

Abstract
Background
In myotonic dystrophy type 1 (DM1), only one FDG-PET study used statistical parametric mapping (SPM) showing frontal reduced FDG-uptake. Our aim was to 1) identify the FDG-PET area with the most severe reduced FDG-uptake using SPM8 in a larger group of patients 2) assess potential correlation between CTG-numbers and FDG-PET.
Methods
FDG-PET was performed in 24 patients and compared to 24 controls. Pearson’s correlation was used to analyse correlation.
Results
SPM8 revealed Brodmann area 8 as the area with the most severe reduced FDG-uptake. Weak, although not statistically significant, correlation was observed between CTG-numbers and reduced FDG-uptake in Brodmann area 8.
Conclusion
In DM1, Brodmann area 8 is the area with the most severe reduced FDG-uptake on FDG-PET. Brodmann area 8 reduced FDG-uptake is correlated –although weakly- to CTG-repeat numbers.
Background
Myotonic dystrophy type 1 (DM1) is caused by an expanded (CTG)n repeat within the noncoding 3’ untranslated region of the myotonic dystrophy protein kinase gene. Cerebral involvement, based on clinical, neuroimaging, and neuropathological evidence, is frequent in DM1 [1–10].
Relatively few reports analysed 18 F-deoxy-glucose positron emission tomography (FDG-PET) in DM1 [1–4]. Only one large study used statistical parametric mapping (SPM), showing bilateral frontotemporal reduced FDG-uptake in the 17 studied patients [1]. In a subset of those 17 patients, a T1-weighted MRI-based partial volume correction was applied. In these patients, bilateral frontal reduced FDG-uptake was observed indicating that frontal reduced FDG-uptake is probably an independent (i.e. not associated with focal brain atrophy) phenomenon in DM1. In their study, no significant correlation was found between FDG-PET metabolism and neuropsychological test results. Correlation between the number of CTG repeats and cerebral metabolism was not analysed directly in that study. One FDG-PET study, using a kinetic modelling to determine the local glucose consumption rate, found that cerebral glucose reduced FDG-uptake was a triplet-size dependent phenomenon. This method, however, requires dynamic PET acquisitions and patient’s blood sampling after 18 F-deoxyglucose injection.
Our aim was to analyse FDG-PET by SPM in a larger group of DM1 patients in order to identify the area with the most severe reduced FDG-uptake and to assess potential correlation between CTG numbers and FDG-uptake. Our study focused on FDG-PET and its relation to CTG-repeats. We did not search for a relationship of these parameters with clinical brain involvement (e.g. cognitive deficit, hypersomnia).
Methods
Patients
In our centre, 48 symptomatic and genetically proven adult DM1 patients are in follow-up. Twenty four of these patients accepted to undergo brain FDG-PET. Ethical approval was not needed for our study, since the ethics committee of CHU Nîmes (France) considered the FDG-PET scan as a standard procedure in DM1, if informed consent was given by the patients. All participants gave their informed consent to publish indirect identifiers such as age and gender. All patients gave their informed consent. Patient’s characteristics are summarized in Table 1. Characteristics of included patients were: men/women ratio = 9/15, mean age of 47 years (SD 12.5, range 26–74) at time of brain imaging, mean age of symptom onset of 27 years (range 5–58, including 6 patients with symptom onset before 18 years of age [range 5–14] and 18 patients with adult onset disease), mean disease duration of 20 years (range 6–41), and mean number of 799 CTG repeats (range 83–2000, including 14 patients with CTG <1000 and 10 patients with CTG of ≥1000). None of our patients had congenital DM1. Six out of 24 DM1 patients had associated diabetes (including five patients treated with oral antidiabetic drugs only and one [patient nr 23] with insulin treatment).
Table 1.
Patient characteristics
| Patient Number | Sex | Age symptom onset | Age at FDG-PET | CTG expansion | Diabetes (glucose level) |
|---|---|---|---|---|---|
| 1 | F | 58 (late onset) | 64 | 83 | No (<120 mg/dl) |
| 2 | M | 35 (late onset) | 74 | 100 | No (<120 mg/dl) |
| 3 | F | 45 (late onset) | 64 | 150 | Yes (120–160 mg/dl) |
| 4 | F | 32 (late onset) | 46 | 176 | No (<120 mg/dl) |
| 5 | F | 25 (late onset) | 41 | 230 | No (<120 mg/dl) |
| 6 | F | 30 (late onset) | 49 | 330 | No (<120 mg/dl) |
| 7 | M | 26 (late onset) | 36 | 400 | No (<120 mg/dl) |
| 8 | M | 30 (late onset) | 50 | 400 | Yes (120–160 mg/dl) |
| 9 | F | 19 (late onset) | 26 | 400 | No (<120 mg/dl) |
| 10 | F | 30 (late onset) | 49 | 400 | No (<120 mg/dl) |
| 11 | M | 40 (late onset) | 58 | 430 | No (<120 mg/dl) |
| 12 | F | 14 (early onset) | 34 | 660 | No (<120 mg/dl) |
| 13 | F | 13 (early onset) | 27 | 660 | No (<120 mg/dl) |
| 14 | F | 14 (early onset) | 31 | 670 | No (<120 mg/dl) |
| 15 | F | 28 (late onset) | 54 | 850 | Yes (120–160 mg/dl) |
| 16 | F | 39 (late onset) | 47 | 1000 | No (<120 mg/dl) |
| 17 | M | 40 (late onset) | 51 | 1168 | Yes (<120 mg/dl) |
| 18 | M | 24 (late onset) | 44 | 1300 | No (<120 mg/dl)c |
| 19 | F | 28 (late onset) | 50 | 1400 | No (<120 mg/dl) |
| 20 | M | 29 (late onset) | 46 | 1500 | No (<120 mg/dl) |
| 21 | F | 38 (late onset) | 58 | 1560 | No (<120 mg/dl) |
| 22 | M | 5 (early onset) | 41 | 1600 | No (<120 mg/dl) |
| 23 | M | 14 (early onset) | 55 | 1700 | Yes (<120 mg/dl) |
| 24 | F | 6 (early onset) | 26 | 2000 | Yes (<120 mg/dl) |
Analysis was done for the total of 24 DM1 patients versus 24 controls. For SPM analysis, the control group consisted of 24 subjects (including 10 women and 14 men; mean age 52, SD 10.5, range 32–70) who had neither a history of neurological and psychiatric illness nor abnormalities on neurological examination. FDG-PET scans in controls were performed for study reasons only.
FDG-PET
All brain FDG-PET scans were done on a PET-CT GEMINI GXL (Philips Medical Systems). After fasting for at least 6 h, blood glucose level was checked and less than 160 mg/dl. The accepted glucose level of 160 mg/dl in our study was relatively high (in contrast with the more commonly used cutoff of 120 mg/dl) because of frequently associated diabetes in DM1 patients. Mean glucose level in our DM1 patients was 103 mg/dl (range 80–159, of whom only 3 patients [i.e. patient nr 3, 8, and 15] had blood glucose levels between 120 and 160 mg/dl). Patients were positioned comfortably in a quiet, dimly lit room before FDG administration and during the uptake phase of FDG (at least 20 min). They received intravenous injection of 185 to 250 MBq (5 to 6.7 mCi: according to the weight) of 18F-FDG by a canula inserted 10 min before. They were instructed not to speak, read or be otherwise active. For imaging, patients were in supine position and their head immobilized in a masthead. Imaging started with a CT surview (view angle 90, Kv 120, 30 mAs), then transmission CT scan for attenuation correction was done (120 KV, Mas/slice 200, Pitch 0.563, Rotation 1.5, thickness 3 mm, filter UB, collimation 16 × 1.5, FOV 600); static emission scan started 30 min after injection, in 3-D mode for 20 min, axial field of view 180 mm, 256 × 256 matrix, voxel size 2 mm3. PET raw data were corrected for attenuation, random effects and scatter, the reconstruction was then done with a three dimensional row-action maximum likehood algorithm LOR-RAMLA resulted in 90 transaxial slices.
Statistical analysis were done using SPM8 software (Wellcome Department of Imaging Neuroscience, London; http://www.fil.ion.ucl.ac.uk/spm) running on Matlab R2012b version (The Mathworks Inc., USA) running on a Windows XP. Images were realigned and spatially normalized into the MNI (Montreal Neurological Institute, McGill University, Montreal, QC, Canada) standard template given by SPM software using affine and non-linear transformation. Images were reformatted to a final voxel size of 2 × 2 × 2 mm and smoothed using an isotropic Gaussian kernel of 12 × 12 × 12 mm FWHM. A global normalization for voxel count was performed using a proportional scaling. Statistical comparisons between groups were performed on a voxel-by-voxel basis t statistics, using "two-sample t-test" design of the SPM8 generating SPM (t) maps. We investigated brain areas with reduced FDG-uptake, a stringent level of significance of p < 0.05 corrected for multiple comparisons (SPM family wise error—FWE) was adopted with an extent threshold of 100 voxels. For visualization the significant voxels were projected onto the 3D rendered brain or a standard MRI template allowing anatomic identification. A MNI to Talairach coordinates conversion was done using Yale Bioimage Suite software (http://www.bioimagesuite.org). On subsequent analyses by lowering p-values successively, we searched for the brain area with the most severe reduced FDG-uptake.
Statistical analyses
Pearson product–moment correlation coefficient was used to analyse potential correlation between the number of CTG repeats and reduced FDG-uptake (by using Z-scores of individual patients). We also compared (using Student’s t-test) uptake between the group of patients with CTG <1000 versus CTG ≥1000 and between the group of patients with early, childhood (age <18) onset versus late, adult (age >18) onset.
The 24 controls were part of a larger control group of 35 cases (age range 32–86). Only the youngest 24 controls were selected in order to avoid possible age-related bias. Difference in age between the 24 DM1 patients and the 24 controls were analyzed by a 2-sample t-test.
In all analyses, when comparing (sub)groups, a threshold of p < 0.05 was used to differentiate significant from non-significant differences.
Results
The age difference between DM1 patients (mean age 47) and controls (mean age 52) was statistically not significant.
SPM8 analysis of cerebral FDG-PET metabolism in our 24 DM1 patients revealed significant bilateral symmetrical reduced FDG-uptake in the lateral part of the frontal lobes (Fig. 1). The area with the most severe reduced FDG-uptake was Brodmann area 8, associated with slight involvement of Brodmann area 9 on the right side. Deep grey matter structures did not show reduced metabolism. When using an extent threshold of 50 voxels, FDG-PET showed no supplementary areas with reduced FDG-uptake compared to when using an extent threshold of 100 voxels.
Fig. 1.
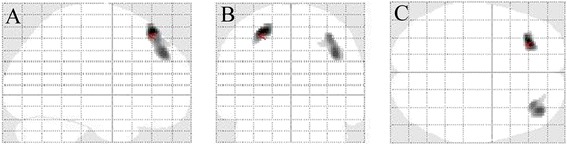
Compared with controls, SPM analysis (a, sagittal view; b, coronal view, and c, axial view) of our 24 DM1 patients showing bilateral reduced FDG-uptake in the lateral part of the frontal lobes, with Brodmann area 8 as the zone with the most severe reduced FDG-uptake
The number of CTG repeats was not correlated with FDG-PET metabolism neither for the entire brain nor for the frontal lobes (considered as a whole). Positive -but statistically not significant- correlation was found between the CTG numbers and the metabolism in Brodmann area 8 (especially for the right side, rho = −0.36; rho = −0.22 for the left side).
When comparing patients with <1000 CTG and ≥1000 CTG, patients with ≥1000 CTG showed lower Z-scores in the Brodmann area 8 (median −0.69 for the right and −0.75 for the left Brodmann area 8) than patients with <1000 CTG (median −0.46 for the right and −0.58 for the left Brodmann area 8) although not statistically significant (p = 0.28 and p = 0.52 for the right and the left side respectively).
When comparing patient with early (childhood) and late (adult) onset DM1, early onset patients showed lower Z-scores (median −0.81 for the right and −0.78 for the left Brodmann area 8) than patients with late onset (median −0.53 for the right and −0.72 for the left Brodmann area 8), without statistically significance (p = 0.085 and p = 0.5 for the right and the left side respectively).
SPM analyses did not show significant differences between diabetic (n = 6) and non-diabetic (n = 18) DM1 patients or between patients with blood glucose levels <120 mg/dl (n = 21) and patients with blood glucose levels between 120 and 160 mg/dl (n = 3).
Discussion
To the best of our knowledge, this is the largest study analysing FDG-PET by SPM in DM1. We found lateral frontal reduced FDG-uptake as predominant FDG-PET pattern, confirming the data of the only existing study using SPM. The zone with reduced FDG-uptake in our study (although not partial volume corrected) was more restricted than the reported area in the earlier study (using partial volume correction). This might be related to the higher number of patients analyzed and/or due the lack of partial volume correction in our study. The area with the most severe reduced FDG-uptake in our study was identified as Brodmann area 8, associated with slight involvement of the right Brodmann area 9. Although statistically not significant, reduced FDG-uptake was more severe in these brain areas in patients with early, childhood (opposed to late, adult) onset and in patients with ≥1000 (opposed to <1000) CTG.
CTG numbers typically differ between different tissues (e.g. between blood and brain tissue). Therefore, the CTG repeat number obtained by blood sample (like in our study) does not necessarily reflect the number of CTG repeats in the brain (and the brain abnormalities found on FDG-PET possibly related to these repeat numbers). The absence of weak relationship between these blood CTG repeat numbers and (clinical or radiological) brain abnormalities in the earlier reports and our study might be explained in part by this phenomenon.
Interestingly, the area with the most severe reduced FDG-uptake in our DM1 patients was Brodmann area 8, an area involved in (or at least very near to zones implicated in) eye movement control (Brodmann area 8A corresponding to the frontal eye field and area 8B corresponding to the premotor ear-eye field, although the exact localisation of these fields are still under debate). A multitude of oculomotor abnormalities have been reported in DM1 [11–13]. Some of these oculomotor abnormalities are thought to be due to oculomotor muscle dysfunction (weakness and/or myotonia) whereas other abnormalities suggest brain dysfunction. Studies are needed to analyse the relationship between oculomotor deficits (especially those related to dysfunction of the frontal eye field and the premotor ear-eye field) and FDG-PET metabolism.
Conclusion
In DM1, Brodmann area 8 is the area with the most severe reduced FDG-uptake on FDG-PET. Brodmann area 8 reduced FDG-uptake is weakly correlated to CTG-repeat numbers.
Abbreviations
DM1, myotonic dystrophy type 1; FDG-PET, 18 F-deoxy-glucose positron emission tomography; SPM, statistical parametric mapping
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Raw data were stored in the secured encrypted central server of the department of Neurology, CHU Nîmes, France. However, because of legal and privacy reasons and due to internal hospital rules, data (other than given in the paper) are not shared.
Authors’ contributions
All authors met the following contributions: made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; been involved in drafting the manuscript or revising it critically for important intellectual content; given final approval of the version to be published. Each author should have participated sufficiently in the work to take public responsibility for appropriate portions of the content; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Authors’ information
Not applicable.
Consent for publication
All participants gave their informed consent to publish indirect identifiers such as age and gender.
Ethics approval and consent to participate
Ethical approval was not needed for our study, since the ethics committee of CHU Nîmes (France) considered the FDG-PET scan as a standard procedure in DM1, if informed consent was given by the patients. All participants gave their informed consent to publish indirect identifiers such as age and gender. All patients gave their informed consent.
Contributor Information
Dimitri Renard, Email: dimitrirenard@hotmail.com.
Laurent Collombier, Email: laurent.collombier@chu-nimes.fr.
Christel Castelli, Email: christel.castelli@chu-nimes.fr.
Jean-Pierre Pouget, Email: jean.pierre.pouget@chu-nimes.fr.
Pierre-Olivier Kotzki, Email: pierre.olivier.kotzki@chu-nimes.fr.
Vincent Boudousq, Email: vincent.boudousq@chu-nimes.fr.
References
- 1.Weber YG, Roebling R, Kassabek J, Hoffmann S, Rosenbohm A, Wolf M, et al. Comparitive analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology. 2010;74:1108–17. doi: 10.1212/WNL.0b013e3181d8c35f. [DOI] [PubMed] [Google Scholar]
- 2.Romeo V, Pegoraro E, Squarzanti F, Sorarù G, Ferrati C, Ermani M, et al. Retrospective study on PET-SPECT imaging in a large cohort of myotonic dystrophy type 1 patients. Neurol Sci. 2010;31:757–63. doi: 10.1007/s10072-010-0406-2. [DOI] [PubMed] [Google Scholar]
- 3.Annane D, Fiorelli M, Mazoyer B, Pappata S, Eymard B, Radvanyi H, et al. Impaired cerebral glucose metabolism in myotonic dystrophy: a triplet-size dependent phenomenon. Neuromuscul Disord. 1998;8:39–45. doi: 10.1016/S0960-8966(97)00144-2. [DOI] [PubMed] [Google Scholar]
- 4.Mielke R, Herholz K, Fink G, Ritter D, Heiss WD. Positron emission tomography in myotonic dystrophy. Pschiatry Res. 1993;50:93–9. doi: 10.1016/0925-4927(93)90014-9. [DOI] [PubMed] [Google Scholar]
- 5.Romeo V, Pegoraro E, Ferrati C, Squarzanti F, Sorarù G, Palmieri A, et al. Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol. 2010;257:1246–55. doi: 10.1007/s00415-010-5498-3. [DOI] [PubMed] [Google Scholar]
- 6.Di Costanzo A, Di Salle F, Santoro L, Tessitore A, Bonavita V, Tedeschi G. Pattern and significance of white matter abnormalities in myotonic dystrophy type 1: an MRI study. J Neurol. 2002;249:1175–82. doi: 10.1007/s00415-002-0796-z. [DOI] [PubMed] [Google Scholar]
- 7.Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain. 2011;134:3530–46. doi: 10.1093/brain/awr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve. 2007;36:294–306. doi: 10.1002/mus.20800. [DOI] [PubMed] [Google Scholar]
- 9.Franc DT, Muetzel RL, Robinson PR, Rodriguez CP, Dalton JC, Naughton CE, et al. Cerebral and muscle MRI abnormalities in myotonic dystrophy. Neuromuscul Disord. 2012;22:483–91. doi: 10.1016/j.nmd.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang L, Anderson T, Migneco OA, Boone K, Mehringer CM, Villanueva-Meyer J, et al. Cerebral abnormalities in myotonic dystrophy. Cerebral blood flow, magnetic resonance imaging, and neuropsychological tests. Arch Neurol. 1993;50:917–23. doi: 10.1001/archneur.1993.00540090024006. [DOI] [PubMed] [Google Scholar]
- 11.Anastasopoulos D, Kimmig H, Mergner T, Psilas K. Abnormalities of ocular motility in myotonic dystrophy. Brain. 1996;119:1923–32. doi: 10.1093/brain/119.6.1923. [DOI] [PubMed] [Google Scholar]
- 12.Versino M, Rossi B, Beltrami G, Sandrini G, Cosi V. Ocular motor myotonic phenomenon in myotonic dystrophy. J Neurol Neurosurg Psychiatry. 2002;72:236–40. doi: 10.1136/jnnp.72.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaunak S, Orrell R, Henderson L, Kennard C. Saccades and smooth pursuit in myotonic dystrophy. J Neurol. 1999;246:600–6. doi: 10.1007/s004150050411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data were stored in the secured encrypted central server of the department of Neurology, CHU Nîmes, France. However, because of legal and privacy reasons and due to internal hospital rules, data (other than given in the paper) are not shared.