in a way in which they can be better compared, bar charts and the table depict arithmetic means calculated in relation to untreated control samples and standard deviations.
FIG 1.

Conjugated bile acids act antivirally against mouse cytomegalovirus (MCMV). (A) Primary mouse hepatocytes were treated for 3 h with 25 μM concentrations of the bile acids taurochenodeoxycholic acid (TCDC), glycochenodeoxycholic acid (GCDC), taurocholic acid (TC), tauroursodeoxycholic acid (TUDC), chenodeoxycholic acid (CDC), taurolithocholic acid (TLC), and taurolithocholic sulfate (TLCS) and were infected with Δm157-MCMV-luciferase (0.1 PFU/cell) for the next 24 h thereafter. Hepatocytes were lysed, and luciferase activity was measured as described in Materials and Methods. The luciferase activity is calculated in relation to untreated control samples. (B) Primary mouse hepatocytes were incubated with the indicated TCDC concentrations for 3 h and subsequently infected with Δm157-MCMV-luciferase (0.1 PFU/cell) for the next 24 h. Cells were lysed, and the luciferase activity was determined. (C) Hepatocytes were incubated with 25 μM TCDC for 3 h and then infected with graded virus doses of Δm157-MCMV-luciferase (0.001 to 10 PFU/cell). Luciferase activity was measured at 24 h postinfection. (D) Hepatocytes were incubated with 25 μM TCDC for 3 h and infected with Δm157-MCMV-HMIEP-eGFP (0.1 PFU/cell) for the next 48 h. MCMV-driven eGFP expression was visualized by fluorescence microscopy. Representative fields are shown. (E) Primary mouse hepatocytes were treated with 25 μM TCDC or 500 U/ml IFN-α for 3 h and infected with Δm157-MCMV-HMIEP-eGFP (0.1 PFU/cell), and virus titers (in PFU/ml) were determined at 48 h postinfection by classical plaque titration. (F) NIH 3T3 and mouse embryonic fibroblast (MEF) cells were incubated for 3 h with 25 μM TCDC and subsequently infected with Δm157-MCMV-luciferase (0.1 PFU/cell). Luciferase activity was determined at 24 h postinfection.
FIG 5.

TCDC reduces IE1 protein amounts, but the antiviral effect of TCDC is independent of IE1 function. (A) Purified MCMV particles were incubated with 25 μM TCDC for 3 h at 37°C and were used for infection of NIH 3T3 and MEF cells. Note that both fibroblast cell types do not support the antiviral activity elicited by bile acids in/on hepatocytes (Fig. 1F). At 24 h postinfection, luciferase activity was quantified. (B) Copy numbers of intracellular genomic viral DNA in relation to cellular DNA were determined by quantitative TaqMan PCR with primers and probes specific for the viral gene region M92 and the cellular β-actin gene. Primary mouse hepatocytes were incubated for 3 h with 25 μM TCDC and then infected with MCMV (0.1 PFU/cell) either at 4°C (which allows virus attachment but precludes virus entry) or at 37°C (which allows virus entry). At 2 h postinfection, extracellular virus particles were removed by multiple rounds of vigorous trypsin washing. Subsequently, the cells were collected and the total (intra)cellular DNA was purified. An incubation of virus with immune serum of MCMV-infected mice (which is known to contain effective concentrations of neutralizing antibodies) served as a positive control for an inhibition of virus entry. (C) Primary mouse hepatocytes were incubated for 3 h with 25 μM and 50 μM TCDC and subsequently infected with MCMV for a further 6 h. Cells were lysed, and RNA was isolated. The abundance of immediate early (IE) ie1 and ie3 mRNAs and housekeeping GAPDH mRNA was determined by semiquantitative one-step reverse transcription-PCR with specific primers. In parallel, cells were lysed and protein lysates were prepared, normalized according to Bradford protein staining, separated by SDS-polyacrylamide gel electrophoresis, and subjected to immunoblotting followed by detection with the indicated antibodies. (D) Primary hepatocytes were treated for 3 h with 25 μM TCDC and infected with MCMV strain pSMA3 or ΔIE1-pSMA3 (0.1 PFU/cell) for 48 h. Virus titers (in PFU/ml) were determined by classic plaque titration.
RESULTS
Bile acids exert an antiviral effect against MCMV in murine hepatocytes.
Physiological serum concentrations of bile acids in healthy individuals range on the orders of approximately 10 μM in humans (42) and 0.4 to 15 μM in mice, whereas murine liver homogenates contain ∼350 μM bile acids (43). To examine the antiviral activities of physiologically relevant concentrations, 25 μM concentrations of the conjugated bile acids TCDC, GCDC, and TC, the hydrophilic conjugated bile acid tauroursodeoxycholic acid (TUDC), the unconjugated bile acid chenodeoxycholic acid (CDC), and the secondary bile acids taurolithocholic acid (TLC) and taurolithocholic acid 3-sulfate (TLCS) were used for the experiments. To assess the effect of bile acids on cytomegalovirus gene expression, we generated primary mouse hepatocytes and made use of a recombinant mouse cytomegalovirus (MCMV) in which the coding sequence of the viral early/late gene m157 has been replaced by the coding sequence of the luciferase gene (derived from the firefly [Photinus pyralis], taxonomy ID 7045).
Treatment with 25 μM TCDC, GCDC, and TC significantly and reproducibly reduced MCMV-driven luciferase expression, albeit to various extents, while an identical concentration of TUDC, CDC, TLC, or TLCS failed to affect luciferase activity (Fig. 1A), indicating that distinct bile acids negatively influence cytomegalovirus gene expression. Increased bile acid concentrations did not further potentiate the antiviral effects of TCDC, GCDC, and TC (Fig. 1B and data not shown). The efficacy of antiviral compounds can critically depend on the multiplicity of infection. Therefore, the effect of 25 μM TCDC was tested against various initial infectious MCMV doses (ranging from 0.001 to 10 PFU/cell). Regardless of the inoculum, a similar effect on viral gene expression was observed (Fig. 1C). To exclude that bile acids act on the luciferase enzyme itself, the inhibitory effect of TCDC was confirmed by fluorescence microscopy using a recombinant MCMV harboring an HCMV MIEP-driven eGFP reporter gene (Fig. 1D). Note that the heterologous insertion of the HCMV-derived MIEP reverts the severe attenuation of an MCMV MIEP deletion in vitro and in vivo (including replication in hepatocytes) (44, 45), indicating the suitability of such reporter viruses.
Based on the evidence that TCDC inhibits the expression of MCMV-driven reporter genes, the effects of TCDC on production of infectious virus progeny were examined. Primary hepatocytes were preincubated for 3 h with 25 μM TCDC and infected with 0.1 PFU/cell MCMV for 48 h, and virus progeny was determined by plaque titration. In contrast to treatment with 500 U/ml IFN-α, TCDC treatment of hepatocytes exhibited significant antiviral activity under this conditions (Fig. 1E).
To test whether bile acids act in a cell type-specific manner, similar experiments were performed in mouse embryonic fibroblasts (MEF) and NIH 3T3 cells, but no antiviral activity of TCDC was detectable (Fig. 1F and data not shown). Taken together, these data document a hepatocyte-specific antiviral activity of physiological concentrations of certain conjugated bile acids (e.g., TCDC) against MCMV.
Inhibition of CMV replication by bile acids is a consequence of neither cytotoxicity nor caspase induction.
High concentrations of bile acids are cytotoxic and can induce apoptosis in hepatocytes (23), and viral infections of the liver, including CMV infections, are also associated with liver cell necrosis and apoptosis (46). Although MCMV expresses several inhibitors of apoptosis and necrosis, treatment of hepatocytes with bile acids prior to infection could still result in reduced virus replication through induction of cell death. Therefore, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) assays were performed. However, no signs of cytotoxicity after 48 h treatment of primary mouse hepatocytes with 25 or even 50 μM TCDC became evident (Fig. 2A).
FIG 2.

TCDC does not inhibit CMV replication by inducing cytotoxicity. (A) Primary mouse hepatocytes were incubated for 48 h with 25 μM and 50 μM TCDC. A potential impact on cell viability was assessed by performing a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) assay. (B) To test the contribution of caspases to the antiviral activity of TCDC, primary mouse hepatocytes were conditioned with a 50 μM concentration of the pan-caspase inhibitor Z-VAD-FMK starting 30 min prior to the 3-h TCDC treatment (25 μM). The cells were infected with Δm157-MCMV-luciferase (0.1 PFU/cell) and lysed 24 h later for the determination of the luciferase activity. Functionality of the pan-caspase inhibitor was confirmed in parallel by coadministration with 100 μM cycloheximide (CHX) and 30 ng/ml TNF-α. Under this condition, Z-VAD-FMK effectively prevented caspase-dependent apoptosis (right panels).
Independence of the antiviral activity of TCDC from caspase activation was shown by coadministration of the pan-caspase inhibitor Z-VAD-FMK, which failed to restore MCMV gene expression. Z-VAD-FMK activity was ensured by its parallel capability to block apoptosis induced by combined treatment with tumor necrosis factor alpha (TNF-α) and cycloheximide (Fig. 2B).
Both of these experiments exclude the possibility that the antiviral effect of TCDC on MCMV is an unspecific effect dependent on cytotoxicity or caspase induction.
None of the established signaling pathways engaged by bile acids is required for the observed antiviral activity against CMV.
Bile acids have far-reaching impacts on cellular signal cascades in hepatocytes. To clarify whether the unknown antiviral effector pathway is a result of altered cellular signaling, a panel of pharmacologic inhibitors was used. However, although several drugs influenced MCMV replication in a TCDC-independent manner, none abrogated the antiviral activity elicited by TCDC (Table 1).
TABLE 1.
The antiviral effect of TCDC is independent of known bile acid signal cascadesa
| Inhibitor | Target | Relative MCMV-dependent luciferase activity (mean ± SD) with: |
|
|---|---|---|---|
| Inhibitor alone | Inhibitor + TCDC | ||
| None | None | 1.0 ± 0.0 | 0.5 ± 0.1 |
| SU6656 | Yes kinase | 1.5 ± 0.3 | 0.6 ± 0.1 |
| PP2 | Src kinase | 0.9 ± 0.4 | 0.4 ± 0.1 |
| Protein kinase inhibitor | Protein kinase A | 1.2 ± 0.3 | 0.4 ± 0.2 |
| PD98059 | Erk kinase | 1.0 ± 0.2 | 0.5 ± 0.2 |
| SB203580 | p38MAPK | 0.9 ± 0.2 | 0.6 ± 0.2 |
| Rapamycin | mTOR | 0.8 ± 0.3 | 0.5 ± 0.1 |
| Sp600125 | JNK1/2 | 0.8 ± 0.2 | 0.3 ± 0.1 |
| Bafilomycin A1 | Autophagy | 1.1 ± 0.3 | 0.6 ± 0.1 |
| LY294002 | PI3 kinase | 0.4 ± 0.1 | 0.1 ± 0.0 |
Hepatocytes were incubated for 30 min with inhibitors of the indicated signal cascades (Yes kinase, SU6656 [10μM]; Src kinase, PP2 [10 μM]; protein kinase A, protein kinase inhibitor [2.3 nM]; Erk kinase, PD98059 [10 μM]; p38MAPK, SB203580 [10 μM]; mTOR, rapamycin [10 nM]; JNK1/2, Sp600125 [10 μM]; autophagy, bafilomycin A1 [250 nM]; PI3 kinase, LY294002 [50 μM]) before being treated for 3 h with 25 μM TCDC prior to infection with Δm157-MCMV-luciferase. After 24 h, cells were lysed and luciferase activity was quantified.
Most effects of bile acids on cellular gene transcription are mediated by activation of the nuclear receptor FXR (47). Consistently, known effects of bile acids on HCV, HBV, and rotavirus replication rely on FXR receptor function (32–34). Therefore, the FXR dependency of the anticytomegalovirus activity of TCDC was tested. Surprisingly, the FXR agonist GW4064 increased luciferase activity induced by Δm157-MCMV-luciferase. Consistent with the lack of antiviral activity of the FXR agonist GW4064, FXR was fully dispensable for the antiviral activity of TCDC as revealed by the usage of FXR-deficient hepatocytes (Fig. 3A). Moreover, GW4064 also induced virus replication in FXR-deficient hepatocytes, indicating that GW4064 modulates MCMV replication independently of FXR activation. Taken together, these findings document that the anticytomegalovirus activity of TCDC is FXR independent.
FIG 3.

Bile acids elicit their antiviral activity independently of FXR, vitamin D receptor, and IFNAR1. (A) Primary hepatocytes of FXR knockout and wild-type control mice were incubated with 25 μM TCDC or GW4064 for 3 h and subsequently infected with Δm157-MCMV-luciferase (0.1 PFU/cell) for the next 24 h before luciferase activity was measured. (B) Primary mouse hepatocytes were incubated for 24 h with 5 μM calcifediol or 0,5 μM calcitriol before being infected with Δm157-MCMV:luciferase (PFU:0.1). Luciferase activity was quantitated at 24 h after infection. (C) Primary mouse hepatocytes were conditioned for 24 h with 500 U/ml IFN-α, 500 U/ml IFN-γ, or a combination of both and, if indicated, for a further 3 h with 25 μM TCDC before the cells were infected with Δm157-MCMV-luciferase (0.1 PFU/cell) for an additional 24 h. Cells were lysed, and luciferase activity was quantified. (D) Primary hepatocytes of IFNAR1-deficient and wild-type mice were incubated for 3 h with 25 μM TCDC before infection with Δm157-MCMV-luciferase (0.1 PFU/cell). At 24 h postinfection, the cells were lysed and the luciferase activity was quantified.
The vitamin D receptor, which can be activated by bile acids (42), can influence innate immunity (48, 49). To test whether the antiviral effect of bile acids is dependent on vitamin D receptor activity, the vitamin D receptor ligand calcitriol and its precursor calcifediol were administered to primary hepatocytes. However, neither calcitriol nor calcifediol showed antiviral activity (Fig. 3B).
Recently, bile acids have been shown to impair IFN signaling (31). Therefore, the impact of IFN signaling on the antiviral effect of TCDC on MCMV was tested. IFN-α did not reduce MCMV-expressed luciferase activity in hepatocytes, while incubation with IFN-γ or a combination of both significantly reduced Δm157-MCMV-dependent luciferase gene expression. Coadministration of TCDC together with IFN-γ resulted in an additive antiviral effect (Fig. 3C). To analyze a potential dependency of the anticytomegalovirus activity of TCDC on type I IFN signaling, hepatocytes from IFN receptor 1 (IFNAR1)-deficient mice were used, with the result that TCDC exhibited antiviral activity identical to that for wild-type mice (Fig. 3D).
The antiviral effect of bile acids depends on NTCP.
Based on the fact that bile acid uptake is restricted to a few cell types such as hepatocytes, which express bile acid-specific transporters (e.g., NTCP [21]), and our observation that the conjugated bile acid TCDC exerts an antiviral effect in hepatocytes but not in the tested murine fibroblasts (Fig. 1F), we presumed that this lack of activity highlights an essential role of bile acid import in the anticytomegalovirus activity of TCDC. Over time in cell culture, hepatocytes continuously lose their ability to transport bile acids by downregulation of NTCP expression (27). Exploiting this fact, primary mouse hepatocytes were cultivated for 4 days prior to bile acid treatment and MCMV infection. These cells no longer took up radioactively labeled TC (data not shown), demonstrating an acquired incapacity for bile acid import. These cells also failed to reduce MCMV-dependent luciferase activity upon bile acid treatment (data not shown). To further substantiate the hypothesis that the import of bile acids is a necessary prerequisite for antiviral activity, an experiment was designed based on the observation that TCDC inhibits MCMV, whereas TUDC does not, and the knowledge that both compounds compete for the same bile acid transporters (50, 51). Graded TUDC concentrations counteracted the antiviral activity of TCDC in a dose-dependent manner (Fig. 4A), supporting the notion that TCDC and TUDC compete at least for one limiting component of the bile acid uptake and/or signaling pathway. Therefore, the newly described NTCP inhibitor irbesartan (52) was used to directly test the role of NTCP. As expected, uptake of radioactively labeled TC into irbesartan-treated mouse hepatocytes was largely reduced (data not shown). Importantly, irbesartan completely abolished the anticytomegalovirus effect otherwise elicited by TCDC (Fig. 4B), indicating that NTCP-dependent bile acid uptake is essential for antiviral activity.
FIG 4.
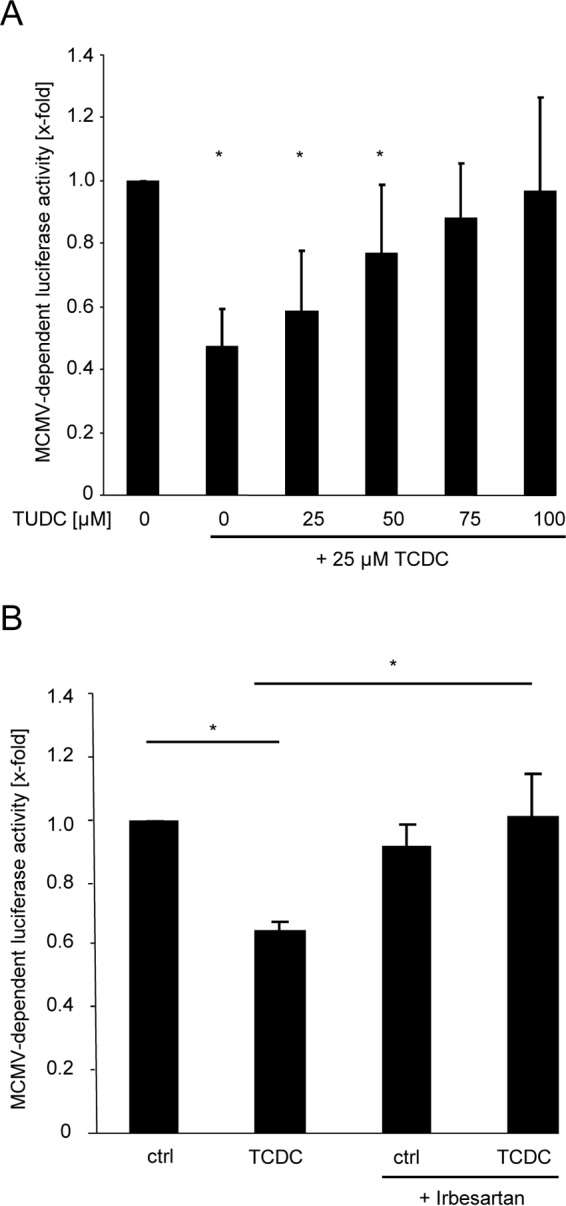
Inhibition of MCMV replication by TCDC is dependent on NTCP-mediated cellular uptake. (A) Primary murine hepatocytes were conditioned for 3 h with 25 μM TCDC. Simultaneously, the indicated concentrations (0 to 100 μM) of TUDC were also added to the cells prior to infection with Δm157-MCMV-luciferase. Note that TUDC does not possess antiviral activity against MCMV (Fig. 1A). At 24 h postinfection, the cells were lysed and luciferase activity was quantified. (B) Primary mouse hepatocytes were incubated for 3 h with 25 μM TCDC, and 50 μM irbesartan was coadministered prior to infection with Δm157-MCMV-luciferase (0.1 PFU/cell). Luciferase activity was quantified at 24 h postinfection.
TCDC affects viral mRNA expression and inhibits global translation.
Since bile acids solubilize lipids and lipid-soluble compounds, potential virolytic effects on the MCMV envelope were evaluated. To this end, purified MCMV particle preparations were incubated with 25 μM TCDC for 3 h prior to infection of cells which do not exhibit antiviral activity upon TCDC treatment, i.e., NIH 3T3 and MEF cells (Fig. 1F). Incubation with TCDC did not affect the infectivity of the purified virions (Fig. 5A), excluding damaging effects of bile acids on cytomegalovirus particles.
To further investigate the influence of bile acid on the viral entry process, primary hepatocytes were incubated with 25 μM TCDC for 3 h before cells were infected with MCMV. Two hours after infection, cells were washed with a diluted trypsin-EDTA solution to remove viral particles that had attached but not entered. The efficiency of the viral entry process was subsequently surveyed by quantification of intracellular viral genome copy numbers (in relation to cellular genomes) by quantitative TaqMan PCR. Efficacy of the trypsin-EDTA washing procedure was controlled upon infection at 4°C, a condition under which viral particles bind but do not enter cells. As expected, raising the incubation temperature from 4°C to 37°C resulted in a substantial increase in the number of intracellular viral genomes (Fig. 5B). As a positive control for inhibition of MCMV entry, the infection was blocked with an immune serum derived from latently infected mice which is known to contain high titers of MCMV-specific neutralizing antibodies. Nevertheless, 25 μM TCDC did not significantly reduce the number of viral genomes within hepatocytes (Fig. 5B), ruling out a prominent effect on viral entry.
To determine the effective time window required to deploy the antiviral effect of TCDC, time-of-addition experiments (from 5 h before to 5 h after infection) were performed; antiviral activity was observed even upon applying TCDC postinfection (data not shown), further corroborating the TCDC resistance of viral entry.
After entry, the MCMV replication cycle starts with the transcription of immediate early (ie) genes from the major IE promoter/enhancer region (MIEP). Transcription of ie genes does not require de novo translation of viral proteins. ie mRNAs constitute the first viral transcripts, encoding nonstructural proteins with prominent roles in the viral replication cycle (53). At 6 h postinfection, ie1 and ie3 transcripts were readily detectable. However, their abundance appeared to be slightly increased by TCDC (Fig. 5C). In clear contrast, an immunoblot analysis revealed a substantial reduction of the viral protein pIE1-pp89 at this time (Fig. 5C), indicating that at this time point the translation but not the transcription of virus-encoded ie1 mRNAs was negatively affected by bile acids. The in vivo replication of an MCMV mutant lacking ie1 coding capacity was shown be significantly reduced (∼1 log10) in the liver (54, 55). However, we observed neither altered replication competence nor different TCDC susceptibility of an ie1-deficient MCMV in primary hepatocytes ex vivo (Fig. 5D), indicating that reduced pIE1 amounts fail to explain the antiviral activity of TCDC.
To assign the antiviral effect of bile acids to distinct phases of viral replication, we quantified mRNA and protein levels of gene products characteristic for the IE (ie1 and ie3), early (e1-M112/M113 and M45), and late (gB-M55) phases of viral gene expression. Primary hepatocytes were incubated with 25 μM TCDC starting 3 h prior to infection with MCMV and were lysed after 6 h, 9 h, 12 h, 24 h, or 48 h. Thereafter, semiquantitative RT-PCRs for ie1, ie3, early 1 (E1), M45, and M55 were performed, in which TCDC-treated cells were compared to untreated control cells. To normalize for potential differences in RNA preparation, reverse transcription, and PCR amplification efficiency, relative changes in viral mRNAs were calculated based on the ΔΔCT method in comparison to the cellular housekeeping transcript sdha. IE and early transcripts could be detected at 6, 9, and 12 h postinfection. TCDC did not reduce the abundance of the respective transcripts and even increased their amounts. Conversely, starting at 24 h postinfection and becoming even more apparent after 48 h, the abundance of viral mRNAs was reduced by bile acids in comparison to that in untreated hepatocytes (Fig. 6A).
FIG 6.

TCDC inhibits MCMV replication at the level of protein translation and modifies viral mRNA expression. (A) Primary mouse hepatocytes were incubated with 25 μM TCDC for 3 h and infected with Δm157-MCMV-luciferase afterwards. Cells were lysed after 6 h, 9 h, 12 h, 24 h, or 48 h, and RNA was isolated. cDNA was synthesized, and quantitative PCR was performed with specific primers for ie1, ie3, early1, M45, and M55. Depicted are the relative RNA expression levels in comparison to that of the cellular transcript sdha. To calculate statistical significance, the results of all transcription values at one time point were considered (except in case of M55, where no gene expression was detectable in the 6- to 12-h period and only later time points were included in the analysis). (B) Primary hepatocytes were treated for 3 h with 25 μM TCDC and infected with MCMV-IE3-HA or Δm157-MCMV-luciferase (multiplicity of infection of 0.1). Hepatocytes were lysed at the indicated time points and pIE1, pIE3-HA, pE1, pM45, gB, and housekeeping GAPDH were detected by SDS-PAGE and immunoblotting with specific antibodies. (C) Primary mouse hepatocytes were preincubated for 3 h with 25 μM TCDC and infected with MCMV (1 PFU/cell). 35S-labeled l-Met/l-Cys (27 μCi) was added to the cells directly after centrifugally enhanced infection. After 6 h, cells were lysed with lysis buffer, proteins were separated by SDS-PAGE, the gel was fixed and dried, and the intensity of radioactively labeled proteins was visualized by autoradiography. The inhibitor of translation CHX was used as a positive control. (D) Intensities of 35S incorporation were quantified (for details, see Materials and Methods). The intensities were calculated in relation to untreated control samples.
Effects of TCDC on viral protein expression were determined by immunoblotting with specific antibodies against pIE1-pp89, pE1, pM45, and gB at 6 h, 9 h, 12 h, 24 h, and 48 h after infection. The preincubation of hepatocytes with TCDC resulted in decreased viral protein levels of IE1 and E1 (Fig. 6B) at all monitored time points. M45 levels were also downregulated at 6 h, 9 h, 12 h, and 24 h after infection in TCDC-treated cells. gB was detectable only after 48 h, and its abundance was also reduced by TCDC. Influences of TCDC on pIE3 expression were tested using a recombinant MCMV harboring an HA epitope insertion at the C-terminal end of the annotated pIE3 (described in reference 39). As shown in Fig. 6C, an immunoblot using an HA-specific antibody confirmed decreased abundance of IE3 upon TCDC treatment between 9 h and 48 h.
Based on the fact that viral mRNA transcript levels rose in the presence of bile acids at the early time points (6 to 12 h), whereas the corresponding protein amounts were markedly reduced at this stage, we concluded that bile acids primarily inhibit the translation process of viral mRNAs. Influences on the posttranscriptional modification and transport of mRNAs affecting their translatability (e.g., capping, polyadenylation, and nuclear export to the ribosomes in the cytoplasm) cannot be formally excluded. However, based on the fact that the primer design ensured amplification of spliced mRNAs in the cases of ie1, ie3, e1, and sdha and the importance of splicing for subsequent mRNA capping (see, e.g., reference 56), as well as the drastic shortening of mRNA half-life upon depolyadenylation (see, e.g., reference 57), such an explanation appears less likely.
In contrast to other herpesviruses (e.g., herpes simplex virus 1 [HSV-1]), cytomegaloviruses do not encode so-called host shutoff proteins, and transcription and translation of cellular genes/gene products proceed during productive viral replication. Consistently, metabolic labeling experiments (e.g., using 35S-labeled l-methionine and/or l-cysteine) and polysome purifications show ongoing host RNA and protein synthesis in CMV-infected cells (58–62; our unpublished data). Therefore, we raised the question whether bile acids generally affect the translation of cellular mRNAs in MCMV-infected cells. Metabolic incorporation assays with 5S-labeled l-Met/l-Cys were performed. As a positive control, cycloheximide was used, which inhibits translation and completely abrogated metabolic labeling of proteins. TCDC treatment slightly reduced the efficiency of translation (as determined by de novo 35S incorporation) in mock-infected cells to 81.9% ± 14% compared to that in untreated control cells. However, in MCMV-infected cells, this effect was significantly increased (to 61.8% ± 13%) (Fig. 6D and E), indicating that the slight impairment of translation of cellular mRNAs is significantly enhanced during MCMV infection. Taken together, our data indicate that bile acids elicit their antiviral activity toward MCMV by altered viral mRNA expression and impaired viral and cellular protein translation.
DISCUSSION
The present study describes an NTCP-dependent antiviral activity of the conjugated bile acids TCDC and GCDC on MCMV replication in hepatocytes. Therefore, the bile acid structure may provide a basis (or the lead compound) for rational design of potent antiviral drugs against cytomegaloviruses. In this context, the exceptionally good (re)absorption of bile acids from the gut and their transport via the portal vein to the liver might be advantageous in comparison to conventional antivirals with a poorer bioavailability in these tissues and might open up a perspective for the topical treatment of CMV disease in bile acid-exposed organs such as the liver and gut, which represent prevalent sites of CMV disease manifestation in immunocompromised patients (4, 6–9). One might argue that the above-described effect on host protein translation in hepatocytes would result in adverse effects. However, certain bile acids, such as ursodeoxycholic acid, are routinely prescribed to patients as medication against cholestatic liver diseases and gallstones (63) and are known to cause few side effects (64). An obvious question is whether our results can be extrapolated to HCMV infection. Unfortunately, in vitro, HCMV replication is not efficient in human hepatoma cells (65), impeding experimental analysis. While bile acid synthesis is a tightly regulated process, some diseases are associated with increased or decreased bile acid levels. Decreased bile acids levels are found after ileal resection (66) or in patients with inflammatory bowel disease such as ulcerative colitis or Crohn's disease (67). In ulcerative colitis, CMV reactivation is more common than in healthy individuals (68, 69). Beside the fact that these patients have an inflammatory disease that is treated with immunosuppressive drugs, reduced bile acid levels could be associated with CMV reactivation in these cases.
The antiviral activity of TCDC is restricted to hepatocytes and was not observed in/on fibroblasts, most likely due to the lack of the bile acid import machinery (e.g., NTCP expression) and other interaction partners of bile acids. Forced expression of NTCP in NIH 3T3 fibroblasts results in overt cell death after treatment with small amounts (in the nanomolar range) of bile acids irrespective of MCMV infection (data not shown), suggesting that additional components are required to establish and phenocopy the situation in primary hepatocytes. A contribution of FXR to the antiviral activity of bile acids was excluded in hepatocytes derived from FXR-deficient mice. Pharmacologic drugs inhibiting known downstream signaling events initiated by bile acid also failed to impair the antiviral effect of TCDC. Based on the inability of a vitamin D receptor ligand to act antivirally, a prominent role of the vitamin D receptor can be excluded. A role of the constitutive androstane receptor or the pregnane X receptor in the antiviral activity of TCDC is also rather unlikely because the natural bile acid ligand of both receptors is lithocholic acid (LCA) (20, 42), which does not exhibit significant anticytomegalovirus properties (see above). Taken together, these findings highlight the distinctiveness of the anticytomegalovirus activity of bile acids.
Interestingly, the antiviral activity elicited by 25 μM TCDC was clearly stronger than that of 500 U/ml IFN-α (which did not have antiviral activity under the conditions analyzed here) and reached a level comparable to that with 500 U/ml IFN-γ. Nevertheless, the effect was independent of IFNAR1, ruling out an involvement of type I IFNs (IFN-α/β) as essential mediators of the antiviral activity against MCMV.
Despite the fact that bile acids can solubilize lipids, TCDC neither impairs the stability of enveloped cell-free virus particles nor affects viral entry or diminishes immediate early gene transcription, but it acts by interfering with the translation of viral and cellular proteins over the entire time course of infection and with viral mRNA expression at late time points (24 to 48 h) (Fig. 6A to D). The inability of TCDC to downregulate the transcription of ie mRNAs at early times of infection indicates that several steps of the CMV replication cycle (entry, transport of the viral capsid to the nucleus, unpackaging, release of the viral double-stranded DNA [dsDNA] genome into the nucleus, and recruitment of the cellular transcription machinery) are resistant to TCDC. Although the exact molecular mechanism by which bile acids affect viral protein translation remains to be elucidated, it is completely different from the described modes of action of established anticytomegalovirus drugs (such as ganciclovir, foscarnet, cidofovir, maribavir, and letermovir), which interfere with different steps of the viral replication cycle (e.g., DNA replication, the activity of the viral kinase pUL97, and DNA cleavage/packaging, respectively). Whether the biphasic effect of bile acids on viral mRNA expression depends on the downregulation of viral proteins or represents an independent effect remains elusive. The effects on ie3, ie1, and e1 mRNA amounts might be explained by diminished pIE3 levels. pIE3 induces the expression of early proteins (e.g., E1) and represses the activity of the major immediate early promoter (MIEP) (70, 71), which in turn drives the expression of ie1 and ie3. Thus, increased ie1 und ie3 mRNA amounts could be explained by diminished pIE3 levels (Fig. 6A and C). Based on the kinetics of protein expression and localization of pIE3 and pE1, it has been concluded that pIE3 amounts “less than that demonstrable with Western blotting or immunohistochemical techniques” seem to suffice to transactivate the m112/113 promoter, whereas larger amounts are required to repress the MIEP (70). Therefore, it is plausible that the amount of pIE3 synthesized under TCDC incubation is enough to induce e1 mRNA expression, while the protein amount is not sufficient for MIEP suppression.
A simple explanation for the impaired transcription at later times of infection is an indirect decrease of proteins which are required for transcription. An ongoing disturbance of translation leads to a gradual loss of proteins, starting with proteins with shorter half-lives. It is tempting to speculate that at least one essential component of the transcriptional machinery has such a low protein stability. Consistently, a global analysis of protein and mRNA stability found that genes implicated in “transcription” and “regulation of transcription” are strongly and significantly enriched in the group with unstable mRNAs and proteins (72). Impaired de novo protein synthesis due to inhibited translation would indirectly affect the global transcription once the levels of an essential component fall below the required concentrations.
As judged by global virus titers in crude organ homogenates, MCMV replication in the liver appears to be rather efficient compared to that in other organs. In this light, it appears at first glance counterintuitive that hepatocytes differ from other cells by a specific mechanism of protection from cytomegalovirus replication which is associated with the ability to import antiviral bile acids. In immunocompetent individuals, CMV infection is normally asymptomatic, since replication is restricted by the immune system to a limited number of virus-producing cells (1, 73). However, under immunocompromised conditions, HCMV replication leads to severe organ manifestation and frequently to HCMV hepatitis in patients (3). Likewise MCMV replicates for prolonged time periods and to higher virus titers in the livers of immunocompromised mice (73), indicating that even in highly permissive organs, mechanisms which restrict CMV replication effectively (e.g., T cells and innate immunity) do exist. In our model, bile acids similarly curtail CMV replication in specific tissues.
Interestingly, patients with acute viral hepatitis due to infections with HBV or HCV show elevated bile acid concentrations (74, 75). It is tempting to speculate that bile acid levels might also be elevated during HCMV hepatitis and that this might provoke a benefit by limiting CMV replication and virus-induced liver tissue damage.
Conversely, during their long history of coevolution with their mammalian hosts, CMV have evolved multiple gene functions that counteract intrinsic antiviral restriction factors as well as mediators of innate and adaptive immunity. It will be interesting to study whether MCMV has adapted to the antiviral activity elicited by bile acids during the long period of coevolution with its host species by acquiring gene functions which confer partial resistance to bile acids.
Taken together, our data reveal a novel aspect of the interaction between cytomegaloviruses and the liver. Conjugated bile acids possess profound anticytomegalovirus activity depending on bile acid import via NTCP. These findings have important implications for the understanding of CMV-mediated liver pathologies, since they document a novel liver-specific intrinsic resistance mechanism and might open new avenues for the design of antiviral drugs against cytomegalovirus disease of the liver.
ACKNOWLEDGMENTS
We express our gratitude to the two anonymous referees for their insightful comments and to Martin Messerle (Hannover Medical School, Germany) for providing Δie1 MCMV. We thank Ursula Kristek and Vanessa Herbertz for excellent technical assistance.
We declare no conflict of interest.
REFERENCES
- 1.Rafailidis PI, Mourtzoukou EG, Varbobitis IC, Falagas ME. 2008. Severe cytomegalovirus infection in apparently immunocompetent patients: a systematic review. Virol J 5:47. doi: 10.1186/1743-422X-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ten Napel CH, Houthoff HJ, The TH. 1984. Cytomegalovirus hepatitis in normal and immune compromised hosts. Liver 4:184–194. [DOI] [PubMed] [Google Scholar]
- 3.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2656–2701. In Knipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Buckner FS, Pomeroy C. 1993. Cytomegalovirus disease of the gastrointestinal tract in patients without AIDS. Clin Infect Dis 17:644–656. doi: 10.1093/clinids/17.4.644. [DOI] [PubMed] [Google Scholar]
- 5.Seehofer D, Rayes N, Tullius SG, Schmidt CA, Neumann UP, Radke C, Settmacher U, Muller AR, Steinmuller T, Neuhaus P. 2002. CMV hepatitis after liver transplantation: incidence, clinical course, and long-term follow-up. Liver Transpl 8:1138–1146. doi: 10.1053/jlts.2002.36732. [DOI] [PubMed] [Google Scholar]
- 6.Lautenschlager I, Hockerstedt K, Jalanko H, Loginov R, Salmela K, Taskinen E, Ahonen J. 1997. Persistent cytomegalovirus in liver allografts with chronic rejection. Hepatology 25:190–194. doi: 10.1002/hep.510250135. [DOI] [PubMed] [Google Scholar]
- 7.Stratta RJ, Shaefer MS, Markin RS, Wood RP, Kennedy EM, Langnas AN, Reed EC, Woods GL, Donovan JP, Pillen TJ, et al. 1989. Clinical patterns of cytomegalovirus disease after liver transplantation. Arch Surg 124:1443–1449. (Discussion, 124:1449–1450.) [DOI] [PubMed] [Google Scholar]
- 8.Theise ND, Conn M, Thung SN. 1993. Localization of cytomegalovirus antigens in liver allografts over time. Hum Pathol 24:103–108. doi: 10.1016/0046-8177(93)90069-S. [DOI] [PubMed] [Google Scholar]
- 9.Paya CV, Hermans PE, Wiesner RH, Ludwig J, Smith TF, Rakela J, Krom RA. 1989. Cytomegalovirus hepatitis in liver transplantation: prospective analysis of 93 consecutive orthotopic liver transplantations. J Infect Dis 160:752–758. doi: 10.1093/infdis/160.5.752. [DOI] [PubMed] [Google Scholar]
- 10.Sinzger C, Bissinger AL, Viebahn R, Oettle H, Radke C, Schmidt CA, Jahn G. 1999. Hepatocytes are permissive for human cytomegalovirus infection in human liver cell culture and In vivo. J Infect Dis 180:976–986. doi: 10.1086/315032. [DOI] [PubMed] [Google Scholar]
- 11.Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, Reddehase MJ, Koszinowski UH. 2008. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3:263–272. doi: 10.1016/j.chom.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Thomas S, Klobuch S, Podlech J, Plachter B, Hoffmann P, Renzaho A, Theobald M, Reddehase MJ, Herr W, Lemmermann NA. 2015. Evaluating human T-cell therapy of cytomegalovirus organ disease in HLA-transgenic mice. PLoS Pathog 11:e1005049. doi: 10.1371/journal.ppat.1005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Podlech J, Reddehase MJ, Adler B, Lemmermann NA. 2015. Principles for studying in vivo attenuation of virus mutants: defining the role of the cytomegalovirus gH/gL/gO complex as a paradigm. Med Microbiol Immunol 204:295–305. doi: 10.1007/s00430-015-0405-2. [DOI] [PubMed] [Google Scholar]
- 14.Lemmermann NA, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H, Karbach A, Wu Y, Jonjic S, Reddehase MJ, Adler B. 2015. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog 11:e1004640. doi: 10.1371/journal.ppat.1004640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seckert CK, Renzaho A, Tervo HM, Krause C, Deegen P, Kuhnapfel B, Reddehase MJ, Grzimek NK. 2009. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83:8869–8884. doi: 10.1128/JVI.00870-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shanley JD, Jordan MC, Cook ML, Stevens JG. 1979. Pathogenesis of reactivated latent murine cytomegalovirus infection. Am J Pathol 95:67–80. [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. 2008. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 18.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, Tanner MS, Kagalwalla AF, Nemeth A, Pawlowska J, Baker A, Mieli-Vergani G, Freimer NB, Gardiner RM, Thompson RJ. 1998. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 19.Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. 2003. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem 278:33920–33927. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 20.Reshetnyak VI. 2013. Physiological and molecular biochemical mechanisms of bile formation. World J Gastroenterol 19:7341–7360. doi: 10.3748/wjg.v19.i42.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alrefai WA, Gill RK. 2007. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res 24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 22.Keitel V, Kubitz R, Haussinger D. 2008. Endocrine and paracrine role of bile acids. World J Gastroenterol 14:5620–5629. doi: 10.3748/wjg.14.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graf D, Kurz AK, Fischer R, Reinehr R, Haussinger D. 2002. Taurolithocholic acid-3 sulfate induces CD95 trafficking and apoptosis in a c-Jun N-terminal kinase-dependent manner. Gastroenterology 122:1411–1427. doi: 10.1053/gast.2002.32976. [DOI] [PubMed] [Google Scholar]
- 24.Graf D, Reinehr R, Kurz AK, Fischer R, Haussinger D. 2003. Inhibition of taurolithocholate 3-sulfate-induced apoptosis by cyclic AMP in rat hepatocytes involves protein kinase A-dependent and -independent mechanisms. Arch Biochem Biophys 415:34–42. doi: 10.1016/S0003-9861(03)00224-8. [DOI] [PubMed] [Google Scholar]
- 25.Chiang JY. 2013. Bile acid metabolism and signaling. Compr Physiol 3:1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidler Pfandler MA, Hochli M, Inderbitzin D, Meier PJ, Stieger B. 2004. Small hepatocytes in culture develop polarized transporter expression and differentiation. J Cell Sci 117:4077–4087. doi: 10.1242/jcs.01279. [DOI] [PubMed] [Google Scholar]
- 27.Tchaparian EH, Houghton JS, Uyeda C, Grillo MP, Jin L. 2011. Effect of culture time on the basal expression levels of drug transporters in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos 39:2387–2394. doi: 10.1124/dmd.111.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rippin SJ, Hagenbuch B, Meier PJ, Stieger B. 2001. Cholestatic expression pattern of sinusoidal and canalicular organic anion transport systems in primary cultured rat hepatocytes. Hepatology 33:776–782. doi: 10.1053/jhep.2001.23433. [DOI] [PubMed] [Google Scholar]
- 29.Correia L, Podevin P, Borderie D, Verthier N, Montet JC, Feldmann G, Poupon R, Weill B, Calmus Y. 2001. Effects of bile acids on the humoral immune response: a mechanistic approach. Life Sci 69:2337–2348. doi: 10.1016/S0024-3205(01)01321-2. [DOI] [PubMed] [Google Scholar]
- 30.Haselow K, Bode JG, Wammers M, Ehlting C, Keitel V, Kleinebrecht L, Schupp AK, Haussinger D, Graf D. 2013. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol 94:1253–1264. doi: 10.1189/jlb.0812396. [DOI] [PubMed] [Google Scholar]
- 31.Graf D, Haselow K, Munks I, Bode JG, Haussinger D. 2010. Inhibition of interferon-alpha-induced signaling by hyperosmolarity and hydrophobic bile acids. Biol Chem 391:1175–1187. doi: 10.1515/BC.2010.108. [DOI] [PubMed] [Google Scholar]
- 32.Ramiere C, Scholtes C, Diaz O, Icard V, Perrin-Cocon L, Trabaud MA, Lotteau V, Andre P. 2008. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRalpha. J Virol 82:10832–10840. doi: 10.1128/JVI.00883-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholtes C, Diaz O, Icard V, Kaul A, Bartenschlager R, Lotteau V, Andre P. 2008. Enhancement of genotype 1 hepatitis C virus replication by bile acids through FXR. J Hepatol 48:192–199. doi: 10.1016/j.jhep.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Chang KO. 2011. Inhibitory effects of bile acids and synthetic farnesoid X receptor agonists on rotavirus replication. J Virol 85:12570–12577. doi: 10.1128/JVI.05839-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. 2000. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744. doi: 10.1016/S0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 36.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 37.Reinehr R, Becker S, Braun J, Eberle A, Grether-Beck S, Haussinger D. 2006. Endosomal acidification and activation of NADPH oxidase isoforms are upstream events in hyperosmolarity-induced hepatocyte apoptosis. J Biol Chem 281:23150–23166. doi: 10.1074/jbc.M601451200. [DOI] [PubMed] [Google Scholar]
- 38.Brune W, Hengel H, Koszinowski UH. 2001. A mouse model for cytomegalovirus infection. Curr Protoc Immunol Chapter 19:Unit 19.17. [DOI] [PubMed] [Google Scholar]
- 39.Rattay S, Trilling M, Megger DA, Sitek B, Meyer HE, Hengel H, Le-Trilling VT. 2015. The canonical immediate early 3 gene product pIE611 of mouse cytomegalovirus is dispensable for viral replication but mediates transcriptional and posttranscriptional regulation of viral gene products. J Virol 89:8590–8598. doi: 10.1128/JVI.01234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trilling M, Le VT, Fiedler M, Zimmermann A, Bleifuss E, Hengel H. 2011. Identification of DNA-damage DNA-binding protein 1 as a conditional essential factor for cytomegalovirus replication in interferon-gamma-stimulated cells. PLoS Pathog 7:e1002069. doi: 10.1371/journal.ppat.1002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski UH. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A 94:14759–14763. doi: 10.1073/pnas.94.26.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monte MJ, Marin JJ, Antelo A, Vazquez-Tato J. 2009. Bile acids: chemistry, physiology, and pathophysiology. World J Gastroenterol 15:804–816. doi: 10.3748/wjg.15.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alnouti Y, Csanaky IL, Klaassen CD. 2008. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Angulo A, Messerle M, Koszinowski UH, Ghazal P. 1998. Enhancer requirement for murine cytomegalovirus growth and genetic complementation by the human cytomegalovirus enhancer. J Virol 72:8502–8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grzimek NK, Podlech J, Steffens HP, Holtappels R, Schmalz S, Reddehase MJ. 1999. In vivo replication of recombinant murine cytomegalovirus driven by the paralogous major immediate-early promoter-enhancer of human cytomegalovirus. J Virol 73:5043–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K. 2014. Molecular mechanisms of hepatic apoptosis. Cell Death Dis 5:e996. doi: 10.1038/cddis.2013.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. 2009. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 48.Hewison M. 2012. An update on vitamin D and human immunity. Clin Endocrinol (Oxf) 76:315–325. doi: 10.1111/j.1365-2265.2011.04261.x. [DOI] [PubMed] [Google Scholar]
- 49.Miller J, Gallo RL. 2010. Vitamin D and innate immunity. Dermatol Ther 23:13–22. doi: 10.1111/j.1529-8019.2009.01287.x. [DOI] [PubMed] [Google Scholar]
- 50.Meier PJ, Eckhardt U, Schroeder A, Hagenbuch B, Stieger B. 1997. Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology 26:1667–1677. doi: 10.1002/hep.510260641. [DOI] [PubMed] [Google Scholar]
- 51.Mita S, Suzuki H, Akita H, Stieger B, Meier PJ, Hofmann AF, Sugiyama Y. 2005. Vectorial transport of bile salts across MDCK cells expressing both rat Na+-taurocholate cotransporting polypeptide and rat bile salt export pump. Am J Physiol Gastrointest Liver Physiol 288:G159–G167. doi: 10.1152/ajpgi.00360.2003. [DOI] [PubMed] [Google Scholar]
- 52.Dong Z, Ekins S, Polli JE. 2013. Structure-activity relationship for FDA approved drugs as inhibitors of the human sodium taurocholate cotransporting polypeptide (NTCP). Mol Pharm 10:1008–1019. doi: 10.1021/mp300453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angulo A, Ghazal P, Messerle M. 2000. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J Virol 74:11129–11136. doi: 10.1128/JVI.74.23.11129-11136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez-Martin S, Kropp KA, Wilhelmi V, Lisnic VJ, Hsieh WY, Blanc M, Livingston A, Busche A, Tekotte H, Messerle M, Auer M, Fraser I, Jonjic S, Angulo A, Reddehase MJ, Ghazal P. 2012. Ablation of the regulatory IE1 protein of murine cytomegalovirus alters in vivo pro-inflammatory TNF-alpha production during acute infection. PLoS Pathog 8:e1002901. doi: 10.1371/journal.ppat.1002901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilhelmi V, Simon CO, Podlech J, Bohm V, Daubner T, Emde S, Strand D, Renzaho A, Lemmermann NA, Seckert CK, Reddehase MJ, Grzimek NK. 2008. Transactivation of cellular genes involved in nucleotide metabolism by the regulatory IE1 protein of murine cytomegalovirus is not critical for viral replicative fitness in quiescent cells and host tissues. J Virol 82:9900–9916. doi: 10.1128/JVI.00928-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cowling VH, Cole MD. 2010. Myc regulation of mRNA cap methylation. Genes Cancer 1:576–579. doi: 10.1177/1947601910378025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wickens M, Anderson P, Jackson RJ. 1997. Life and death in the cytoplasm: messages from the 3′ end. Curr Opin Genet Dev 7:220–232. doi: 10.1016/S0959-437X(97)80132-3. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka S, Furukawa T, Plotkin SA. 1975. Human cytomegalovirus stimulates host cell RNA synthesis. J Virol 15:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stinski MF. 1977. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J Virol 23:751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. 2014. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep 6:9–17. doi: 10.1016/j.celrep.2013.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chantler JK. 1978. The use of hypertonicity to selectively inhibit host translation in murine cytomegalovirus-infected cells. Virology 90:166–169. doi: 10.1016/0042-6822(78)90346-X. [DOI] [PubMed] [Google Scholar]
- 62.Chantler JK, Hudson JB. 1978. Proteins of murine cytomegalovirus: identification of structural and nonstructural antigens in infected cells. Virology 86:22–36. doi: 10.1016/0042-6822(78)90004-1. [DOI] [PubMed] [Google Scholar]
- 63.Trauner M, Graziadei IW. 1999. Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment Pharmacol Ther 13:979–996. doi: 10.1046/j.1365-2036.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 64.Hempfling W, Dilger K, Beuers U. 2003. Ursodeoxycholic acid—adverse effects and drug interactions. Aliment Pharmacol Ther 18:963–972. doi: 10.1046/j.1365-2036.2003.01792.x. [DOI] [PubMed] [Google Scholar]
- 65.Benz C, Reusch U, Muranyi W, Brune W, Atalay R, Hengel H. 2001. Efficient downregulation of major histocompatibility complex class I molecules in human epithelial cells infected with cytomegalovirus. J Gen Virol 82:2061–2070. doi: 10.1099/0022-1317-82-9-2061. [DOI] [PubMed] [Google Scholar]
- 66.Aldini R, Roda A, Festi D, Sama C, Mazzella G, Bazzoli F, Morselli AM, Roda E, Barbara L. 1982. Bile acid malabsorption and bile acid diarrhea in intestinal resection. Dig Dis Sci 27:495–502. doi: 10.1007/BF01296727. [DOI] [PubMed] [Google Scholar]
- 67.Gnewuch C, Liebisch G, Langmann T, Dieplinger B, Mueller T, Haltmayer M, Dieplinger H, Zahn A, Stremmel W, Rogler G, Schmitz G. 2009. Serum bile acid profiling reflects enterohepatic detoxification state and intestinal barrier function in inflammatory bowel disease. World J Gastroenterol 15:3134–3141. doi: 10.3748/wjg.15.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garrido E, Carrera E, Manzano R, Lopez-Sanroman A. 2013. Clinical significance of cytomegalovirus infection in patients with inflammatory bowel disease. World J Gastroenterol 19:17–25. doi: 10.3748/wjg.v19.i1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen M, Bradford K, Zhang X, Shih DQ. 2011. Cytomegalovirus reactivation in ulcerative colitis patients. Ulcers 2011. doi: 10.1155/2011/282507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Q, Li L, Maul GG. 2005. Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate-early promoter. J Virol 79:257–263. doi: 10.1128/JVI.79.1.257-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martinez FP, Cosme RS, Tang Q. 2010. Murine cytomegalovirus major immediate-early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J Gen Virol 91:2664–2676. doi: 10.1099/vir.0.022301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 73.Podlech J, Holtappels R, Wirtz N, Steffens HP, Reddehase MJ. 1998. Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J Gen Virol 79:2099–2104. doi: 10.1099/0022-1317-79-9-2099. [DOI] [PubMed] [Google Scholar]
- 74.Sugita T, Amano K, Nakano M, Masubuchi N, Sugihara M, Matsuura T. 2015. Analysis of the serum bile acid composition for differential diagnosis in patients with liver disease. Gastroenterol Res Pract 2015:717431. doi: 10.1155/2015/717431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Makino I, Nakagawa S, Mashimo K. 1969. Conjugated and unconjugated serum bile acid levels n patients with hepatobiliary diseases. Gastroenterology 56:1033–1039. [PubMed] [Google Scholar]