

Abstract
The disorders of large granular lymphocytes include reactive proliferation as well as indolent or aggressive neoplasms of cytotoxic T cells, γδ T cells, and natural killer (NK) cells. They are associated with autoimmune and infectious disorders and have varied immunophenotypic features. We report a case, which highlights this complex association of autoimmune and infectious diseases with large granular lymphocytosis, the overlapping spectrum of large granular lymphocyte leukemias, and γδ T cell lymphomas as well as the difficulties in the diagnosis and management of these indolent T cell lymphomas in the usual clinical settings.
KEY WORDS: Celiac disease (CD), large granular lymphocyte (LGL), pure red cell aplasia (PRCA), γδ T cell
Introduction
Large granular lymphocytes (LGLs) are a distinct subpopulation of lymphocytes, which constitute 10-15% of peripheral blood mononuclear cells (0.1-0.3 × 109 /l) and are derived from cytotoxic T cells, γδ T cells, or natural killer (NK) cells. The disorders of LGLs include reactive proliferation, indolent neoplasms such as T-cell LGL (T-LGL) leukemia, and a chronic lymphoproliferative disorder of NK cells as well as their aggressive variants. They are associated with various autoimmune and infectious disorders.[1,2,3] These disorders, especially the indolent ones being uncommon and having overlapping features with reactive proliferations, lead to diagnostic dilemmas in routine clinical practice. Here, we report such a case of complex association of large granular lymphocytosis with pure red cell aplasia (PRCA), celiac disease (CD), and chronic hepatitis B virus (HBV) infection.
Case Report
A 46-year-old male presented with severe transfusion dependent anemia. He was diagnosed with CD and was asymptomatic on a gluten-free diet. One year back, he had an episode of fever and anemia during which he was found to have chronic HBV infection for which he was treated with entecavir 0.5 mg daily. During the present admission, he had no significant physical examination findings except for severe pallor and mild hepatomegaly. He was managed with blood transfusions and hematinics and evaluated for the cause of anemia. The investigation results are summarized in Table 1. There was anemia with reticulocytopenia. Peripheral blood (PB) smear showed absolute lymphocytosis (6993/μl) with >90% of them being LGLs [Figure 1a]. Bone marrow examination (BME) revealed normocellular bone marrow (BM) with erythroid hypoplasia, the myeloid to erythroid ratio being 40:1 and without any dyserythropoiesis or any viral cytopathic change. BM trephine biopsy showed multiple reactive lymphoid nodules composed of both B cells and T cells, as revealed by immunohistochemistry [Figure 1b–f]. A multiparameter flow cytometric immunophenotyping (MFCI) performed from the PB using a limited panel of antibodies showed an excess of CD8+CD16+ T cells. Serological tests for immunoglobulin M (IgM) Parvovirus antibodies were negative. Considering the possibility of PRCA, the patient was treated with prednisolone 1 mg/kg/day, along with entecavir. The subsequent clinical course is summarized in Figure 2. A repeat BME, 16 months after the initial BME showed PRCA, increased stainable iron (Perls stain), and multiple reactive lymphoid nodules. MFCI of PB lymphocytes [Figure 3] showed that 92% of them were T cells. There was an increase in γδ T cells (CD2+ CD5- CD7+ CD4- CD8variable CD16+ CD56- CD57variable), which constituted approximately 48% of T cells. The last follow-up (20 months after initial BME) revealed the persistence of large granular lymphocytosis and refractory anemia being supported with blood transfusions and iron chelation. The patient was asymptomatic of CD and HBV infection.
Table 1.
Summary of investigations during initial workup of anemia
| Hemoglobin (after transfusion) | 64 g/L | Serum iron | 145 µg/dL |
| Total leukocyte count | 11.1×109/L | Serum ferritin | 1123 ng/mL |
| Platelet count | 4.65×109/L | Total iron binding capacity | 322 µg/dL |
| Reticulocyte count (corrected) | 0.99% | Transferrin saturation | 45% |
| Plasma hemoglobin | Not raised | Serum vitamin B12 | 1346 pg/mL |
| Urine hemoglobin | Negative | Stool for occult blood | Negative |
| HBV DNA | <20 IU/L | Liver function tests | Normal |
| Renal function tests | Normal | Thyroid function tests | Normal |
| IgA tissue transglutaminase | <5 U/ml | Antinuclear antibodies (ANAs) | Negative |
| Antiliver kidney microsome (LKM) antibodies | Negative | Antimitochondrial antibodies (AMAs) | Negative |
| Antiparietal cell antibodies (PCAs) | Negative | Antismooth muscle actin (SMA) antibodies | Positive |
Figure 1.
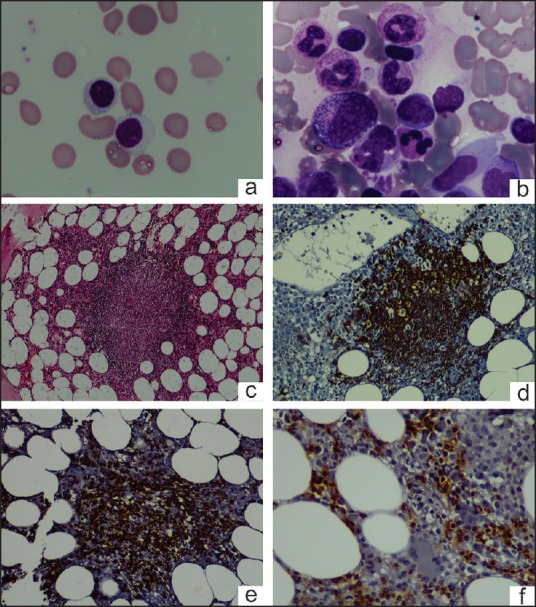
Peripheral blood smear shows large granular lymphocyte (a) [100x, May-Grünwald-Giemsa (MGG) stain]; Bone marrow aspirate shows erythroblastopenia and large granular lymphocytes (b) (100x, MGG stain); trephine biopsy shows reactive lymphoid nodules (c) (20x, H&E stain) composed of CD20+ cells (d) (20x) and CD3+ cells (e) (20x); CD8+ cells in the interstitium (f) (20x)
Figure 2.
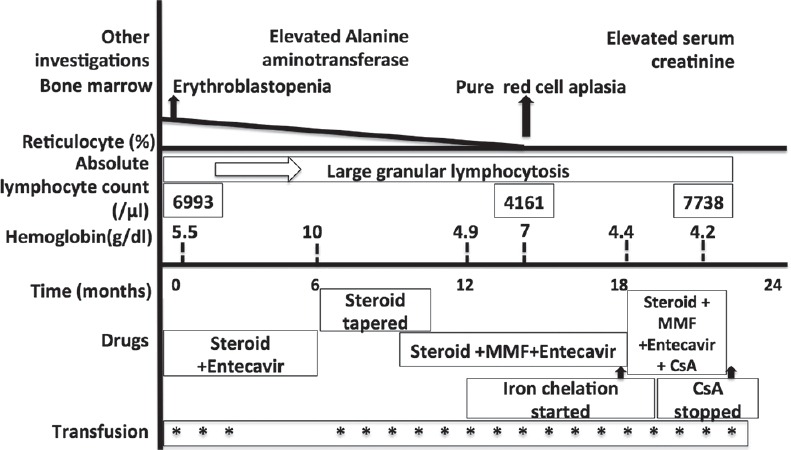
Clinical course of the patient. (MMF - mycophenolate mofetil; CsA - cyclosporine)
Figure 3.

Flow cytometry performed on peripheral blood. CD3+ cells (92% of lymphocytes) were gated. 48% of T cells were γδ TCR+ (3a), which were CD2+CD7+ (not shown), CD8+ (variable) (3a), CD5− (3b), CD4− (3c), CD57+(variable), CD56− (3d), and CD16+ (not shown)
Discussion
The association of PRCA with large granular lymphocytosis is reported in the literature, which could be either nonclonal as in idiopathic PRCA or clonal as in LGL leukemias. There are isolated cases of PRCA associated with CD[4] and HBV infection.[5] Expansion of LGLs can be transient or chronic reactive proliferation, neoplastic, or borderline between reactive and leukemic disorders. Reactive causes include various autoimmune disorders, splenectomy, post allogenic stem cell or solid organ transplantation, HIV and other viral infections.[2] LGL leukemias are indolent disorders with a survival of more than 10 years in usual settings and characterized by persistent (>6 months), large granular lymphocytosis in PB, usually of 2-20 × 109 /L without a clearly identifiable cause. They are often associated with infections, various hematologic manifestations such as PRCA, neutropenia, and thrombocytopenia as well as various autoimmune disorders and autoantibodies.[2,6] BM shows subtle interstitial infiltrate with intravascular or linear pattern highlighted by immunohistochemistry and often associated with reactive nodular aggregates of B and T cells.[2] Normal LGLs are either CD3+ CD8+ cytotoxic T cells (T LGL) or CD3- NK cells. Normal T LGLs are CD3+ CD2+ CD4- CD5+ CD7+ CD8+αβTCR+γδTCR- while LGL leukemias show abnormal immunophenotype such as abnormal expression of CD2, CD5, and CD7 and coexpression of CD16, CD56, and CD57.[2] Most of the LGL leukemias show CD3+ CD8+ CD4-αβTCR+ phenotype. Other phenotypes include CD3+ CD8- CD4+αβTCR+, CD3+ CD8+ CD4-γδTCR+, and CD3+ CD8- CD4-γδTCR+ with CD16 and CD57 expression in a variable number of cases.[6] Our patient had CD and chronic HBV in the background, and had smooth muscle antibodies (SMAs). There have been previous reports of LGL leukemia associated with CD and HBV;[3,7] there has also been an occasional report of reactive expansion of LGLs in HBV infection.[8] In our patient, autoimmune features could be a manifestation of LGL leukemia or the expansion of LGLs might be secondary to CD or HBV or both. The differentiation of the clonal process from reactive proliferation requires analysis of T cell receptor (TCR) rearrangement by flow cytometry or Southern blot analysis or polymerase chain reaction (PCR). More commonly used PCR has a sensitivity of only 70-80%.[1,2] Our patient showed a persistent LGL (>2000/μl) for nearly 2 years and had an abnormal immunophenotype of CD2+ CD4- CD5- CD7+ CD8variable CD16+ CD56- CD57variable γδ TCR+. The BM involvement was similar to that described in the literature [Figure 1] and was refractory to various modalities of treatment, all of which favor, though does not prove an indolent leukemic process over a reactive condition. Compared to αβ T cell LGL leukemia, γδ type is rare with up to 50% of them showing CD3+ CD8+ CD4− CD16+ CD57+ phenotype, slightly more common than CD4- CD8- phenotype. Both subtypes have a similar clinical behavior.[3,9] Both αβTCR+ and γδTCR+ LGL leukemias are included under LGL leukemia in the World Health Organization (WHO) 2008 classification but γδ TCR+ LGL leukemias are a distinct class of γδ T cell lymphomas with indolent clinical course in contrast to most other γδ T cell lymphomas.[10] Various treatment options that are useful for LGL leukemias include steroids, low dose methotrexate, cycyclophosphamide, purine analogs, alemtuzumab (anti-CD52 monoclonal antibody), and stem cell transplantation.[1]
In our case, large granular lymphocytosis probably would have contributed significantly to the development of PRCA. However, the role of HBV and CD in the expansion of LGLs and the subsequent development of PRCA are debatable. The clonality studies could not be performed in our case but it has to be highlighted that the demonstration of clonality may not always support a malignant process as oligoclonal and sometimes clonal expansions can be seen in various benign conditions including viral infections.[2] The management of PRCA remains complicated by association with multiple diseases, especially HBV infection.
Our case highlights the uncommon and complex association of multiple immunologically related diseases and large granular lymphocytosis; the overlapping spectrum of indolent LGL and γδ T cell proliferations, and the difficulties in the diagnosis of indolent T cell lymphomas in the usual clinical settings as reflected in few case reports and a lack of any large series in the Indian literature.
Financial support and sponsorship
Nil.
Conflicts of interest
Authors declare that they have no conflict of interests.
References
- 1.Dearden C. Large granular lymphocytic leukaemia pathogenesis and management. Br J Haematol. 2011;152:273–83. doi: 10.1111/j.1365-2141.2010.08494.x. [DOI] [PubMed] [Google Scholar]
- 2.O’Malley DP. T-cell large granular leukemia and related proliferations. Am J Clin Pathol. 2007;127:850–9. doi: 10.1309/A8FHDA0VVRJ05GJP. [DOI] [PubMed] [Google Scholar]
- 3.Bourgault-Rouxel AS, Loughran TP, Jr, Zambello R, Epling-Burnette PK, Semenzato G, Donadieu J, et al. Clinical spectrum of gammadelta+ T cell LGL leukemia: Analysis of 20 cases. Leuk Res. 2008;32:45–8. doi: 10.1016/j.leukres.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Couderc AL, Costello R, Bagnères D, Rossi P, Vitton V, Demoux AL, et al. Is pure red cell aplasia a new extra digestive manifestation of celiac disease? Rev Med Interne. 2006;27:336–9. doi: 10.1016/j.revmed.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Ide T, Sata M, Nouno R, Yamashita F, Nakano H, Tanikawa K. Clinical evaluation of four cases of acute viral hepatitis complicated by pure red cell aplasia. Am J Gastroenterol. 1994;89:257–62. [PubMed] [Google Scholar]
- 6.Chan WC, Foucar K, Morice WG, Catovsky D. T cell large granular lymphocytic leukemia. In: Swerldlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. pp. 272–3. [Google Scholar]
- 7.Malamut G, Meresse B, Verkarre V, Kaltenbach S, Montcuquet N, Duong Van Huyen JP, et al. Large granular lymphocytic leukemia: A treatable form of refractory celiac disease. Gastroenterology. 2012;143:1470–2.e2. doi: 10.1053/j.gastro.2012.08.028. [DOI] [PubMed] [Google Scholar]
- 8.Agostini C, Zambello R, Pontisso P, Alberti A, Trentin L, Siviero F, et al. Lymphoproliferative disease of granular lymphocytes in a patient with concomitant hepatitis B virus infection of CD4 lymphocytes. J Clin Immunol. 1989;9:401–8. doi: 10.1007/BF00917105. [DOI] [PubMed] [Google Scholar]
- 9.Sandberg Y, Almeida J, Gonzalez M, Lima M, Bárcena P, Szczepañski T, et al. TCRgammadelta+ large granular lymphocyte leukemias reflect the spectrum of normal antigen-selected TCR gammadelta+ T-cells. Leukemia. 2006;20:505–13. doi: 10.1038/sj.leu.2404112. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad E, Kingma DW, Jaffe ES, Schrager JA, Janik J, Wilson W, et al. Flow cytometric immunophenotypic profiles of mature gamma delta T-cell malignancies involving peripheral blood and bone marrow. Cytometry B Clin Cytom. 2005;67:6–12. doi: 10.1002/cyto.b.20063. [DOI] [PubMed] [Google Scholar]