

Abstract
Pheochromocytoma is a catecholamine secreting tumor that originate from chromaffin cells. Usually, it is solid neoplasm of the adrenal medulla, however cystic pheochromocytoma is a rare neuro-endocrine tumour that is frequently asymptomatic and often diagnosed incidentally on imaging or intra-operatively. Only a few cases of cystic pheochromocytomas have been reported in the world literature. We present a case of giant cystic pheochromocytoma in a 65 years old lady who presented with a large retroperitoneal lump, which is probably the world's third largest pheochromocytoma as per the available indexed literature.
Keywords: Cystic, giant pheochromocytoma, immunohistochemistry
INTRODUCTION
First described in 1886 by Fränkel,[1] pheochromocytomas are tumors of pronounced clinical importance because of the catecholamines they secrete. Its incidence is 0.1% in the general population.[2] Most pheochromocytomas are benign, and it is almost impossible to distinguish a benign from a malignant tumor only by its histological criteria. Many attempts have been made to find markers that would predict the future behaviour of a nonmetastatic pheochromocytoma.[3] Usually, it is a solid neoplasm of the adrenal medulla. However cystic pheochromocytoma is a rare neuroendocrine tumor that is often asymptomatic and diagnosed incidentally on imaging or intraoperatively.[4] Only a few cases of cystic pheochromocytomas have been reported in the world literature. We present a case of giant cystic pheochromocytoma in a 65-year-old female who presented with a large retroperitoneal lump, which is the world's third largest pheochromocytoma and turned out to be a nonfunctional tumor with the silent presentation as per the available indexed literature, and that forms the basis for present communication.
CASE REPORT
A 65-year-old female presented to the surgical clinic with complaints of left upper abdominal lump which she noticed 6 months ago and it progressively increased in that duration. She had no history of pain, hypertension, headache, palpitation, and excessive sweating. On examination, her pulse was 76/min and blood pressure was 126/84 mmHg with a large non-tender lump predominantly in the left upper abdomen. Laboratory investigations were normal except hemoglobin of 9.6 g%. Computed tomography (CT) revealed a large retroperitoneal mass which was predominantly multi-loculated cystic lesion with punctate calcifications [Figure 1]. In the radiological imaging however, the organ of origin could not be identified.
Figure 1.
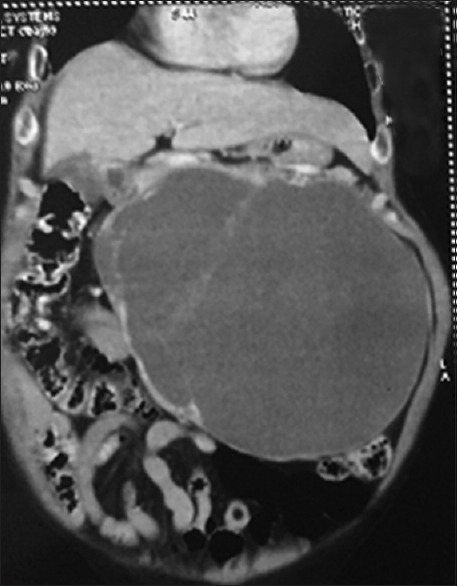
Coronal section of contrast enhanced CT scan showing multi-loculated cystic retroperitoneal mass lesion
The pancreas, spleen, and kidneys were normal. There was no note on CT scan about adrenal or lymph nodes. Because of the thereotical risk of tumor seedling in fine needle aspiration cytology track preoperative fine needle aspiration or biopsy was not performed. The patient was taken up for exploratory laparotomy. Intraoperatively a giant cystic mass was found arising from the left adrenal gland and was densely adherent to left renal pedicle and aorta [Figure 2]. Manipulation of the mass resulted in extreme blood pressure fluctuations. The patient underwent removal of this adrenal tumor with left nephrectomy. The resected specimen of tumor measured 25 cm × 17 cm × 15 cm, weighing 2750 g containing areas of hemorrhage and cysts of varying sizes.
Figure 2.
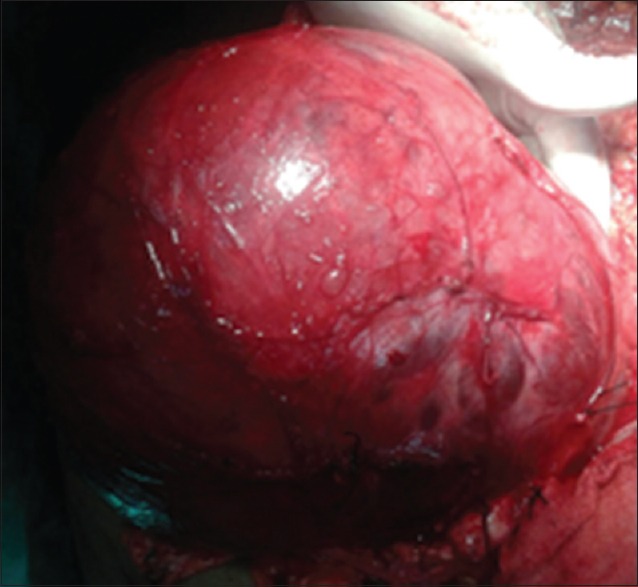
Intra-operative photograph of the giant cystic mass arising from left adrenal gland
On histopathological examination grossly it was a cystic mass densely attached to the hilum of the left kidney. Kidney measured 11 cm × 5 cm × 4 cm and appeared unremarkable. The external surface of the tumor was bosselated, smooth and grey-brown in color. On sectioning cyst was multi-loculated and filled with a thick brownish fluid. Multiple cysts varying in size from 0.5 to 20 cm in diameter were present. Wall thickness varied from 0.2 to 1 cm and appeared grey tan in color. Inner surface of cysts was smooth to granular. Sections stained with hematoxylin and eosin stain from various areas revealed a tumor enclosed within a fibrous capsule. The tumor cells were round to polygonal arranged in nests bound by delicate fibrovascular stroma forming a zellballen. The cells showed mild to moderate variation in size and had finely granular amphophilic cytoplasm. A single round to oval nucleus was present with salt and pepper chromatin. Occasional bizarre cells were also present. Mitotic activity was inconspicuous [Figure 3a]. A diagnosis of pheochromocytoma was made. Immunohistochemistry was performed for confirmation. The tumor cells were reactive of neuron-specific enolase [Figure 3b], synaptophysin and S-100 confirming the diagnosis of pheochromocytoma.
Figure 3.
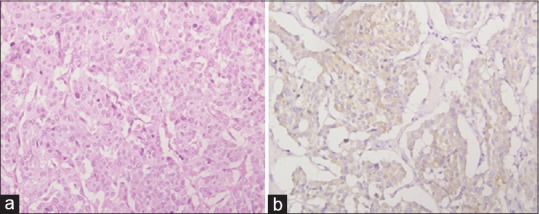
(a) Histopathological section showing round to polygonal cells forming a zellballen (H and E, ×400). (b) Tumor cells positive for neuron specific enolase
The patient had an uneventful recovery and was discharged on 7th postoperative day. She is doing well at 2 months of follow-up.
DISCUSSION
Pheochromocytoma is an infrequent tumor, originating from the adrenal medulla and sympathoadrenal neuroendocrine system chromaffin cells. They produce and secrete catecholamines; the triad of headache, sweating, and palpitations in patients with hypertension is diagnostic, with a 94% specificity and 91% sensitivity. Of all the adrenal pheochromocytoma, 20–30% of them are asymptomatic; they are called clinically silent pheochromocytoma.[5] They are bilateral, malignant, pediatric, or asymptomatic in 10% of cases. Hereditary pheochromocytoma is associated with von Hippel–Lindau syndrome, multiple endocrine neoplasia Type 2 (2A/2B), neurofibromatosis Type 1, and hereditary pheochromocytoma-paraganglioma (due to mitochondrial succinate dehydrogenase gene mutations. Different factors contribute to the diagnosis: (1) Extensive necrosis cystic region at the centre of the mass may significantly decrease the number of cells producing catecholamine; (2) interstitial tissue without bioactivity may be the main ingredient of the tumor; considerable blood sinus around the cystic area is also demonstrated; (3) most of the catecholamines and metabolic product may be stored in the capsular mass, which infuses intravenously into the blood circulation when isolating the mass; as such, a hypertension crisis is possible.[6] Preoperative management is essential to prevent hemodynamic instability and hypertensive crisis during intraoperative or postoperative period. Once the adrenal tumor is diagnosed, surgical resection is the only curative treatment. Although some reports have concluded that laparoscopic adrenalectomy is safe, effective, and minimally invasive, it is considered a gold standard in the surgical management of small benign adrenal tumors. Essential intraoperative surgical steps include early isolation of the tumor's venous drainage with minimal manipulation of the mass followed by complete resection of the tumor. In immunohistochemistry, the tumor cells are reactive for neuron specific enolase, synaptophysin and S-100 confirming the diagnosis of pheochromocytoma. Postoperatively, patients should be followed yearly for at least 10 years, as 16% of patients develop recurrence within 10 years. There are 20 case reports of pheochromocytomas more than 10 cm in the worldwide literature.[7,8,9] On extensive search of indexed literature, it turned out to be the first largest cystic pheochromocytoma from India and third largest such tumor in the world.
CONCLUSION
Giant pheochromocytoma with prominent cystic change is a rare entity and is mostly asymptomatic. Silent pheochromocytoma is one of the exceptions that does not exhibit classic symptoms and therefore, silent pheochromocytoma often remains undiagnosed until surgical excision occurs and the anesthesia teams face a greater challenge. It should be considered in differential diagnosis of a retroperitoneal tumor of patients with nonspecific symptoms and given adequate treatment to promote the perioperative safety. Surgical resection is the only curative treatment for such tumors.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Frankel F. Classics in oncology. A case of bilateral completely latent adrenal tumor and concurrent nephritis with changes in the circulatory system and retinitis. CA Cancer J Clin 1886. 1984:93–106. doi: 10.3322/canjclin.34.2.93. [DOI] [PubMed] [Google Scholar]
- 2.Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: Current approaches and future directions. Oncologist. 2008;13:779–93. doi: 10.1634/theoncologist.2008-0043. [DOI] [PubMed] [Google Scholar]
- 3.Glodny B, Winde G, Herwig R, Meier A, Kühle C, Cromme S, et al. Clinical differences between benign and malignant pheochromocytomas. Endocr J. 2001;48:151–9. doi: 10.1507/endocrj.48.151. [DOI] [PubMed] [Google Scholar]
- 4.Reisch N, Peczkowska M, Januszewicz A, Neumann HP. Pheochromocytoma: Presentation, diagnosis and treatment. J Hypertens. 2006;24:2331–9. doi: 10.1097/01.hjh.0000251887.01885.54. [DOI] [PubMed] [Google Scholar]
- 5.Bush WH, Elder JS, Crane RE, Wales LR. Cystic pheochromocytoma. Urology. 1985;25:332–4. doi: 10.1016/0090-4295(85)90346-2. [DOI] [PubMed] [Google Scholar]
- 6.Minei S, Yamashita H, Koh H, Satoh T, Kobayashi S, Furuhata M, et al. Giant cystic pheochromocytoma: A case report. Hinyokika Kiyo. 2001;47:561–3. [PubMed] [Google Scholar]
- 7.Suga K, Motoyama K, Hara A, Kume N, Ariga M, Matsunaga N. Tc-99m MIBG imaging in a huge clinically silent pheochromocytoma with cystic degeneration and massive hemorrhage. Clin Nucl Med. 2000;25:796–800. doi: 10.1097/00003072-200010000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Basiri A, Radfar MH. Giant cystic pheochromocytoma. Urol J. 2010;7:16. [PubMed] [Google Scholar]
- 9.Pradeep PS. Pheochromocytoma: A composite resection. Indian J Surg. 2006;68:163–4. [Google Scholar]