

Abstract
Pseudomonas aeruginosa (PsA) is a highly prevalent bacterial organism recovered from the lungs of cystic fibrosis (CF) patients and chronic PsA infection is linked to progressive pulmonary function decline. The eradication and treatment of this organism from CF airways is particularly challenging to CF care providers. Aerosolized antibiotics that target PsA help to slow down growth, maintain lung function and reduce the frequency of pulmonary exacerbations. In this review, we discuss the currently available inhaled antibiotics for management of PsA lung infections in CF patients, with a focus on liposomal amikacin for inhalation (LAI). LAI is a unique formulation of amikacin under development that enhances drug delivery and retention in CF airways via drug incorporation into neutral liposomes. Factors such as once-daily dosing, mucus and biofilm penetration and potentially prolonged off-drug periods make LAI a potentially attractive option to manage chronic PsA lung infections in CF patients.
KEYWORDS : amikacin, arikace, arikayce, cystic fibrosis, LAI, Pseudomonas aeruginosa
Cystic fibrosis (CF) is a life-limiting hereditary disorder occurring in virtually all ethnic groups with a prevalence of approximately 30,000 patients in the USA and over 70,000 worldwide. CF is caused by mutations in a chromosome 7 gene that encodes the CF transmembrane conductance regulator (CFTR) protein [1–4]. CFTR is a chloride and bicarbonate channel that regulates epithelial ion transport and hydration [5,6]. Mutations in CFTR affect the balance of epithelial ion and water transport, causing dehydrated and viscous mucus secretions in most CF-affected organs [7]. CF is a multisystem disease with prominent symptoms in the sinopulmonary tree, GI tract, pancreas, hepatobiliary system and male reproductive tract. Pulmonary infection and subsequent pulmonary decline is the leading cause of CF morbidity and mortality. A deficiency in mucociliary clearance makes the airways susceptible to bacterial and other infections that typically become chronic and lifelong [8]. Therefore, focusing efforts on treatment of lung infections in CF patients is a central component of CF care.
The burden of Pseudomonas aeruginosa pulmonary infection in CF patients
The microbial milieu of CF lungs changes with age. Haemophilus influenzae and Staphylococcus aureus (including both methicillin-sensitive and more recently methicillin-resistant strains) are more frequent pathogens in younger pediatric patients. With advancing age or disease, additional pathogens such as Stenotrophomonas maltophila, Achromobacter xyloxidans, Burkholderia cepacia complex, nontuberculous mycobacteria and other pathogens with inherent resistance to many antibiotics often predominate [9]. CF registry data confirm that Pseudomonas aeruginosa (PsA) typically becomes the predominant organism cultured from the respiratory tract by adulthood, with the prevalence rising from 20% in patients less than 5 years of age up to as high as 70% by 18 years of age [9]. It also is well established that chronic lung infection with PsA causes more-rapid pulmonary decline and contributes to early death in CF patients [10]. Thus, treatment of PsA infection has become an integral part of CF care. In addition to the ‘traditional’ CF pulmonary pathogens, recent research provides evidence for a highly complex microbiome in the CF airway. A better understanding of these organisms and the host:pathogen relationship in CF is believed to be critical to improved management and appropriate treatment of CF airway infections [11].
PsA isolates frequently possess features that contribute to their adaptation and persistence in the CF lung. They often possess a hypermutable genetic background that leads to the emergence of variants capable of long-term persistence, such as a mucoid phenotype, modified antigenic structures and antimicrobial resistance [12,13]. The mucoid phenotype is caused by extracellular alginate production, which can reduce antimicrobial effectiveness by limiting drug penetration. Evidence also suggests that PsA can grow as a biofilm in the lower respiratory tract of CF patients, rendering the organism resistant to many antimicrobials [14,15]. In-depth reviews of this topic have been published previously [16,17].
Although the definition of chronic PsA infection is still under debate [18,19], chronic PsA colonization is associated with a more-rapid decline in lung function [20]. The complex CF lower-airway environment (alginate production, neutrophilic inflammation and thick mucus) makes it particularly challenging to clear mucoid PsA from CF airways and thus management of chronic infection is a mainstay of current CF care.
• The goals of treatment of PsA infection in CF patients
The goals of chronic PsA treatment can be broadly categorized into two aims: the treatment of acute pulmonary exacerbations and the management of chronic PsA infection during periods of relative clinical stability. Acute pulmonary exacerbations in CF typically include an increase in pulmonary and systemic symptoms (e.g., increased cough, chest congestion and sputum production accompanied by malaise and weight loss) coupled with physical findings (e.g., crackles), as well as a reduction in lung function as measured by the forced expiratory volume in 1 s (FEV1) [21–25]. Treatment includes an increase in airway clearance and the addition of antibiotics that target known lower airway pathogens. In patients with chronic PsA infection, this usually includes the addition of anti-PsA antibiotics via oral, inhaled and/or intravenous routes. Although this approach is beneficial, a significant percentage of patients (˜25%) fail to fully recover the lost lung function [26,27]. As this decline in lung function can be accumulative over the lifespan of CF patients, it is imperative to develop strategies to restore lung function in all affected patients.
Management of established PsA infections during clinical stability is a common point of CF care, and typically includes the cycling of antibiotics. Aerosolized antibiotics have the advantage of achieving high intrapulmonary concentrations with the potential for fewer associated systemic side effects. The use of inhaled antibiotics for chronic PsA infection has become a standard of care for CF. The goal of this therapy is the reduction of PsA density and host inflammation, maintenance of lung function and reduction in the frequency of acute pulmonary exacerbations [28,29].
PsA infections can manifest in different ways, with early PsA infections being mostly transient. Clearance of organisms can occur either spontaneously or after treatment with anti-PsA antibiotics [30,31]. These infections are typically caused by organisms that are sensitive to a variety of antibiotics, and evidence suggests that these transient infections are not associated with long-term decline in pulmonary function [10,32]. Several European CF care centers have adapted a strategy to aggressively treat early PsA infections, resulting in delay of the onset of chronic PsA colonization [33–35]. Two large controlled studies have examined similar strategies to eradicate early PsA infection (Early Inhaled Tobramycin for Eradication, and Early Pseudomonas Infection Control) [31,36–38]. These studies demonstrated that treatment of early PsA infection with tobramycin inhalational solution (TIS; TOBI®, Novartis Pharmaceuticals Corporation, USA) twice daily for 28 days (with or without oral ciprofloxacin) leads to successful eradication of PsA in up to 85% of CF patients, with >60% of patients remaining PsA negative at 6 months post-treatment.
As stated previously, chronic PsA infection (frequently with mucoid strains) is an important complication in CF, and is therefore the prime target of long-term inhaled antibiotic therapy in these patients. The remainder of this review will focus on established therapies and therapies under development to manage chronic PsA infection in CF.
• Overview of the market: currently available therapies to manage chronic PsA infection
Tobramycin inhalational solution
Tobramycin is an aminoglycoside antibiotic produced by Streptomyces tenebrarius as a component of the nebramycin antibiotic complex [39]. It is bactericidal mechanism of action is disruption of bacterial protein synthesis. Tobramycin inhalational solution (TIS) is a sterile, clear aqueous solution with a pH and salinity specifically modified for administration by a reusable air-driven nebulizer (PARI LC Plus). TIS is formulated as single-use 5 ml ampules containing 300 mg tobramycin and 11.25 mg sodium chloride in sterile water. A landmark study evaluating TIS in CF patients chronically infected with PsA (FEV1 of 25–75% of predicted) demonstrated that three 28-day TIS-treatment cycles (alternating with 28 days off treatment) over 6 months produced sustained improvements in lung function, reductions in PsA density and pulmonary exacerbations, and improvements in weight compared with placebo [40]. Based primarily on these results, current Cystic Fibrosis Foundation pulmonary treatment guidelines recommend TIS for the management of chronic PsA infections in CF patients 6 years of age and older. The cycled dosing is frequently used (particularly in the USA), and has been proposed to allow the airways to remain largely colonized with susceptible organisms, thereby maintaining tobramycin efficacy over time [41].
Tobramycin inhalational powder
Tobramycin inhalational powder (TIP) is delivered by a hand-held dry-powder inhaler device (T-326 and together known as a TOBI Podhaler™, Novartis Pharmaceuticals Corporation, USA). TIP (112 mg administered as four powder capsules twice daily) is licensed for use in 28-day cycles similar to TIS. A large multicenter study (EAGER trial) showed that the safety profile of TIP and TIS were similar and they had comparable effects on lung function and sputum PsA density in CF patients chronically infected with PsA [42]. The incidence of cough, dysphonia and dysgeusia was slightly higher in patients receiving TIP. The administration time for TIP was approximately 14 min faster than TIS (excluding time required to set up, clean and disinfect the TIS nebulizer), and patient-reported satisfaction was significantly higher for TIP compared with TIS [42].
Concentrated tobramycin solutions for inhalation
Concentrated solutions of tobramycin for inhalation (75 mg/ml formulated in 4 ml) enable delivery of a more highly concentrated solution with the same total dose of tobramycin compared with TIS (300 mg/5 ml of tobramycin). This allows for delivery in a somewhat shorter period of time (approximately 15 min), achieving a higher peak sputum concentration compared with TIS [43]. Concentrated tobramycin solution for inhalation is currently marketed in multiple countries (Bramitob®; Chiesi Farmaceutici, Parma, Italy [44] and Bethkis®; Chiesi USA, Inc., NC, USA [45]).
In the USA, the concentrated formulation of tobramycin (4 ml single-use ampules containing 300 mg of tobramycin) is administered by inhalation using a hand-held nebulizer (PARI LC PLUS® Reusable Nebulizer; PARI Respiratory Equipment, Inc., VA, USA) with an air compressor (PARI Vios®).
The European counterpart has a concentration, formulation and delivery time similar to the USA version. When compared with TIS, both concentrated tobramycin formulations lead to a reduction in the density of PsA in sputum after both short-term (4 weeks) and long-term (over 56 weeks) intermittent therapy in patients with CF [43]. FEV1% predicted increased from baseline and non-inferiority between treatments was met and maintained throughout the extension phase of the study over 56 weeks [43]. No remarkable safety issues were identified throughout both study phases, with similar percentages of patients reporting adverse events (AEs) in the two treatment groups during the 8-week core phase (Bramitob®/Bethkis [31.4%]; TIS [28.0%]).
Aminoglycosides are an important antibiotic class to manage PsA lung infections in CF [10]. Inhaled tobramycin is well tolerated in CF patients and in a study by Ramsey et al. was shown to be more effective than bronchodilators or chest physiotherapy in improving lung function [4,5,40,46]. As recently summarized in a Cochrane review, inhaled antibiotics, particularly tobramycin, improve lung function and decrease pulmonary exacerbations when compared with placebo or usual treatment. Aerosolized tobramycin at a 600 mg daily dose (300 mg twice daily) has been shown to be safe and without clear renal or audiologic complications associated with either short- or long-term use [4,5,47,48].
Nebulized concentrated tobramycin
Nebulized concentrated tobramycin (NCT; Vantobra™, PARI Pharma GmbH, Starnberg, Germany) was recently approved in Europe. NCT includes 170 mg tobramycin/1.7 ml delivered via a drug-specific nebulizer handset (TOLERO®, operated with eBase controller; PARI Pharma) that is more concentrated than TIS with 100 mg tobramycin per ml (T100). The chemical formula of NCT is identical to that of TIS (C18H37N5O9 for tobramycin). Pulmonary exposure to tobramycin is similar to TIS due to the delivery device used with the concentrated product. Since the composition of NCT is quite similar to TIS, bioequivalence to the TIS reference product has been the focus of NCT development for regulatory agencies.
Sands and colleagues compared the T100 product to the standard combination of TIS/PARI LC Plus in a randomized, two-period, multicenter, cross-over 14-week Phase II study. Fifty-eight patients ≥4 years of age with chronic PsA infection were enrolled.
Pharmacokinetics showed a nonsignificant trend toward higher sputum concentrations of T100 (potentially reflective of higher local pulmonary concentrations) and lower serum concentrations (consistent with a lower systemic exposure). There was a reduction in PsA CFU after treatment with both drug/device combinations, with no difference between combinations at the end of Treatment period 1, but a statistically significant difference in log10 CFU pre- versus post-treatment between T100 and TIS at the end of Treatment period 2 favoring T100 (p = 0.0225). Effect on FEV1% predicted was similar for both groups for Treatment period 1; T100 (8.2 ± 9.49%; difference pre- vs post-treatment: p < 0.0001) and TIS (4.8 ± 9.58%; difference pre- vs post-treatment: p = 0.0132). The treatment effect similar for NCT and TIS after Treatment period 2 (2.4 ± 10.64% p = 0.2436 vs -0.44 ± 8.10% p = 0.7862).
AEs were noted in 29 out of 58 (50%) patients in both groups; three were severe and none were felt to be drug related (hospitalizations). Tinnitus was observed in two patients treated with T100 (3.4% of all patients) [49]. The nebulization times for the T100 drug/device combination was a mean of 4.4 min per treatment compared with the standard TIS mean of 24.3 min per treatment [49], with lowest reported duration of 14.6 min [50].
Comparable overall efficacy over the entire study supports therapeutic equivalence, and T100 had lower systemic exposure along with a similar safety profile compared with TIS.
Aztreonam solution for inhalation
Aztreonam solution for inhalation (AZLI; Cayston®, Gilead, CA, USA) is delivered via a nebulizer system (Altera® Nebulizer System; PARI Respiratory Equipment, Inc., VA, USA), utilizing a rapid delivery device (e-Flow®, PARI Pharma). It is a monobactam antibacterial dry mixture of aztreonam and arginine that is dosed three-times daily for 28-day treatment cycles. Delivery time is typically 2–3 min, and it is approved in the USA and many other countries for the management of PsA infection in CF patients. The AIR–CF1 trial published by McCoy and colleagues demonstrated that a 28-day course of AZLI given three-times daily led to delayed time to need inhaled or intravenous anti-PsA antibiotics, improved respiratory-symptom scores, increased FEV1 and decreased sputum density of PsA [51]. The AIR-CF2 trial confirmed that 28 days of AZLI (with either twice or thrice daily dosing) immediately following a 28-day course of TIS delayed the time to need for additional inhaled or systemic anti-PsA antibiotics [52]. Subsequently, the AIR-CF3 trial showed that repeated cycled treatment with AZLI was well tolerated and demonstrated clinical benefits in respiratory-symptom scores, FEV1 and weight gain [53]. A recently completed open-label trial in CF patients chronically treated with TIS showed that AZLI (three-times daily) had superior effects on lung function when compared with TIS over three 28-day courses [54]. The effects of both agents on FEV1 were relatively small compared with earlier nebulized antibiotic trials. The smaller treatment effect and AZLI treatment advantage may reflect the impact of prior long-term nebulized TIS use in this population.
Inhaled colistimethate sodium
Colistin is an antibiotic belonging to the polymixin family that exerts its action through disruption of the cell-wall membrane of Gram-negative rod bacteria [55,56]. The narrow spectrum of colistin activity targets several potential CF-related Gram-negative pathogens, such as Pseudomonas, Enterobacter, Acinetobacter, Citrobacter and Klebsiella. Given the low rates of resistance to colistin, it is an attractive choice to treat multidrug resistant isolates of PsA. Inhaled colistin (Colobreathe™, Forest Laboratories, Inc., NY, USA) is a capsule containing 1,662,500 IU of colistimethate sodium (approximately equal to 125 mg) that is administered with a portable inhaler (Turbospin®, PH&T, Milan, Italy) that relies on the patients’ inspiratory flow to drive dry-powder delivery. It is dosed as one capsule twice a day and administration time is less than 60 s. The device requires little cleaning or maintenance and is disposable at the end of each 28-day pack. The Freedom study investigated the efficacy and safety of three 28-day cycles of inhaled dry-powder colistimethate sodium compared with TIS in patients with CF aged ≥6 years chronically infected with PsA and reported non-inferiority to TIS over 24 weeks of active cycled treatment [57]. Both study arms showed only modest effects on lung function over the course of the study. However, the study subjects rated inhaled colistin higher than TIS for ease of delivery. AEs were similar in both groups except for cough and dysgeusia, which were more frequently reported in the colistin group [57]. Inhaled colistin has gained regulatory approval in several countries to treat chronic PsA infection in CF, but it is not currently FDA approved in the USA.
It is worth noting that in a recent network meta-analysis, Littlewood and colleagues concluded that tobramycin inhalational preparations, AZLI and inhaled colistin had comparable efficacy in CF patients chronically infected with PsA when assessing improvement in FEV1% predicted at 4 weeks [58].
• Therapies in development to manage chronic PsA infection
Fosfomycin/tobramycin for inhalation
Fosfomycin/tobramycin for inhalation (FTI) is a 4:1 combination of fosfomycin and tobramycin for nebulized delivery [59]. Fosfomycin is an inhibitor of bacterial cell-wall synthesis that is active against Gram-negative, Gram-positive and anaerobic bacteria [60]. Its potent activity against S. aureus (both methicillin sensitive [MSSA] and methicillin resistant [MRSA]) is of particular relevance to CF, since chronic MRSA infection is associated with higher mortality in CF patients [61]. Trapnell and colleagues reported that in a Phase II study of CF patients ≥8 years of age infected with PsA, FTI administered twice daily for 28 days was well tolerated and maintained the substantial improvements in FEV1% predicted that were achieved following a run-in period with a different inhaled antibiotic (AZLI), when compared with placebo. In addition, there were fewer AE reported with the lower FTI 80/20 mg dose compared with the FTI 160/40 mg dose [59].
Levofloxacin
Levofloxacin is a fluoroquinolone used extensively in its oral and parenteral form for bronchopneumonia caused by PsA in patients with CF. MP-376 (Aeroquin™; Forest Pharmaceuticals, Inc, NJ, USA) [62] is a solution of levofloxacin (240 mg/2.4 ml) administered via a customized nebulizer (eFlow®; PARI Pharma) [63]. Results from a Phase II study of nebulized levofloxacin in CF patients chronically infected with PsA demonstrated reduced sputum PsA density at Day 28 and improvement in FEV1, with the 240 mg twice daily dose demonstrating greater efficacy relative to other doses or placebo regimens [64]. Results of two Phase III studies were announced in a news brief by Aptalis Pharma in January 2013 [65]. In a randomized placebo-controlled single treatment-cycle study (Study 207) of 330 CF patients, comparing 240 mg of MP-376 nebulized twice daily failed to demonstrate statistical significance for time to first pulmonary exacerbation. However, MP-376 demonstrated efficacy in important secondary end points (e.g., lung function, PsA in sputum). Another Phase III study (Study 209) was a randomized open-label study comparing MP-376 with TIS for three 28-day on/off treatment cycles in 282 CF patients. Levofloxacin demonstrated noninferiority to TIS in relative change from baseline in FEV1% predicted after the first treatment cycle of 28 days with a maintained effect on lung function over all three treatment cycles. Levofloxacin was also similar or superior to TIS in clinically significant secondary end points (quality-of-life measures and time to pulmonary exacerbation) [65]. Detailed results from these two trials have not yet been published.
• Liposomal amikacin for inhalation (LAI) for management of chronic PsA infection
Introduction to the compound
Amikacin is an aminoglycoside derived from kanamycin that is active against most CF-relevant gram-negative bacteria, including PsA [66]. LAI (Arikayce™, previously Arikace™; Insmed, NJ, USA) is a unique formulation that encapsulates aqueous amikacin in charge-neutral, highly biocompatible liposomes composed of dipalmitoyl phosphatidyl choline and cholesterol [67,68]. These lipids, which are found within the recycling pool of natural surfactant, are proposed to ‘shield’ positively charged amikacin from the polyanionic mucins that are characteristic of CF sputum [69]. A summary of LAI has been provided in Table 1.
Table 1. . Summary table of liposomal amikacin for inhalation.
| Drug name | Liposomal amikacin, Arikayce™ |
|---|---|
| Phase |
Phase III |
| Indication |
Chronic Pseudomonas aeruginosa infection in CF |
| Pharmacology description |
Aminoglycoside |
| Route of administration |
Inhaled |
| Chemical structure | 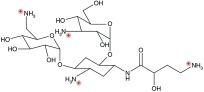 |
| |
Pharmaprojects – copyright to Citeline Drug Intelligence (http://informa.citeline.com) |
| Pivotal trial(s) | [67,69,71,76] |
Pharmacokinetics & pharmacodynamics
Okusanya and colleagues evaluated the pharmacokinetic and pharmacodynamic (PK–PD) properties of LAI in a cohort of 105 CF patients chronically infected with PsA. PK–PD relationships between efficacy end point measures (change in lung function from baseline) and PsA burden (change in log10 CFU from baseline) and amikacin exposure (dose, Day 1 AUC, dose/MIC ratio and Day 1 AUC/MIC ratio) were observed. An LAI dose of 560 mg once daily for a total of 14 days was associated with statistically significant improvements in FEV1 and FEV1% predicted and a reduction in log10 CFU of PsA from baseline. Doses >560 mg per day were not studied, and thus extrapolations of drug effects beyond this dose level are not available. These data serve to support the possible efficacy of a once-daily dose of 560 mg LAI for the treatment of CF patients with chronic infections due to PsA [67].
Clinical efficacy
LAI is delivered using an optimized investigational nebulizer (eFlow® Nebulizer System, PARI Pharma, Munich, Germany) [70] that rapidly delivers drug throughout the respiratory tree. Due to their uniform size, LAI (˜300 nm) particles have been demonstrated to penetrate the meshwork of CF sputum where they are retained and subsequently lyzed by local PsA-derived products (rhamnolipids) [71,72]. Thus, LAI has the capacity to deliver drug deeply into CF sputum, where active drug is released at the site of bacteria. LAI has been shown to have a prolonged lung half-life (several hours) relative to liposome-free antibiotics [71–73], which allows for once-daily dosing and drug accumulation in the lower airway of CF sputum. In a preclinical rat model of PsA lung infection, once-daily nebulized LAI compared favorably with liposome-free amikacin and tobramycin at more frequent dosing (twice daily) to reduce PsA density [49].
Extensive preclinical studies also demonstrated that while LAI can concentrate in alveolar macrophages (thought to be part of the natural clearance response to inhaled lipids), this was not associated with macrophage defects in cytokine signaling or pathogen-killing function [74]. These studies point toward the potential use of LAI to treat intracellular pathogens such as nontuberculous mycobacteria (NTM) infection in patients with and without CF (ClinicalTrials.gov identifier: NCT01315236). Indeed, treatment of pulmonary NTM infection is one of the greatest current challenges in CF care and therapeutic regimens typically include prolonged (months to years) treatment with intravenous amikacin combined with other antibiotics [32,75].
Recently, the results of a multinational Phase II, randomized, double-blind placebo-controlled trial of LAI in CF patients chronically infected with PsA was published [76]. The inclusion criteria were as follows: CF patients ≥6 years of age, FEV1 ≥40% predicted, chronic PsA infection, at least 28 days of clinical stability off of inhaled or intravenous antibiotics prior to enrollment and no known allergy to amikacin. Subjects harboring Burkholderia cepacia or nontuberculous mycobacteria, active allergic bronchopulmonary aspergillosis or significant comorbid medical conditions placing the subject at potential risk from enrollment in the study were excluded. A total of 105 subjects were enrolled and dosed across 32 international sites (19 in the USA and 13 in Europe), and cohorts were treated with different doses of LAI (70, 140, 280 and 560 mg) once daily for 28 days compared with placebo. Mucoid PsA infection rates were >80% at screening, and chronic TIS use was somewhat less common in the European versus USA subjects (19 and 35%, respectively). The European CF subjects were also younger than those enrolled in the USA (median age of 16.5 vs 30.5 years for the US subjects), but with similar lung function (median FEV1% predicted of the European subjects was 65.7% [±20%, SD] vs 65.3% [±19%, SD] for the USA subjects). Due to the large number of sites enrolling a relatively small number of subjects, and the disparity in age of subjects enrolled from Europe versus the USA, the potential for bias and/or confounders cannot be excluded.
Patients treated with 280 mg (n = 21) or 560 mg of LAI (n = 36) had significant improvements in lung function relative to the placebo controls (n = 36) after 28 days of treatment (p < 0.05 for both dose cohorts). In the 560 mg dose group, FEV1 improvements at Day 28 compared with placebo were 0.081 l ± 0.161 versus 0.011 l ± 0.101; p = 0.033. Consistent with a prolonged and efficacious depot effect of study drug, a prolonged treatment effect (relative difference in FEV1 of 12.5%) was observed 1 month after discontinuing LAI compared with the placebo controls (day 56, p < 0.01). This was accompanied by a modest increase in systemic exposure and renal clearance, further supporting prolonged deposition of LAI in the lung following nebulized delivery. There was a significant improvement in patient-reported respiratory symptoms associated with LAI treatment; 67% of subjects in the high-dose LAI group (560 mg) reported improved respiratory symptoms compared with 36% of subjects treated with placebo (p < 0.01). These improvements in respiratory symptoms correlated with FEV1 improvements at the end of treatment (Day 28, p < 0.001). There was a rapid and sustained reduction in PsA sputum density of 1–2 logs in the higher dose groups, including mucoid PsA strains of enrolled subjects.
Safety & tolerability
In the Phase II study of LAI, the LAI-treated cohorts demonstrated a similar safety and AE profile relative to controls over the course of the study, and AEs were consistent with underlying CF lung disease. There were no clear dose-dependent effects of LAI on a variety of pulmonary AEs (e.g., cough, dyspnea, dysphonia, pulmonary congestion, wheezing, hemoptysis) in the placebo-controlled trial. The median sputum concentration of amikacin prior to and at 1 h after dosing on days 1, 14 and 28 indicated that the airway clearance of amikacin was consistent over the 28-day dosing cycle.
During an open-label extension study completed in Europe (n = 49 subjects), LAI 560 mg administered once daily for 28 days followed by 56 days off study drug for six cycles (18 months total extension) was well tolerated, with an AE profile similar to that observed during the placebo-controlled study [76]. A total of 48:49 subjects reported an AE during the open label extension, with approximately one third experiencing an SAE (none were categorized as related to study drug). Of the 351 AEs reported, 33 were felt to be possibly or probably related to study drug Importantly, FEV1 at the end of the sixth treatment cycle (end of treatment and 56 days off of study drug) remained above baseline values. This prolonged off-drug period has not typically been examined in CF studies of inhaled antibiotics, but it is provocative to consider whether prolonged off-treatment periods would translate into improved drug safety and/or efficacy. Together, the results of this study provided the necessary support for pivotal Phase III trials of LAI in CF patients chronically infected with PsA. Preliminary results from a randomized open-label Phase III study of once daily LAI compared with twice daily TIS have been reported. Key enrollment criteria included age >6 years, FEV1 >25% predicted, confirmed chronic PsA infection and no acute use of antibiotics for 28 days prior to initiating study drug. The preliminary results of this 6-month, three-cycle trial (28 day on/off LAI vs TIS), suggest that LAI is noninferior to TIS to maintain lung function in CF patients chronically infected with PsA [77]. In data presented in abstract and poster form, 371 patients were screened and 302 were randomized to LAI (n = 148) or TIS (146) arms. Safety data indicated that 84.5% of LAI and 78.8% of TIS subjects reported treatment emergent AEs (TEAEs). A total of 38.5% were categorized as drug related in the LAI group compared with 14.4% in the TIS group. A total of 95% of TEAEs were graded as mild or moderate in the two groups, and 10.1% of subjects in the LAI arm experienced an AE that led to discontinuation of study drug compared with 4.8% in the TIS group. The relative change in FEV1 percent predicted from study baseline ranged from 4 to 8% (end of cycle) across the three cycles for both study groups. Both LAI and TIS reduced PsA sputum concentrations by 1.0–1.5 logs (end of cycle) across the three cycles, and trends in improved patient-reported respiratory symptoms in the LAI group were observed. Interim analysis from a subsequent rollover study reported that cycled LAI treatment was associated with sustained improvement in pulmonary function and a favorable tolerability profile with exposure up to 1 year [78]. Table 2 details the characteristics comparing LAI to other inhaled antibiotics for treatment of chronic PsA in CF patients.
Table 2. . Comparison of dosing characteristics of liposomal amikacin for inhalation with other inhaled antibiotic drugs for treatment of Pseudomonas aeruginosa in cystic fibrosis.
| Antibiotic | Drug class | Dosing and delivery |
|---|---|---|
| Liposomal amikacin for inhalation (LAI; Arikayce™) |
Aminoglycoside |
560 mg QD by nebulization with eFlow® |
| Tobramycin inhalation solution (TIS) |
Aminoglycoside |
300 mg/5 ml BID by nebulization with PARI LC Plus nebulizer |
| Tobramycin inhalation powder (TIP) |
Aminoglycoside |
112 mg BID by dry-powder inhaler with T-326 device |
| Nebulized concentrated tobramycin (NCT; T100; Vantobra) |
Aminoglycoside |
170 mg/1.7 ml BID by nebulization with TOLERO operated with eBase controller |
| Aztreonam solution for inhalation (AZLI; Cayston®) |
Beta-lactam |
75 mg TID by nebulization with eFlow |
| Colistimethate sodium (Colobreathe™) |
Polymixin |
1,662,500 IU inhalation powder, hard capsules. One capsule inhaled BID |
| Fosfomycin/tobramycin for inhalation (FTI) |
Phosphoenolpyruvate synthetase inhibitor |
80 mg/20 mg TID by nebulization with eFlow |
| Levofloxacin (Aeroquin™) | Fluoroquinolone | 240 mg BID by nebulization with eFlow |
BID: Twice daily; QD: Once a day; TID: Three-times daily; IU: International units.
Regulatory affairs
Of note, the studies described in this section were performed predominantly in Europe, but may provide support for approval of LAI use in relevant countries. The Phase III trial achieved the primary end point of a noninferiority margin of 5% for LAI compared with TIS. A follow-up open label study in patients who completed the Phase III randomized trial is currently ongoing [79]. In addition, LAI has been granted Orphan Drug status in the US and Europe for Pseudomonas lung infections in CF patients [80].
Conclusion
There is a remarkable amount of active clinical research focused on developing new inhaled antibiotics to manage PsA lung infections in patients with CF. Several tobramycin-based preparations are currently available, including various liquid and dry powder formulations that have potential product-specific advantages as discussed in this review. In addition, AZLI has been approved for approximately five years in the USA and Europe, and is an established option to manage chronic PsA infection in CF. Colistimethate dry power has recently gained regulatory approval in Europe and is another convenient option. Efforts to develop quinolone- and tobramycin/fosfomycin-based inhaled antibiotics are ongoing, with antimicrobial targets that may extend beyond PsA. Compared with drugs that are approved or in development, LAI is a unique potential therapy for use in patients chronically infected with PsA for a number of reasons. First, it has a tolerable safety profile coupled with clear evidence of efficacy across a number of end points in Phase II clinical trials (including lung function, sputum bacterial density and patient-reported outcomes). Second, it has been developed with an interest in reducing the treatment burden in CF patients, including the use of a rapid-delivery nebulizer and a once-daily dosing regimen. It is well known that adherence to CF-care regimens is suboptimal, and the establishment of chronic PsA infection typically adds another level of complexity to a CF patients’ short-term and long-term care plans. Furthermore, sputum microbiology is an important aspect of care that is negatively influenced by factors such as poor adherence to prescribed antimicrobials. Thus, reducing the time and frequency of drug delivery offers the potential to optimize inhaled antibiotic adherence by simplifying chronic treatment regimens.
The fundamental difference between LAI and all other inhaled antibiotics that are approved or in development is the incorporation of liposomal technology. This novel concept also has several potential advantages, including shielding of aminoglycoside from charged sputum, delivery of antibiotic cargo deep into CF sputum, release of active drug at the site of desired action, potential activity in complex CF lung infections (e.g., nontuberculous mycobacteria) and prolonged deposition in the lung. This final attribute raises the potential for prolonged off-treatment regimens.
The availability of different inhaled-antibiotic options raises a number of questions for CF care providers, including how to best match patients to optimal treatment choices, whether to incorporate multiple inhaled antibiotics into individualized patient-treatment plans and whether continuously cycled or cotherapies are appropriate for select patients. Addressing these questions will be a significant challenge in the coming years, but will be facilitated tremendously by robust patient registries coupled with comparative effectiveness research across the CF community.
EXECUTIVE SUMMARY.
Mechanisms of action
The active ingredient, amkacin, is an aminoglycoside antibiotic. It acts by irreversibly binding to the 30S subunit of bacterial ribosomes; blocking a recognition step in protein synthesis and therefore causing growth inhibition.
Arikayce™ is a sustained-release lipid formulation of amikacin for inhalation.
Charge neutral biocompatible liposomes (˜0.3 µm) packed with amikacin enable penetration of the drug into biofilm and release of drug at the site of bacteria.
Pharmacokinetic properties
High lung Cmax, AUC and half- life results in an improved AUC:MIC ratio.
PsA virulence factors can facilitate release of amikacin from Arikayce™ lipsome.
Primarily excreted by the kidneys.
Clinical efficacy
Prolonged deposition in the lung following treatment course resulting in maintained effect on lung function when off treatment.
Improvement in respiratory symptoms, forced expiratory volume in 1 s and Pseudomonas aeruginosa burden.
Safety & tolerability
Side-effect profile consistent with underlying cystic fibrosis lung disease and similar to placebo.
Dysphonia observed with higher doses of drug.
Drug interactions
Drug interactions for the inhaled formulation of amikacin have not been reported.
Dosage & administration
Five hundered and ninety milligrams once daily delivered by eFlow® Nebulizer System.
Acknowledgements
The authors thank J. Denise Wetzel, medical writer, division of pulmonary medicine, CCHMC, for reviewing the manuscript.
Footnotes
Financial & competing interests disclosure
JP Clancy has received contracted research support from Vertex, N30, Gilead, Insmed and Bayer, has participated in educational programs with Vertex and Genentech, and has received grant support from Gilead, CFF and the NIH. Grants: 5R01HL116226–02 MR predictors of infection, inflammation, and structural lung damage in CF and R457-CR11 Cystic Fibrosis Foundation Research Development Program. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
• of interest – as pivotal clinical trials that support the use of approved inhaled antibiotics and inhaled antibiotics in development; •• of considerable interest – as pivotal studies for the use of liposomal amikacin to treat chronic PsA infections in CF patients
- 1.Ramsey BW, Dorkin HL, Eisenberg JD, et al. Efficacy of aerosolized tobramycin in patients with cystic fibrosis. N. Engl. J. Med. 1993;328(24):1740–1746. doi: 10.1056/NEJM199306173282403. [DOI] [PubMed] [Google Scholar]
- 2.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N. Engl. J. Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 3.Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139(6):1480–1490. doi: 10.1378/chest.10-2077. [DOI] [PubMed] [Google Scholar]
- 4.Touw DJ, Jacobs FA, Brimicombe RW, Heijerman HG, Bakker W, Briemer DD. Pharmacokinetics of aerosolized tobramycin in adult patients with cystic fibrosis. Antimicrob. Agents Chemother. 1997;41(1):184–187. doi: 10.1128/aac.41.1.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinkamp G, Tummler B, Gappa M, et al. Long-term tobramycin aerosol therapy in cystic fibrosis. Pediatr. Pulmon. 1989;6(2):91–98. doi: 10.1002/ppul.1950060207. [DOI] [PubMed] [Google Scholar]
- 6.Mogayzel PJ, Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann. Am. Thorac. Soc. 2014;11(10):1640–1650. doi: 10.1513/AnnalsATS.201404-166OC. [DOI] [PubMed] [Google Scholar]
- 7.Tiddens HA, Bos AC, Mouton JW, Devadason S, Janssens HM. Inhaled antibiotics: dry or wet? Eur. Respir. J. 2014;44(5):1308–1318. doi: 10.1183/09031936.00090314. [DOI] [PubMed] [Google Scholar]
- 8.Doring G, Flume P, Heijerman H, Elborn JS Consensus Study G. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J. Cystic Fibrosis. 2012;11(6):461–479. doi: 10.1016/j.jcf.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Cystic Fibrosis Foundation Patient Registry Annual Data Report. 2015 (9 June 2015) 2013.
- 10.Ratjen F, Brockhaus F, Angyalosi G. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: a review. J. Cystic Fibrosis. 2009;8(6):361–369. doi: 10.1016/j.jcf.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Schloss PD, Kalikin LM, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc. Natl Acad. Sci. USA. 2012;109(15):5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288(5469):1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 13.Kenna DT, Doherty CJ, Foweraker J, Macaskill L, Barcus VA, Govan JR. Hypermutability in environmental Pseudomonas aeruginosa and in populations causing pulmonary infection in individuals with cystic fibrosis. Microbiology. 2007;153(Pt 6):1852–1859. doi: 10.1099/mic.0.2006/005082-0. [DOI] [PubMed] [Google Scholar]
- 14.Ciofu O, Mandsberg LF, Wang H, Hoiby N. Phenotypes selected during chronic lung infection in cystic fibrosis patients: implications for the treatment of Pseudomonas aeruginosa biofilm infections. FEMS Immunol. Med. Microbiol. 2012;65(2):215–225. doi: 10.1111/j.1574-695X.2012.00983.x. [DOI] [PubMed] [Google Scholar]
- 15.Drenkard E, Ausubel FM. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature. 2002;416(6882):740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 16.Hogardt M, Heesemann J. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr. Topics Microbiol. Immunol. 2013;358:91–118. doi: 10.1007/82_2011_199. [DOI] [PubMed] [Google Scholar]
- 17.Sousa AM, Pereira MO. Pseudomonas aeruginosa diversification during Infection development in cystic fibrosis lungs – a review. Pathogens. 2014;3(3):680–703. doi: 10.3390/pathogens3030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pressler T, Bohmova C, Conway S, et al. Chronic Pseudomonas aeruginosa infection definition: EuroCareCF Working Group report. J. Cystic Fibrosis. 2011;10(Suppl. 2):S75–S78. doi: 10.1016/S1569-1993(11)60011-8. [DOI] [PubMed] [Google Scholar]
- 19.Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J. Cystic Fibrosis. 2003;2(1):29–34. doi: 10.1016/S1569-1993(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 20.Schaedel C, De Monestrol I, Hjelte L, et al. Predictors of deterioration of lung function in cystic fibrosis. Pediatr. Pulmon. 2002;33(6):483–491. doi: 10.1002/ppul.10100. [DOI] [PubMed] [Google Scholar]
- 21.Flume PA, Mogayzel PJ, Jr, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009;180(9):802–808. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 22.Kraynack NC, Gothard MD, Falletta LM, McBride JT. Approach to treating cystic fibrosis pulmonary exacerbations varies widely across US CF care centers. Pediatr. Pulmon. 2011;46(9):870–881. doi: 10.1002/ppul.21442. [DOI] [PubMed] [Google Scholar]
- 23.Regelmann WE, Schechter MS, Wagener JS, et al. Pulmonary exacerbations in cystic fibrosis: young children with characteristic signs and symptoms. Pediatr. Pulmon. 2013;48(7):649–657. doi: 10.1002/ppul.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabin HR, Butler SM, Wohl ME, et al. Pulmonary exacerbations in cystic fibrosis. Pediatr. Pulmon. 2004;37(5):400–406. doi: 10.1002/ppul.20023. [DOI] [PubMed] [Google Scholar]
- 25.Bilton D, Canny G, Conway S, et al. Pulmonary exacerbation: towards a definition for use in clinical trials. Report from the EuroCareCF Working Group on outcome parameters in clinical trials. J. Cystic Fibrosis. 2011;10(Suppl. 2):S79–S81. doi: 10.1016/S1569-1993(11)60012-X. [DOI] [PubMed] [Google Scholar]
- 26.Maiz L, Giron RM, Olveira C, et al. Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: systematic review of randomised controlled trials. Expert Opin. Pharmacother. 2013;14(9):1135–1149. doi: 10.1517/14656566.2013.790366. [DOI] [PubMed] [Google Scholar]
- 27.Sanders DB, Hoffman LR, Emerson J, et al. Return of FEV1 after pulmonary exacerbation in children with cystic fibrosis. Pediatr. Pulmon. 2010;45(2):127–134. doi: 10.1002/ppul.21117. [DOI] [PubMed] [Google Scholar]
- 28.Lipuma JJ. Microbiological and immunologic considerations with aerosolized drug delivery. Chest. 2001;120(3 Suppl.):118S–123S. doi: 10.1378/chest.120.3_suppl.118s. [DOI] [PubMed] [Google Scholar]
- 29.Flume PA, Vandevanter DR. Clinical applications of pulmonary delivery of antibiotics. Adv. Drug Deliv. Rev. 2015;85:1–6. doi: 10.1016/j.addr.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011;9:32. doi: 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treggiari MM, Retsch-Bogart G, Mayer-Hamblett N, et al. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch. Pediatr. Adolesc. Med. 2011;165(9):847–856. doi: 10.1001/archpediatrics.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study comparing cycled versus culture based treatment of PsA.
- 32.Esther CR, Jr, Henry MM, Molina PL, Leigh MW. Nontuberculous mycobacterial infection in young children with cystic fibrosis. Pediatr. Pulmon. 2005;40(1):39–44. doi: 10.1002/ppul.20222. [DOI] [PubMed] [Google Scholar]
- 33.Hoiby N, Frederiksen B, Pressler T. Eradication of early Pseudomonas aeruginosa infection. J. Cystic Fibrosis. 2005;4(Suppl. 2):49–54. doi: 10.1016/j.jcf.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 34.Taccetti G, Bianchini E, Cariani L, et al. Early antibiotic treatment for Pseudomonas aeruginosa eradication in patients with cystic fibrosis: a randomised multicentre study comparing two different protocols. Thorax. 2012;67(10):853–859. doi: 10.1136/thoraxjnl-2011-200832. [DOI] [PubMed] [Google Scholar]
- 35.Lee TW. Eradication of early Pseudomonas infection in cystic fibrosis. Chron. Respir. Dis. 2009;6(2):99–107. doi: 10.1177/1479972309104661. [DOI] [PubMed] [Google Scholar]
- 36.Brodt AM, Stovold E, Zhang L. Inhaled antibiotics for stable non-cystic fibrosis bronchiectasis: a systematic review. Eur. Respir. J. 2014;44(2):382–393. doi: 10.1183/09031936.00018414. [DOI] [PubMed] [Google Scholar]
- 37.Ratjen F, Munck A, Kho P, Angyalosi G, Group ES. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax. 2010;65(4):286–291. doi: 10.1136/thx.2009.121657. [DOI] [PubMed] [Google Scholar]; • Pivotal study of TIS assessing optimal duration of early treatment of PsA.
- 38.Rosenfeld M, Emerson J, Mcnamara S, et al. Risk factors for age at initial Pseudomonas acquisition in the cystic fibrosis epic observational cohort. J. Cystic Fibrosis. 2012;11(5):446–453. doi: 10.1016/j.jcf.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer RD, Young LS, Armstrong D. Tobramycin (nebramycin factor 6): in vitro activity against Pseudomonas aeruginosa . Appl. Microbiol. 1971;22(6):1147–1151. doi: 10.1128/am.22.6.1147-1151.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N. Engl. J. Med. 1999;340(1):23–30. doi: 10.1056/NEJM199901073400104. [DOI] [PubMed] [Google Scholar]; • Pivotal study of TIS leading to regulatory approval.
- 41.Gilligan PH. Microbiology of airway disease in patients with cystic-fibrosis. Clin. Microbiol. Rev. 1991;4(1):35–51. doi: 10.1128/cmr.4.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konstan MW, Flume PA, Kappler M, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: the EAGER trial. J. Cystic Fibrosis. 2011;10(1):54–61. doi: 10.1016/j.jcf.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study of TIP supporting regulatory approval.
- 43.Mazurek H, Chiron R, Kucerova T, et al. Long-term efficacy and safety of aerosolized tobramycin 300 mg/4 ml in cystic fibrosis. Pediatr. Pulmon. 2014;49(11):1076–1089. doi: 10.1002/ppul.22989. [DOI] [PubMed] [Google Scholar]
- 44.Chiesi Farmaceutici s.PA. Product portfolio. 2015. 2014. http://www.chiesi.uk.com/product_portfolio/
- 45.Chiesi USA Inc. Bethkis? (tobramycin inhalation solution): Concentrated formulation for efficient nebulization. 2015 (Web page accessed April 6, 2015), (2015) 6 April 2015.
- 46.Geller DE, Pitlick WH, Nardella PA, Tracewell WG, Ramsey BW. Pharmacokinetics and bioavailability of aerosolized tobramycin in cystic fibrosis. Chest. 2002;122(1):219–226. doi: 10.1378/chest.122.1.219. [DOI] [PubMed] [Google Scholar]
- 47.Smith AL, Ramsey BW, Hedges DL, et al. Safety of aerosol tobramycin administration for 3 months to patients with cystic fibrosis. Pediatr. Pulmon. 1989;7(4):265–271. doi: 10.1002/ppul.1950070413. [DOI] [PubMed] [Google Scholar]
- 48.Ryan G, Singh M, Dwan K. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2011;3:CD001021. doi: 10.1002/14651858.CD001021.pub2. [DOI] [PubMed] [Google Scholar]; • Systematic review of inhaled antibiotics in CF.
- 49.Sands D, Sapiejka E, Gaszczyk G, Mazurek H, Group TS. Comparison of two tobramycin nebuliser solutions: pharmacokinetic, efficacy and safety profiles of T100 and TNS. J. Cystic Fibrosis. 2014;13(6):653–660. doi: 10.1016/j.jcf.2014.04.006. [DOI] [PubMed] [Google Scholar]; • Pivotal study of T100 comparing it to TNS.
- 50.(Chmp) CFMPFHU. European Medicine Agency Assessment report: Vantobra. 2015.
- 51.Mccoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2008;178(9):921–928. doi: 10.1164/rccm.200712-1804OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study of AZLI supporting regulatory approval.
- 52.Retsch-Bogart GZ, Quittner AL, Gibson RL, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest. 2009;135(5):1223–1232. doi: 10.1378/chest.08-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study of AZLI supporting regulatory approval.
- 53.Oermann CM, Retsch-Bogart GZ, Quittner AL, et al. An 18 month study of the safety and efficacy of repeated courses of inhaled aztreonam lysine in cystic fibrosis. Pediatr. Pulmon. 2010;45(11):1121–1134. doi: 10.1002/ppul.21301. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study of AZLI supporting regulatory approval.
- 54.Assael BM, Pressler T, Bilton D, et al. Inhaled aztreonam lysine vs. inhaled tobramycin in cystic fibrosis: A comparative efficacy trial. J. Cystic Fibrosis. 2012 doi: 10.1016/j.jcf.2012.07.006. [DOI] [PubMed] [Google Scholar]; • Study demonstrating superiority of AZLI over TNS.
- 55.Yahav D, Farbman L, Leibovici L, Paul M. Colistin: new lessons on an old antibiotic. Clin. Microbiol. Infect. 2012;18(1):18–29. doi: 10.1111/j.1469-0691.2011.03734.x. [DOI] [PubMed] [Google Scholar]
- 56.Nation RL, Li J. Colistin in the 21st century. Curr. Opin. Infect. Dis. 2009;22(6):535–543. doi: 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schuster A, Haliburn C, Doring G, Goldman MH Freedom Study G. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study. Thorax. 2013;68(4):344–350. doi: 10.1136/thoraxjnl-2012-202059. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Pivotal study of colistimethate sodium supporting European regulatory approval.
- 58.Littlewood KJ, Higashi K, Jansen JP, et al. A network meta-analysis of the efficacy of inhaled antibiotics for chronic Pseudomonas infections in cystic fibrosis. J. Cystic Fibrosis. 2012;11(5):419–426. doi: 10.1016/j.jcf.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 59.Trapnell BC, Mccolley SA, Kissner DG, et al. Fosfomycin/tobramycin for inhalation in patients with cystic fibrosis with Pseudomonas airway infection. Am. J. Respir. Crit. Care Med. 2012;185(2):171–178. doi: 10.1164/rccm.201105-0924OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Phase II study of FTI in CF patients.
- 60.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. The mechanism of action of fosfomycin (phosphonomycin) Ann. NY Acad. Sci. 1974;235(0):364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 61.Dasenbrook EC, Checkley W, Merlo CA, Konstan MW, Lechtzin N, Boyle MP. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303(23):2386–2392. doi: 10.1001/jama.2010.791. [DOI] [PubMed] [Google Scholar]
- 62.Forest Pharmaceuticals I. Forest laboratories: Product website. 2015 (Date accessed October 26, 2015), (2014) 2014.
- 63.Geller DE, Flume PA, Griffith DC, et al. Pharmacokinetics and safety of MP-376 (levofloxacin inhalation solution) in cystic fibrosis subjects. Antimicrob. Agent. Chemother. 2011;55(6):2636–2640. doi: 10.1128/AAC.01744-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geller DE, Flume PA, Staab D, et al. Levofloxacin inhalation solution (MP-376) in patients with cystic fibrosis with Pseudomonas aeruginosa . Am. J. Respir. Crit. Care Med. 2011;183(11):1510–1516. doi: 10.1164/rccm.201008-1293OC. [DOI] [PubMed] [Google Scholar]; • Phase II study of levofloxacin inhalation solution in CF patients.
- 65.Aptalis Pharma I. Trial of Aeroquin Versus Tobramycin Inhalation Solution (TIS) in Cystic Fibrosis (CF) PatientsAptalis Pharma, Inc. Announces Results of Phase 3 Studies of Aeroquin™ (Levofloxacin Solution for Inhalation) Among Patients With Cystic Fibrosis and Chronic Lung Infection. 2013. NCT01270347 Study Mpex 209 and 207.
- 66.Young DC, Zobell JT, Stockmann C, et al. Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: V. Aminoglycosides. Pediatr. Pulmon. 2013 doi: 10.1002/ppul.22813. [DOI] [PubMed] [Google Scholar]
- 67.Okusanya OO, Bhavnani SM, Hammel J, et al. Pharmacokinetic and pharmacodynamic evaluation of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infection. Antimicrob. Agent. Chemother. 2009;53(9):3847–3854. doi: 10.1128/AAC.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Phase Ib/II studies of LAI in CF patients.
- 68.Meers P, Neville M, Malinin V, et al. Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. J. Antimicrob. Chemother. 2008;61(4):859–868. doi: 10.1093/jac/dkn059. [DOI] [PubMed] [Google Scholar]; •• Demonstrated that LAI liposomes penetrate CF sputum and locally release amikacin driven by PsA products.
- 69.Weers J, Metzheiser B, Taylor G, Warren S, Meers P, Perkins WR. A gamma scintigraphy study to investigate lung deposition and clearance of inhaled amikacin-loaded liposomes in healthy male volunteers. J. Aerosol Med. Pulmon. Drug Deliv. 2009;22(2):131–138. doi: 10.1089/jamp.2008.0693. [DOI] [PubMed] [Google Scholar]; •• Demonstrated the prolonged deposition of nebulized LAI in the lungs of normal subjects.
- 70.Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst. Rev. 2014;11:CD004197. doi: 10.1002/14651858.CD004197.pub4. [DOI] [PubMed] [Google Scholar]
- 71.Li Z, Zhang Y, Wurtz W, et al. Characterization of nebulized liposomal amikacin (Arikace) as a function of droplet size. J. Aerosol Med. Pulmon. Drug Deliv. 2008;21(3):245–254. doi: 10.1089/jamp.2008.0686. [DOI] [PubMed] [Google Scholar]; •• Examined aerosolization characteristics of LAI.
- 72.Mckeage K. Tobramycin inhalation powder: a review of its use in the treatment of chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Drugs. 2013;73(16):1815–1827. doi: 10.1007/s40265-013-0141-0. [DOI] [PubMed] [Google Scholar]
- 73.Stanojevic S, Waters V, Mathew JL, Taylor L, Ratjen F. Effectiveness of inhaled tobramycin in eradicating Pseudomonas aeruginosa in children with cystic fibrosis. J. Cystic Fibrosis. 2014;13(2):172–178. doi: 10.1016/j.jcf.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 74.Doring G, Conway SP, Heijerman HG, et al. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur. Respir. J. 2000;16(4):749–767. doi: 10.1034/j.1399-3003.2000.16d30.x. [DOI] [PubMed] [Google Scholar]
- 75.Waters V, Ratjen F. Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis. Cochrane Database Syst. Rev. 2012;12:CD010004. doi: 10.1002/14651858.CD010004.pub2. [DOI] [PubMed] [Google Scholar]
- 76.Clancy JP, Dupont L, Konstan MW, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax. 2013;68(9):818–825. doi: 10.1136/thoraxjnl-2012-202230. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Summarized the entire Phase II program for LAI development to treat chronic PsA infection in CF patients.
- 77.Bilton D, Pressler T, Fajac I, et al. 2013 North American Cystic Fibrosis Conference (NACFC) 2013. Phase 3 efficacy and safety data from randomized, multicenterstudy of liposomal amikacin for inhalation (Arikace®) compared with Tobi® incystic fibrosis patients with chronic infection due to Pseudomonas aeruginosa . Presented at. [Google Scholar]; •• Abstract reporting results of Phase III study comparing LAI with TOBI.
- 78.Bilton D, Pressler T, Fajac I, et al. 2014 North American Cystic Fibrosis Conference. Atlanta, GA, USA: 9–11 October 2014. Analysis of long-term liposomal amikacin for inhalation in patients with cysticfibrosis and chronic infection from Pseudomonas aeruginosa . Poster presented at. [Google Scholar]; • Abstract reporting results of study on long-term efficacy, safety, and tolerability of LAI.
- 79.Acquire Media online news brief. Arikace meets primary endpoint of non-inferiority to Tobi in Phase 3 clinical trial in Europe and Canada to treat Pseudomonas aeruginosa in cystic fibrosis patients (2013) 2013. 6 July 2015.; • New brief reporting results of Phase III clinical trial of LAI achieving noninferiority to TOBI for change in lung function from baseline.
- 80.US Food and Drug Administration. Products Designating an orphan product: Drugs and biological products. 2015. http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation