

Dermal fibroblasts from patients harboring mutations in the gap junction connexin43 gene have distinctly different mechanistic causes for the clinical presentations of oculodentodigital dysplasia.
Abstract
Oculodentodigital dysplasia (ODDD) is a rare genetic disease that affects the development of multiple organs in the human body. More than 70 mutations in the gap junction connexin43 (Cx43) gene, GJA1, are associated with ODDD, most of which are inherited in an autosomal dominant manner. Many patients exhibit similar clinical presentations. However, there is high intrafamilial and interfamilial phenotypic variability. To better understand this variability, we established primary human dermal fibroblast cultures from several ODDD patients and unaffected controls. In the present study, we characterized three fibroblast lines expressing heterozygous p.L7V, p.G138R, and p.G143S Cx43 variants. All ODDD fibroblasts exhibited slower growth, reduced migration, and defective cell polarization, traits common to all ODDD fibroblasts studied so far. However, we found striking differences in overall expression levels, with p.L7V down-regulated at the mRNA and protein level. Although all of the Cx43 variants could traffic to the cell surface, there were stark differences in gap junction plaque formation, gap junctional intercellular communication, Cx43 phosphorylation, and hemichannel activity among Cx43 variants, as well as subtle differences in myofibroblast differentiation. Together these findings enabled us to discover mutation-specific pathologies that may help to predict future clinical outcomes.
INTRODUCTION
Mutations in the gene encoding connexin43 (Cx43), GJA1, are definitively linked to the pleiotropic disease known as oculodentodigital dysplasia (ODDD; Paznekas et al., 2003). More than 70 mostly missense mutations have been linked to ODDD (Paznekas et al., 2009; Laird, 2014), which result in several common clinical presentations: syndactyly and camptodactyly of the fourth and fifth fingers, enamel hypoplasia, microcornea and microphthalmos, and craniofacial abnormalities, including narrow, pinched noses with hypoplastic alae nasi and bone deformities (Judisch et al., 1979). However, these presentations are not completely conserved across all mutations in the GJA1 gene, since some patients present with only a few symptoms, whereas others exhibit symptoms including neurological defects (Gutmann et al., 1991; Loddenkemper et al., 2002; De Bock et al., 2013), such as conductive hearing loss, trichosis, and bladder incontinence (Loddenkemper et al., 2002; Paznekas et al., 2009). Our understanding of how these mutations lead to particular pathologies in some individuals but not in others is unclear.
Cx43 is a member of the gap junction family of proteins and is the most widely expressed connexin in the human body (Laird, 2006). Classically, connexins form hexameric oligomers called connexons that traffic to the cell surface and dock with compatible connexons on adjacent cells to form gap junction channels (Musil and Goodenough, 1990). These channels allow the passage of ions and small molecules up to 1 kDa in size directly between cells in a process termed gap junctional intercellular communication (GJIC; Alexander and Goldberg, 2003). Evidence now also suggests that connexons can function as undocked channels on the plasma membrane called hemichannels (Goodenough and Paul, 2003; D’Hondt et al., 2014). Although the functional role of hemichannels in vivo is incompletely understood, data suggest that particular connexin mutations can lead to disease via “leaky” hemichannels (Essenfelder et al., 2004; Stong et al., 2006; Mhaske et al., 2013; Berger et al., 2014; Levit and White, 2015; Shuja et al., 2016). In particular, previous reports suggested that the Cx43 G138R and G143S ODDD mutants exhibit “leaky” hemichannels when expressed in HeLa cells (Dobrowolski et al., 2007) or in cell cultures obtained from Cx43-G138R mutant mice (Dobrowolski et al., 2008).
The majority of ODDD patients inherit one copy of the mutated GJA1 gene, although recessive cases have been reported (Paznekas et al., 2009; Laird, 2014). Therefore most patients express a 1:1 ratio of wild-type Cx43 to mutated Cx43. This inheritance predicts that the Cx43 mutants might act in a dominant-negative manner on wild-type Cx43. However, it is also plausible that the wild-type allele might rescue some of the mutant Cx43 functions or that the expressed mutants might have no effect on the coexpressed wild-type Cx43. Still further, it is possible that the Cx43 mutant will exhibit a gain-of-function phenotype (Shapiro et al., 1997), as reported for several connexin-related diseases (Kelly et al., 2015b). To address these possibilities and reduce the problems linked to overexpression studies in which a 1:1 ratio of mutant to wild-type Cx43 expression is hard to achieve, we obtained and cultured human dermal fibroblasts from three ODDD patients and two unaffected controls. To the best of our knowledge, we are the only laboratory that has successfully cultured cells from ODDD patients, which allows us to “drill down” on how defined mutations exhibit specific pathological effects. To this end, we previously showed that fibroblasts expressing the D3N and V216L Cx43 mutants reduce GJIC, proliferation, and migration and may therefore negatively affect wound healing (Churko et al., 2011b). In addition, the D3N mutant expresses less Cx43 mRNA transcript, whereas both D3N and V216L trigger gene-reprogramming events for certain extracellular matrix–associated proteins (Esseltine et al., 2015). Here we characterize three novel dermal fibroblast cell lines that express L7V, G138R, and G143S amino acid substitutions in the Cx43 polypeptide chain. We find that overall Cx43 protein levels, GJIC, ATP release, cell growth, cell migration, and cell differentiation into myofibroblasts are differentially affected in mutant fibroblasts in a mutation-dependent manner.
RESULTS
Profile of ODDD
Skin biopsies were obtained from three patients exhibiting many classical ODDD symptoms (see Table 1 for symptoms). All patients gave informed consent to participate in the study, and investigations were conducted in accordance with the Declaration of Helsinki. As described previously (Churko et al., 2011b), dermal fibroblasts were cultured from punch biopsies taken from ODDD patients and unaffected controls, and the coding region of the GJA1 gene was sequenced (Figure 1A). We confirmed that patient #1 had a missense mutation (19T > G) leading to a leucine-to-valine substitution at position 7 (p.L7V) in the amino acid sequence, found within the N-terminal domain of Cx43, as reported previously (Paznekas et al., 2009). Patient #2 had a missense mutation (412G > C) leading to a glycine-to-arginine substitution at position 138 (p.G138R) within the intracellular loop of Cx43, as previously reported (Paznekas et al., 2003), and patient #3 had a missense mutation (427G > A) leading to a glycine-to-serine substitution at position 143 (p.G143S), also within the intracellular loop of Cx43 (Figure 1B), as reported here for the first time. Of interest, both G138R and G143S mutations are found within a calmodulin-binding domain (Zhou et al., 2007). All patients exhibited heterozygosity for the Cx43 mutant allele, in agreement with previous reports that ODDD is mostly inherited as an autosomal dominant disease (Laird, 2014). Clinical presentations reported for ODDD patients harboring the L7V, G138R, and G143S mutants revealed many common and classical conditions, with mutant- specific presentation variability in the teeth, eyes, bladder, skin, and hearing (Table 1).
TABLE 1:
Clinical symptoms described for ODDD patients with L7V, G138R, and G143S amino acid substitutions.
| Classical ODDD symptoms | Additional symptoms | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Amino acid change | Location in protein | Craniofacial | Eye | Teeth | Digits | Neuro/bladder | Ear | Hair/skin | Other | Reference |
| 19T>G (S) [1] | p.L7V | N-terminal domain | Characteristic facies with dysmorphic nose and hypoplastic nares Cleft palate Small chin | Microphthalmia | Syndactyly (figures) Clinodactyly | Premature Hypospadias | Paznekas et al. (2009) | ||||
| 412G>C (F) [5] | p.G138R | Intracellular loop/CaM-binding domain | Characteristic facies with dysmorphic nose and hypoplastic nares | Microcornea | Enamel hypoplasia Small | Syndactyly (figures and toes) Aplasia and hypoplasia | Neural spasticity/hyperreflexia White matter changes Urinary incontinence | Middle ear infections Conductive hearing loss | Hyperkeratosis of hands and feet | Gracile build | Paznekas et al. (2003), Shapiro et al. (1997) |
| 427G>A (F) [9] [1]* | p.G143S | Intracellular loop/CaM-binding domain | Characteristic facies with dysmorphic nose and hypoplastic nares Macrocephaly Brachycephaly | Microcornea* | Small (1 case only) Normal enamel | Syndactyly (figures) Camptodactyly Aplasia | Spasticity and hyperreflexia of lower limbs* Mild urinary incontinence* | Unilateral conductive hearing loss secondary to infection | Normal hair | Brueton et al. (1990), Richardson et al. (2004) | |
CaM, calmodulin; F, familial; S, sporadic; numeral in square brackets indicates number of patients.
Italicized symptoms are highlighted where we specifically note that these phenotypes were missing or normal.
Information adapted from Paznekas et al. (2009), unless otherwise noted.
*Reported here for the first time, from a patient in this study.
FIGURE 1:
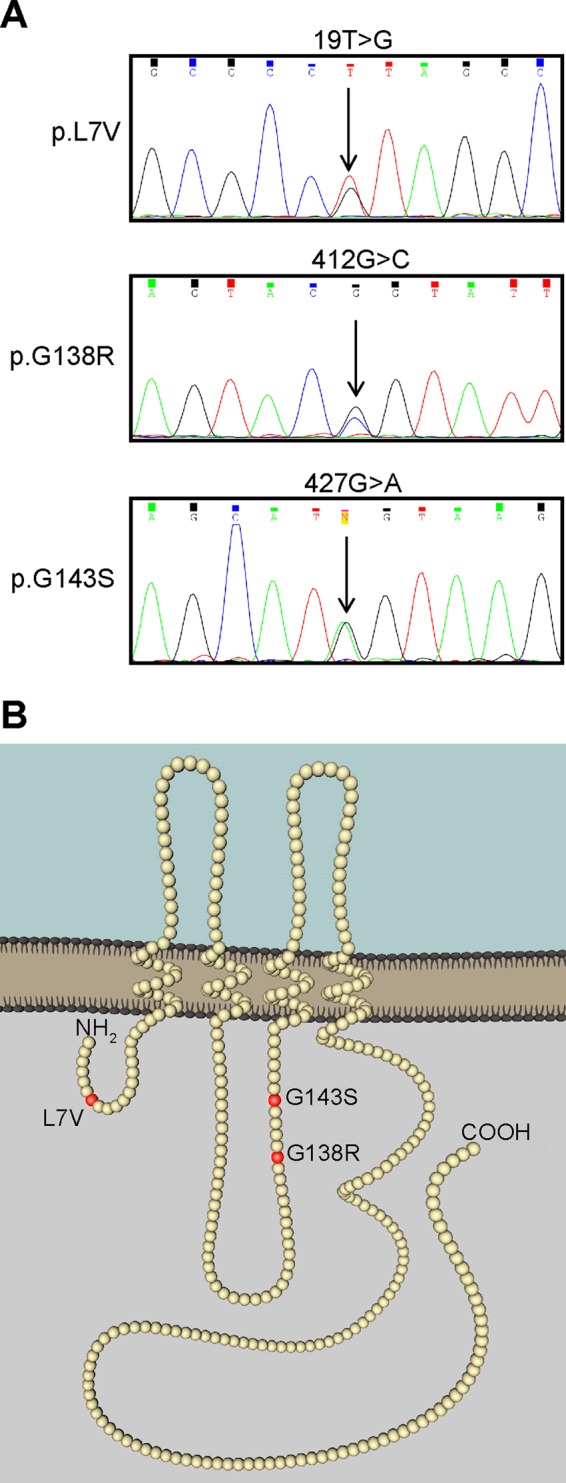
Patient genotyping and model depicting the location of Cx43 amino acid changes. (A) Chromatogram of cDNA sequencing revealed heterozygous nucleotide substitutions that translated into single–amino acid substitutions in the Cx43 protein sequence. (B) Sites of the amino acid substitutions (red balls) in the Cx43 polypeptide chain.
Cx43 levels differ among ODDD fibroblasts
Fibroblasts immunolabeled for Cx43 and counterstained for actin (to denote the cellular architecture) revealed Cx43 gap junction plaques at putative cell–cell interfaces in both control and ODDD mutant–expressing cells (Figure 2A, insets, arrows), indicating that the L7V, G138R, and G143S mutants do not inhibit Cx43 trafficking to the cell surface. Although all the mutants exhibited some degree of cell surface localization in plaque-like structures, there was a striking decrease in overall Cx43 levels in L7V fibroblasts and to a lesser extent G138R (Figure 2, B and C). All of the mutant fibroblast lines were phosphorylated to the P1 Cx43 species, although these levels were slightly reduced in the L7V- and G138R-expressing fibroblasts. In addition, the L7V and G138R fibroblasts had significantly less of the P2 phosphorylated form of Cx43 when normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Figure 2D), and all three mutant fibroblast lines had significantly reduced P2 Cx43 levels as a function of the total amount of Cx43 protein (Figure 2E). We examined whether the mRNA levels varied in the L7V, G138R and G143S fibroblasts by quantitative PCR (qPCR) and found that Cx43 mRNA transcripts were reduced in L7V fibroblasts compared with control cells (Figure 2F).
FIGURE 2:

ODDD fibroblasts form gap junction–like plaques but have variable Cx43 expression levels. (A) Dermal fibroblasts from unaffected (control [Ctrl]) and ODDD patients were immunolabeled for Cx43 (green) and counterstained for actin (red). Insets highlight the presence of gap junction–like plaques (arrows). Scale bars, 20 μm. (B) Immunoblotting for Cx43 revealed total Cx43 content as well as the various phosphorylated species (P0/1/2). GAPDH was run as a loading control. (C–E) Quantification (N = 5) of total Cx43 (C) and the P2 species (D) normalized to GAPDH and relative P2 phosphorylation as a function of total Cx43 (E). (F) Quantitative real-time PCR of Cx43 mRNA isolated from control and ODDD patient fibroblasts as normalized to 18S rRNA (N = 3).
Cx43 can traffic to the cell surface but is differentially regulated in ODDD fibroblasts
To confirm that Cx43 was trafficking to the cell surface in ODDD fibroblasts, we performed cell surface biotinylation followed by pull-down assays using NeutrAvidin beads and immunoblotting for Cx43 (Figure 3A). The amount of biotinylated Cx43 as a function of the total amount of Cx43 was significantly higher in the L7V- and G143S-expressing fibroblasts, suggesting that these cells had proportionally higher amounts of Cx43 at the cell surface (Figure 3A). This was a striking finding, given that L7V had generally less total Cx43. The amount of Cx43 in L7V lysates after pull down (cleared lysates) was significantly reduced compared with control cells, confirming that overall Cx43 levels were lower in L7V-expressing fibroblasts. In addition, the lack of accumulated Cx43 for any of the mutants in the cleared lysates indicated that there was no abnormal Cx43 buildup or retention within the cell (Figure 3A). The amount of Cx43 assembled into gap junction plaques, and possibly inaccessible to biotinylation, was not specifically measured in each fibroblast line. Because all the fibroblasts exhibited Cx43 trafficking to the cell surface, we next used high-resolution Airyscan microscopy to investigate its cell surface distribution. Of interest, we found that individual gap junction plaque lengths were significantly smaller in cells expressing L7V and G138R mutants than with Cx43 in control cells (Figure 3B). Finally, we investigated the Cx43 banding profile in Triton X-100–soluble or –insoluble fibroblast lysates. Consistent with earlier results, overall L7V levels were reduced and the G138R-expressing fibroblasts had reduced P2 but increased P0 species in the insoluble fraction (Figure 3C). In contrast, the G143S profile exhibited more pronounced P2 phosphorylation in the Triton-X–insoluble fraction, indicating the presence of mature Cx43 gap junction plaques at the cell surface.
FIGURE 3:
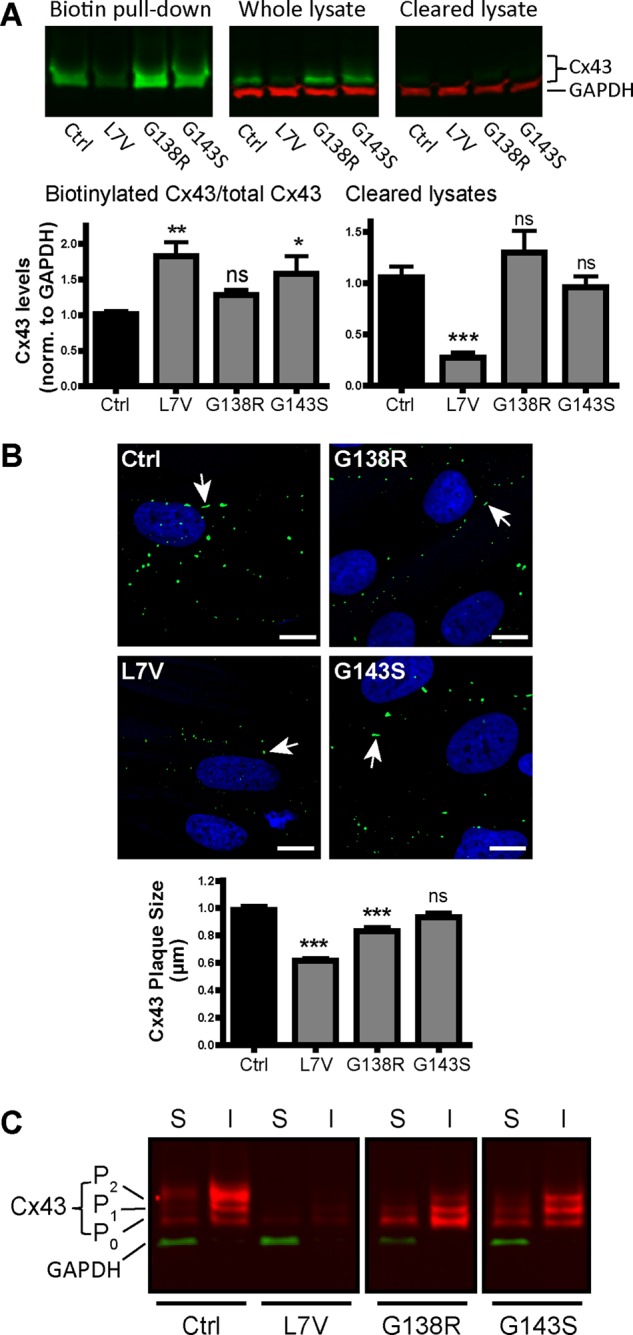
Analysis of cell surface Cx43 using biotinylation pull down, high-resolution microscopy, and Triton X-100 solubility. (A) Representative Western blots of control and patient fibroblasts immunolabeled for Cx43 (green) and GAPDH (red). Biotinylated cell surface Cx43 precipitated with NeutrAvidin beads (Biotin pull down), total Cx43 in the input lysate (Whole lysate) and Cx43 after biotin pull down (Cleared lysate). Graphs represent quantification of biotinylated (cell surface) Cx43 as a fraction of total Cx43 and Cx43 found in cleared lysates (N = 3). (B) High-resolution microscopy of Cx43 immunofluorescence labeling in control and mutant fibroblasts and quantification of average plaque size (N = 3; Ctrl, n = 953; L7V, n = 473; G138R, n = 406; G143S, n = 530). Arrows highlight representative Cx43 gap junction plaques. Scale bars, 10 μm. (C) Representative Western blot of Cx43 (red) in Triton X-100–soluble (S) and –insoluble (I) fractions. GAPDH (green) as a control is present only in soluble fractions.
GJIC varies among ODDD fibroblasts
Because all of the fibroblasts exhibited some level of cell surface localization of Cx43, we next examined whether they could form functional intercellular gap junction channels. To do this, we microinjected fully confluent fibroblasts with a Cx43-permeable dye (Alexa Fluor 350) and quantified the extent of dye spread. Of interest, the G143S-expressing fibroblasts exhibited a higher order of dye transfer than the L7V- and G138R-expressing fibroblasts (Figure 4, A and B). Dual whole-cell patch-clamp experiments were then performed to determine the coupling status and junctional conductance between contacting cell pairs. These data confirmed that the G143S-expressing fibroblasts were surprisingly able to form functional gap junctions, with an average junctional conductance Gj ≈ 10 nS, similar to ∼8 nS for control cells (Figure 4C). In contrast, L7V and G138R Cx43–expressing fibroblasts exhibited negligible (<0.1 nS) junctional conductance, suggesting that these mutants act in a strong dominant-negative manner on coexpressed endogenous Cx43 to inhibit overall gap junction channel function (Figure 4C).
FIGURE 4:

Patient fibroblasts exhibit variable gap junction function. (A) Representative images of Alexa Fluor 350 transfer between cells after microinjection. Red asterisk, injected cell. Scale bars, 20 μm. (B) Incidence and degree of dye transfer, categorized into the percentage of cells exhibiting no transfer or first- or second-order transfer. (C) Gj as measured by dual whole-cell patch clamp. n, number of injections from three independent experiments (B) or number of cell pairs recorded (C).
L7V fibroblasts exhibit increased ATP release
We next sought to determine whether hemichannel regulation was affected in ODDD fibroblasts. We used a bioluminescent ATP release assay to quantify the amount of ATP released from fibroblasts bathed in normal extracellular solution (ECS), where the presence of extracellular calcium and magnesium normally keep hemichannels in a closed conformation, or in a divalent cation–free solution (DCFS) previously shown to open Cx43 hemichannels (Tong et al., 2007). Surprisingly, the L7V- but not G138R- or G143S-expressing fibroblasts released significantly more ATP into the ECS than did control cells (Figure 5A) despite L7V cells expressing ∼80% less Cx43 protein. In DCFS, there was no significant difference in ATP release between the mutant and control fibroblasts (Figure 5, A and B), suggesting that the hemichannels are functional despite the dominant-negative effects by L7V and G138R on GJIC. The increase in ATP release after DCFS treatment was significantly reduced in L7V-expressing fibroblasts compared with control cells but only as a result of the higher-than-normal basal (ECS) ATP levels (Figure 5B). Of interest, when treated with DCFS in the presence of the hemichannel blocker carbenoxolone (CBX; 100 μM), the released ATP levels remained high for L7V, whereas those from control and G138R- and G143S-expressing fibroblasts were reduced to ECS baseline levels (Figure 5, A and C). There was no fold change in ATP release for L7V fibroblasts after CBX treatment, in contrast to the 25–50% decrease in ATP release from control and G138R- and G143S-expressing fibroblasts (Figure 5C). These data suggest fibroblasts expressing the L7V mutant may exhibit “leaky” characteristics that that are insensitive to CBX.
FIGURE 5:
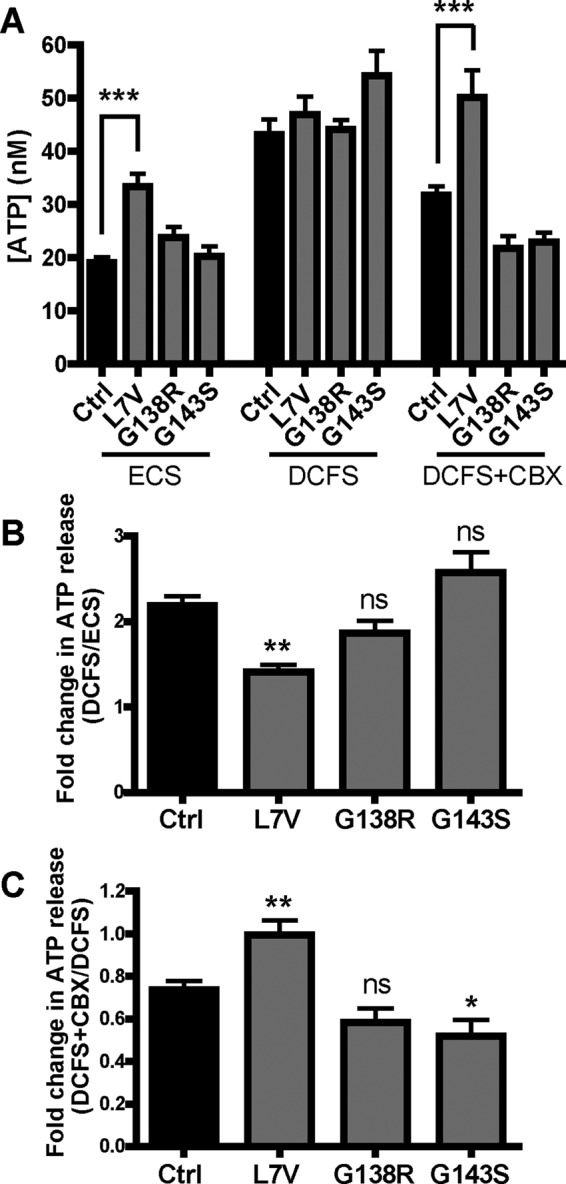
ATP release is dysregulated in L7V-expressing fibroblasts. (A) ATP concentrations in the extracellular medium were measured in response to ECS, DCFS, and DCFS supplemented with 100 μM CBX from three independent experiments (n = 7–18). (B) Fold change in ATP release upon hemichannel opening in DCFS as a fold increase over baseline (ECS). (C) Fold change in ATP release upon hemichannel blockage with CBX compared with DCFS.
Cell growth, migration, and polarization are dysregulated in ODDD mutant fibroblasts
Processes such as proliferation and migration are highly correlated with Cx43 expression. Abnormal expression or function of Cx43 has therefore been suggested to influence dermal fibroblast response to injury (Churko et al., 2011b; Francis et al., 2011; Mendoza-Naranjo et al., 2012). We investigated these properties in control and ODDD fibroblasts and found that all three ODDD cell lines exhibited significantly reduced growth rates compared with control (Figure 6A). In addition, migration through a transwell insert toward a platelet-derived growth factor (PDGF) chemoattractant was modestly but consistently reduced for all ODDD cells (Figure 6B). An important step for directional migration is for cells to polarize their Golgi apparatus toward their leading edge (Magdalena et al., 2003). We investigated this for ODDD fibroblasts in an in vitro scratch wound assay and found that all three cell lines had reduced ability to correctly polarize their Golgi (stained with GM130) toward the leading edge (Figure 6C). These data confirm that Cx43 plays an important role in cell growth, migration, and polarization, which are impaired in ODDD fibroblasts.
FIGURE 6:

ODDD patient fibroblasts exhibit reduced growth, less migration, and dysregulated polarization. (A) Cell counts performed at days 3, 6, and 9 after seeding (N = 3, n = 6–12). (B) Transwell migration shown as a percentage of cells migrating through an 8-μm porous membrane in response to no serum, 20% FBS, or 100 ng/ml PDGF-BB in the lower chamber (N = 3, n = 12–24) (C) Scratch-wound assay of confluent fibroblasts stained for the Golgi marker GM130 (green) and the nuclear stain Hoescht (blue). Representative control (Ctrl) and G143S images. Filled arrowheads represent cells with correctly aligned Golgi apparatus; open arrowheads represent defective Golgi alignment. The ratio of leading-edge cells (denoted above the dashed white line) with a correctly aligned Golgi apparatus was quantified (three experiments, 14–35 images). Scale bar, 20 μm.
ODDD fibroblasts exhibit altered responses to wound-healing cytokines
One of the key roles of dermal fibroblasts is to form and maintain the connective tissue layer of the skin by secreting and remodeling extracellular matrix proteins, especially during wound healing. Thus we investigated the effect of the wound-healing cytokines, including PDGF-BB and transforming growth factor-β1 (TGF-β), on ODDD fibroblasts with respect to Cx43 expression and myofibroblast differentiation. First, PDGF and TGF-β induced ∼1.2- to 2.8-fold increases in Cx43 levels compared with untreated cells (Figure 7, A and B), which was not significantly different between control and ODDD fibroblasts (Figure 7B). However, the G138R- and G143S-expressing fibroblasts, but not those expressing L7V, had a significantly lower fold increase of the P2 Cx43 species after TGF-β treatment than did control cells (Figure 7C). Despite fold increases in Cx43 and Cx43 P2 species after TGF-β treatment, however, the actual amount of P2 Cx43 protein in mutant fibroblasts compared with control cells remained significantly low (Figure 7D), suggesting that the fully mature P2 form of Cx43 is not rescued.
FIGURE 7:

Effect of PDGF and TGF-β on Cx43 and α-SMA expression. (A) Representative Western blot analysis of lysates obtained from fibroblasts treated with 50 ng/ml PDGF-BB or 5 ng/ml TGF-β for 3 d. Membranes were stained for Cx43, α-SMA, and GAPDH as a loading control. (B) Total Cx43 fold change in response to PDGF and TGF-β, with 1.0 being the untreated baseline for each genotype. (C) P2 Cx43 fold change from untreated cells, with 1 being the untreated baseline. (D) Total P2 Cx43 levels normalized to GAPDH after TGF-β treatment. (E) Quantification of α-SMA levels, normalized to GAPDH, in fibroblasts before and after TGF-β treatment. Here a and b denote significant differences between untreated ODDD and Ctrl fibroblasts; for b, p < 0.05. The letters y and z denote significant differences between Ctrl and ODDD fibroblasts treated with TGF-β, where, for z, p < 0.05. (F) Fold increase in α-SMA after TGF-β treatment. N = 3 for all cases.
α-Smooth muscle actin (α-SMA) is a marker for fibroblast-to-myofibroblast differentiation during wound healing and is up-regulated in response to TGF-β. Our data showed that the control, L7V, and G143S fibroblasts exhibited a significant increase in α-SMA levels upon treatment with TGF-β, whereas G138R fibroblasts responded less well (Figure 7, A and E). Both the L7V and G143S but not the G138R cells had significantly higher basal levels of α-SMA than did control, whereas the G138R-expressing fibroblasts had lower levels of α-SMA after TGF-β treatment (Figure 7E). However, all three ODDD fibroblast lines had significantly reduced fold changes in α-SMA after TGF-β treatment compared with control (Figure 7F). Of interest, although the L7V and G143S mutants had higher basal levels of α-SMA, this did not translate into an increase in stress fiber formation, suggesting the α-SMA was mostly cytosolic (Figure 8). On treatment with TGF-β, the G138R-expressing fibroblasts appeared to form α-SMA–rich stress fibers in distinct populations of cells, whereas this was more homogeneous in the other fibroblast lines (Figure 8). These data suggest that regulation of myofibroblast differentiation, which is tightly correlated with α-SMA up-regulation, may be affected in ODDD-expressing fibroblasts.
FIGURE 8:

Myofibroblast differentiation is inconsistent in G138R mutant–expressing fibroblasts. Fibroblasts untreated or treated with TGF-β (5 ng/ml) for 3 d were fixed and immunolabeled for nonmuscle myosin IIA (red) and α-SMA (green). Asterisks for G138R indicate fibroblasts devoid of α-SMA stress fibers. Scale bar, 20 μm.
DISCUSSION
Since the discovery that ODDD is definitively linked to mutations in the gene encoding Cx43, >70 mostly autosomal dominant mutations have been reported (Paznekas et al., 2009). Remarkably, ODDD patients rarely present with identical clinical symptoms. Whereas some Cx43-rich organs are routinely spared from disease or abnormalities (e.g., heart; Molica et al., 2014), other organs are moderately or severely affected (e.g., bone). Still further, some ODDD patients have anomalies involving eight or more tissues/organs, whereas others may have only four affected tissues/organs. Attempts to correlate specific Cx43 gene mutations to cellular phenotypes, and ultimately to clinical presentations, have been limited to mutant overexpression and characterization in reference cell cultures or to three genetically modified mouse models of ODDD (Flenniken et al., 2005; Kalcheva et al., 2007; Dobrowolski et al., 2008; Stewart et al., 2013; Huang et al., 2014). Although mouse models of ODDD overcome the limitations of obtaining the appropriate 1:1 ratio of wild-type to mutant protein expected of an autosomal dominant disease, which is difficult to obtain in overexpression systems, it still does not fully mimic the human condition. A deeper understanding of the complex mechanisms that cause specific pathologies requires the use of models that most closely mimic or are genetically identical to the human condition. In the past few years, we have been fortunate to obtain dermal fibroblasts from a variety of ODDD patients and unaffected controls to assess the effect of specific Cx43 gene mutations on channel function and cellular phenotypes. Here, using these novel human models of ODDD, we revealed three distinct causal mechanisms. First, in cells harboring the L7V mutant, Cx43 was poorly expressed and phosphorylated, and upon trafficking to the cell surface, it resulted in greater ATP release, possibly as a result of dysregulated hemichannels, with limited capacity to make functional gap junction channels. Second, in cells containing the G138R mutant, Cx43 was normally expressed but found at lower and less phosphorylated steady-state levels at the cell surface, with reduced gap junction size and reduced ability for GJIC. Finally, cells harboring the G143S mutant were the least affected mutant, expressing normal levels of Cx43 that were only slightly underphosphorylated, resulting in GJIC being identical to controls. Even though the functional status of Cx43 was highly variable among the mutant fibroblasts, all cells grew more slowly, responded less well to the chemotaxis reagent PDGF, exhibited impaired polarization, and exhibited somewhat similar responses to TGF-β.
As reported elsewhere (Brueton et al., 1990; Paznekas et al., 2003, 2009; Shuja et al., 2016), the clinical features associated with patients expressing L7V, G138R, and G143S Cx43 mutants have some common ground (Table 1). For example, all three patients exhibit characteristic facies that includes thin and narrow noses, underdeveloped/thin nostrils, and/or prominent bridges in addition to syndactyly of the fourth and fifth digits of the hands. Both the L7V and G138R patients have small eye/cornea defects, whereas microcornea was found in only one patient (in this study) from 10 reported with the G143S variant. The G138R and G143S patients have reported middle ear infections, resulting in conductive hearing loss. Surprisingly, only patients expressing the G138R mutant exhibit enamel hypoplasia, which is usually a common hallmark of ODDD. Patients reported in the literature with the G138R form of Cx43 appear to exhibit greater disease burden since they present with neurological symptoms, including neural spasticity, hyperreflexia, urinary incontinence, syndactyly, and hypoplasia of hands and feet and hyperkeratosis (Paznekas et al., 2003; Shuja et al., 2016). Although some of these symptoms, namely spasticity and hyperreflexia of lower limbs and mild urinary incontinence, are reported here for the p.G143S patient, a previous study of nine patients from four generations of a family had no report of neural or urinary problems (Brueton et al., 1990; Richardson et al., 2004).
Intriguingly, our studies here using patient fibroblasts suggest that the G138R mutation does not affect Cx43 expression and function as severely as the L7V mutation. The L7V mutation resides in the N-terminal domain of Cx43, which is predicted from the crystal structure of Cx26 to insert into the pore-lining segment of the channel (Maeda et al., 2009; Bennett et al., 2016) and is proposed to play a role in transjunctional voltage-dependent gating and ion permeability (Harris, 2001; Ek Vitorin et al., 2016). Previously we examined another mutation in this same domain (D3N) and found that human fibroblasts exhibited a lower expression of Cx43 and impaired Cx43 function (Churko et al., 2011b; Esseltine et al., 2015). Of interest, both D3N and L7V mutants are located in the N-terminal domain, and both mutants lead to reduced Cx43 RNA levels, which suggests that this part of the sequence is important for Cx43 RNA transcription and/or stability and may be affected by important regulatory elements (Kandouz et al., 2013; Oyamada et al., 2013). L7V-expressing fibroblasts can deliver low levels of Cx43 to the cell surface, where they may contribute to increased (“leaky”) ATP release and form few gap junction channels. Thus the L7V mutant could be construed as a gain-of-function mutation since Cx43 hemichannels that do exist at the cell surface should primarily be maintained in a closed state (Shao et al., 2012). In addition, ATP release is much higher in unstimulated L7V- expressing cells despite expressing ∼80% less Cx43 protein than control cells. Of interest, another heterozygous mutation (p.G8V) in the N-terminal domain of Cx43 was recently reported to have enhanced hemichannel activities, leading to keratoderma-hypotrichosis-leukonychia totalis syndrome (Wang et al., 2015). In addition, a recent study (Ek Vitorin et al., 2016) revealed that the N-terminal domain of Cx43 plays a crucial role in channel regulation and that Cx43 gap junction and hemichannels are more resistant to closure when the N-terminal is replaced with that of Cx37. This suggests that the predicted role of the N-terminus inserting into the channel pore and regulating channel permeability is dysregulated in both L7V and G8V Cx43 variants and, for L7V, may lead to resistance of CBX-induced hemichannel blockage. However, we did not find cytotoxic effects in fibroblasts expressing L7V, as was reported for the G8V mutant (Wang et al., 2015). Thus it is possible that down-regulation of Cx43 and L7V mutant expression levels functions as a feedback survival mechanism, since a high level of overactive hemichannels at the cell surface would most assuredly be cytotoxic. We propose that overall down-regulation of total Cx43 may protect L7V-harboring patients from having even more disease, given the severity of the mutation. However, it is important to note that linking ATP release directly to hemichannel function/dysfunction is technically challenging and has been controversial (Spray et al., 2006). Because the L7V fibroblasts had higher ATP release under normal calcium and CBX blockage conditions, both of which should normally close hemichannels, we cannot rule out other, indirect routes of ATP leakiness. Future studies will need to determine whether the L7V mutant alone leads to leaky ATP release and whether the mutant directly confers resistance to CBX blockage.
Somewhat surprisingly, patients harboring the G138R mutant exhibit the most severe disease of the mutants examined in this report. These patients harboring the G138R mutant appear to express slightly lower levels of total Cx43 with no GJIC and no dysregulated increase in Cx43 hemichannel function. Previously the overexpression of the G138R mutant in HeLa cells was found to result in an increase in hemichannel function (Dobrowolski et al., 2007), which appears not to be the case when the mutant and endogenous Cx43 are expressed at patient-relevant levels. Nonetheless, the present finding that the G138R mutant inhibits the gap junction channel function of coexpressed endogenous Cx43 is in keeping with studies in which the G138R was overexpressed in cultured cells (Gong et al., 2007). Still other studies showed that the overexpression of the G138R mutant did not impair keratinocyte differentiation into organotypic epidermis (Churko et al., 2010). Genetically modified mice globally expressing one copy of the G138R mutant (Cx43G138R/+) mimicked ODDD and, similar to the human dermal fibroblasts reported here, failed to phosphorylate Cx43 to the P2 species, indicative of fully assembled gap junction plaques (Dobrowolski et al., 2008). Of importance, ATP release increased in cardiomyocytes obtained from G138R mutant mice, suggesting that Cx43 hemichannels may be hyperactive. Given that the present study is the only one in which the G138R mutant was examined in the human context, we suggest that the hemichannel dysfunction reported by others is less likely to cause the clinical phenotype and that instead the reduction of Cx43 levels and GJIC is at the root of the disease in these patients.
Our functional analysis of the G143S mutant is among the most puzzling. Given that total Cx43 levels, hemichannel function, and intercellular channel function all appear normal in fibroblasts expressing the G143S mutant, one might predict that this mutant would not cause clinical disease. In fact, patients harboring the G143S mutant do not present with all of the classical symptoms of ODDD (Churko et al., 2011a). In one family, nine individuals exhibited syndactyly/camptodactyly and facial abnormalities. However, ocular and dental defects, which are common hallmarks of ODDD patients, were not present in any of the affected family members (Brueton et al., 1990), and only one case (in this study) reported microcornea. Of interest, when the G143S mutant was expressed in connexin-deficient HeLa cells, GJIC was completely inhibited, hemichannel function was increased, and the half-life of the mutant was increased by approximately 1 h (Dobrowolski et al., 2007). However, it is important to point out that those studies were performed in an overexpression system with HeLa cells expressing mutant protein only. Although we now know that the G143S mutant has no inhibitory effects on either hemichannel or gap junction channel function in the human context, in which mutant Cx43 is expressed in a 1:1 ratio to normal Cx43, it is possible that the mutant delays the turnover of Cx43, thus introducing a cellular pathology. This effect could be linked to the reduced ratio of P2 isoform to total Cx43, since increased phosphorylation is linked to Cx43 internalization and degradation (Guan and Ruch, 1996; Laird, 2005). Alternatively, it is of note that the G143S mutation resides in a domain of Cx43 where pH gating may be dysregulated as the potential intramolecular binding site for the C-terminal tail of Cx43 (Hirst-Jensen et al., 2007). Nevertheless, it is intriguing that the G138R and the G143S mutations reside in the same intracellular loop domain and both result in clinical ODDD, with little (G143S) or drastic (G138R) effects on Cx43 function.
Phosphorylation of Cx43 plays a critical role in regulating Cx43 channel function, as well as in key stages of its life cycle (Laird, 2005; Solan and Lampe, 2014). The extent of phosphorylation in ODDD fibroblasts was highly variable among the Cx43 mutants. All fibroblasts exhibited some P1 species expression, suggesting that Cx43 was phosphorylated. This might explain why all of the mutants retained the ability to traffic to the cell surface (Solan and Lampe, 2009) and exhibited ATP release under hemichannel-activating conditions. In G143S-expressing cells, the higher amount of P1 species under normal conditions and the presence of P2 species in Triton X-100–insoluble fractions might account for why GJIC was not affected. For example, P1 phosphorylation at specific sites (such as S365) might be more prominent in G143S-expressing cells, thus permitting cell surface localization and channel function (Solan and Lampe, 2007). Conversely, the absence of P2 phosphorylation and reduced P1 expression in L7V- and G138R-expressing fibroblasts might render Cx43 gap junctions nonfunctional (Solan and Lampe, 2007, 2009). It will be interesting to see in future studies whether inducing phosphorylation at specific Cx43 sites could rescue Cx43 function in ODDD cells.
Although patient fibroblasts exhibited differences in their ability to synthesize Cx43 and assemble functional gap junctions, they all changed the phenotype of the cell. All patient dermal fibroblasts grew more slowly in culture, similar to what we observed for human patient fibroblasts harboring the D3N and V216L mutations (Churko et al., 2011b). Moreover, the internal compass of all patient fibroblasts was altered, as cells failed to fully polarize during cell migration assays, which might be partially related to a suboptimal response to growth factors like PDGF or problems in microtubule organization (Francis et al., 2011). It is plausible, then, that the symptoms associated with the G143S mutant are simply due to reduced ability of cells to proliferate and migrate efficiently, since these were the main anomalies found in our assays. Similarly, patient fibroblasts did not fully respond to TGF-β, as normal Cx43 phosphorylation levels were never achieved, and cells had variable ability to differentiate into myofibroblasts, as revealed by the expression of α-SMA. The L7V and G143S fibroblasts had higher basal levels of α-SMA, suggesting a more differentiated basal state, whereas the G138R mutant fibroblasts had a significant reduction in α-SMA expression after TGF-β treatment, which suggests an inability to fully differentiate. Collectively, whereas all three patient fibroblasts exhibited dysregulated Cx43, they shared common anomalies related to cell migration and response to growth factor stimulation. It will be interesting to determine why all Cx43 mutants studied to date negatively regulate cell growth and migration. In addition, although patient-derived dermal fibroblasts offer an excellent model system for investigating the consequences of endogenous wild-type and mutant Cx43 expression, ODDD rarely manifests in the skin. Going forward, it will also be useful to investigate the consequences of altered Cx43 function in cell types associated with ODDD symptoms, such as osteoblasts. Given limitations in which tissues can be readily biopsied from ODDD patients, this will be best achieved by reprogramming patient dermal fibroblasts into inducible pluripotent stem cells and differentiating cell types that are known to be compromised in ODDD.
In summary, we discovered that the variability in the clinical presentation of ODDD can be explained in part by how specific missense mutations affect Cx43 expression, trafficking, hemichannel function, and gap junctional intercellular communication. Even mutations that are located within the same functional domain of Cx43 can exhibit radically distinct effects on Cx43 function. Future genomic studies may also reveal modifying variants in other genes, including other connexins that could modulate the phenotype. Finally, these findings may have future therapeutic relevance when considering whether to reduce the inhibitory effects of Cx43 mutant levels in the disease state or use drugs that may block hyperactive hemichannels, stabilize gap junction plaque formation, and/or increase gap junctional intercellular communication.
MATERIALS AND METHODS
All consumables were obtained from Invitrogen/Thermo Fisher Scientific (Waltham, MA) unless stated otherwise.
Primary human fibroblasts
Fibroblast cultures were created from human ODDD patient skin biopsies as described previously (Churko et al., 2011b), with informed patient consent and approval by the Western University Health Science Research Ethics Board, the Johns Hopkins Joint Committee on Clinical Investigations, and the Institutional Review Board (00040092) from the University of Utah, as well as in accordance with the Declaration of Helsinki. For controls, we used two human dermal fibroblast lines obtained from unaffected relatives of patients expressing Cx43 p.D3N and p.V216L variants, as we reported previously (Churko et al., 2011b). For the purpose of this study, data from both cell lines (referred to as control [Ctrl]) were combined for quantification and statistical analysis and to minimize any significant variability among control groups. Although patient samples were not age or sex matched, all of the fibroblasts were standardized to the same passage number (3–7) for each assay. The fibroblasts were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine and incubated at 37°C with 5% CO2. The medium was changed once a week for 3–4 wk. Cells were subcultured once they reached confluence with trypsin (0.05% trypsin with 0.25 mM EDTA).
One-step reverse transcriptase-PCR and sequencing
Cells were grown to confluence, and RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The RNA was converted to cDNA, and a region of the Cx43 sequence was amplified using a SuperScript One-Step RT-PCR with Platinum Taq DNA Polymerase kit. The following primers were used to amplify an 840–base pair region of Cx43 that incorporated all three mutation sites: forward, 5′-AGCAAAAGAGTGGTGCCCAG-3′; reverse, 5′-GAGCAGCCATTGAAATAAGC-3′. PCR products were run on a 1.5% agarose gel, bands were extracted using the QIAquick Gel Extraction Kit (Qiagen), and purified cDNA was sent for sequencing with the following primers: 5′-ACTTCTTTATCTCAATCTGC-3′ (reverse primer 1), 5′-AGTCCACCTGATGTGGGCAG-3′ (reverse primer 2), and 5′-GAGCAGCCATTGAAATAAGC-3′ (reverse primer 3).
Quantitative real-time PCR
Total RNA was isolated from cultured fibroblasts using the RNeasy Mini Kit and then used for real-time PCR as described previously (Esseltine et al., 2015). Briefly, real-time qPCR was performed with iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) with the following cycle conditions: 95°C for 3 min, then 95°C for 10 s, 55°C for 10 s, and 72°C for 30 s for 39 cycles, followed by a melt curve from 65°to 95°C to analyze primer quality. Samples were run using a Bio-Rad Laboratories CFX96 Touch Real-Time PCR Detection System. The following primers were used to amplify Cx43: forward, 5′-AGAGGAAGAACTCAAGGTTGCC-3′; reverse, 5′-AGGCCACCTCAAAGATAGACT-3′. Total RNA was controlled for with 18S rRNA: forward, 5′-GTAACCCGTTGAACCCCATT-3′; reverse, 5′-CCATCCAACGGTAGTAGCG-3′.
Immunofluorescence labeling
Fibroblasts were fixed with 10% neutral-buffered Formalin, permeabilized, and blocked with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) and 3% bovine serum albumin (Santa Cruz Biotechnology, Dallas, TX) in phosphate-buffered saline (PBS). The following primary antibodies were left on the cells for 2 h at room temperature or at 4°C overnight: rabbit polyclonal anti-Cx43 (1.4 μg/ml; C6219; Sigma-Aldrich), mouse monoclonal anti-GM130 (0.63 μg/ml; 610822; BD Biosciences, Franklin Lakes, NJ), mouse monoclonal anti–αSMA (4 μg/ml; A5228, Sigma-Aldrich), and rabbit polyclonal anti–nonmuscle myosin IIA (2 μg/ml; PRB-440P; Covance, Princeton, NJ). After being washed in PBS, cells were incubated for 1 h with the following secondary antibodies: goat anti-mouse Alexa Fluor 488 or 555 (A11017/A21425; 1:500 dilution; Invitrogen) or goat anti-rabbit Alexa Fluor 488 or 555 (1:500 dilution; A11008/A21429; Invitrogen). F-actin filaments were labeled with phalloidin conjugated to Alexa Fluor 568 (1:500 dilution, A12380; Invitrogen). Nuclei were stained with Hoechst 33342 (10 μg/ml; 62249; Invitrogen). Cells were washed with PBS, mounted on slides, and imaged on a Zeiss 510 Meta confocal microscope using a 63× oil immersion lens with 1.4 numerical aperture (Zeiss, Oberkochen, Germany).
Cell surface biotinylation
Fibroblasts were placed on ice and washed in ice-cold Hank’s balanced salt solution, and cell surface proteins were labeled with 1.5 mg/ml EZLink Sulfo-NHS-SS-biotin (21331; Thermo Fisher) for 1 h. Biotin was subsequently quenched with 100 mM glycine for 30 min, and cells were lysed. Cleared supernatants containing 150–250 μg of protein were incubated with 25 μl of NeutrAvidin affinity beads (29200; Thermo Fisher) overnight, with rotation at 4°C to precipitate biotin-labeled proteins. After incubation, the beads were washed twice with PBS, and precipitated proteins were separated by SDS–PAGE, transferred to a nitrocellulose membrane, and immunoblotted. Total Cx43 and GAPDH levels were determined by immunoblotting 20 μg of protein from each cell lysate for biotinylation, input, and cleared lysate.
Gap junction measurements
Fibroblasts were immunofluorescently labeled for Cx43 and imaged with a 63× oil immersion lens on an LSM 800 Airyscan confocal microscope (Zeiss) with a resolution limit of 140 nm. The length of each plaque was measured using ZEN Blue software (Zeiss). The data represent SEM of three independent experiments for a total of 27 separate images and >400 measurements for each cell line. A one-way analysis of variance (ANOVA) with a Tukey post hoc test was performed to determine statistical significance (***p < 0.001) compared with control.
Triton solubility assay
Protein lysates from control and ODDD fibroblasts were divided into Triton X-100–soluble and –insoluble fractions as described previously (Kelly et al., 2015a). Briefly, cells were gently collected into cold PBS, pelleted, and resuspended in a 1% Triton X-100 buffer. Cells were left for 30 min on ice before pelleting and collecting the Triton-X–soluble supernatant. The remaining Triton-X–insoluble pellet was then dissolved in an equal volume of lysis buffer containing 1% SDS. Equal volumes of Triton X-100–soluble and –insoluble fractions were separated by SDS–PAGE and subjected to Western blotting. GAPDH was used to verify the quality of the isolated fractions.
Microinjection/dye transfer
Fully confluent fibroblast cultures were microinjected with the Cx43 permeable dye Alexa Fluor 350 (10 mM) as described previously (Berger et al., 2014). The degree of transfer was quantified into three categories: no transfer, in which the dye was confined to the injected cell; first order, in which the dye spread to the first layer of cells surrounding the injected cell; or second order, in which the dye spread to cells farther than the first layer.
Patch-clamp electrophysiology
Functional gap junction coupling between paired human fibroblasts was assessed using the dual whole-cell patch-clamp technique as previously described (Gong et al., 2006; Churko et al., 2011b). The cells were subcultured 2–3 h before the experiments. Isolated cell pairs were bathed in an external solution containing 140 mM NaCl, 5 mM KCl, 2 mM CsCl2, 2 mM CaCl2, 1 mM MgCl2, 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 5 mM d-glucose, 2 mM pyruvate, and 1 mM BaCl2, pH 7.4. The junctional current was measured with Axopatch 200B patch-clamp amplifiers (Axon Instruments, Union City, CA) at room temperature (22–25°C). Patch pipettes had a resistance of 2.5–5.5 MΩ and were backfilled with an internal solution containing 130 mM CsCl2, 10 mM ethylene glycol tetraacetic acid (EGTA), 0.5 mM CaCl2, 3 mM MgATP, 2 mM Na2ATP, and 10 mM HEPES, pH titrated to 7.2 with CsOH. Data were low-pass filtered at 2 kHz, acquired using pClamp9 software (Axon Instruments), and digitized at 10-kHz sampling rate. Each cell of a pair was initially held at a common holding potential of 0 mV. To evaluate junctional coupling, we applied 7-s hyperpolarizing pulses from the holding potential of 0 to −20 mV to one cell to establish a transjunctional voltage gradient, and we measured the junctional current in the second cell. Macroscopic Gj was calculated from Gj = Ij/Vj, where Ij is the measured junctional current and Vj is transjunctional voltage. One-way ANOVA with a Bonferroni post hoc test was performed to determine statistical significance (**p < 0.01). Gj is presented as mean ± SEM.
Drug treatments
PDGF-BB was used because it can activate both the α and β receptors (Heldin and Westermark, 1999). PDGF-BB was reconstituted in 100 mM acetic acid supplemented with 0.1% BSA and used at a working concentration of 50–100 ng/ml. TGF-β1 (R&D Systems, Minneapolis, MN) was reconstituted in 4 mM HCl supplemented with 1% BSA and used at a working concentration of 5 ng/ml.
Western blotting
Fibroblast protein lysates were obtained and processed as described previously (Kelly et al., 2015a). Briefly, a 1% Triton X-100/0.5% Nonidet P-40 extraction buffer was used to lyse the cells. After clearing the lysates, 20–30 μg protein/lane was separated by electrophoresis on 10% SDS–PAGE gels and transferred to nitrocellulose membranes for immunoblotting. The same antibodies were used as for immunofluorescent labeling, with the addition of mouse monoclonal anti-GAPDH (0.1 μg/ml; Millipore, Billerica, MA). Primary antibodies were detected using goat anti-rabbit IRdye 800 (1:10,000; LI-COR Biosciences, Lincoln, NE) and Alexa Fluor 680 goat anti-mouse (1:10,000, Invitrogen) secondary antibodies. Membranes were scanned and quantified using the Odyssey Infrared Imaging System (LI-COR Biosciences).
Growth curve analysis
Fibroblasts were plated at 1 × 104/well in a six-well dish and allowed to grow for 9 d in DMEM complete. Replicate samples were collected on days 3, 6, and 9 and viable cell numbers calculated with a Countess Cell Counter. Data from three independent experiments were collated and assessed using GraphPad Prism 4.0 (GraphPad, La Jolla, CA). A two-way ANOVA (repeated measures) from data points on days 3, 6, and 9 with a Bonferroni post hoc test was performed to determine statistical significance (**p < 0.01, ***p < 0.001).
Chemotaxis migration assay
HTS FluoroBlok Transwell inserts with an 8-μm porous polyethylene terephthalate membrane (Corning, Corning, NY) were placed into 24-well plates containing DMEM without FBS, DMEM with 20% FBS, or DMEM with 50 ng/ml PDGF-BB. Fibroblast suspensions were plated into the upper chamber of the Transwell insert at a final concentration of 5 × 104 cells/well. After 24 h, the membranes were fixed in methyl alcohol for 5 min, washed with PBS, and stained with Hoechst 33342 (10 μg/ml in water) for 5 min. Membranes were cut out with a scalpel blade and mounted in 90% glycerol under a glass coverslip on slides. Four to six random images were taken of the upper and lower sides of each membrane, and the percentage of cell migration was calculated.
Golgi polarization scratch assay
Fibroblasts were grown to full confluency on glass coverslips. A single scrape with a P20 pipette tip was made down the middle of each coverslip. The cells were left for 3 h and then fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). The Golgi apparatus was stained with an antibody directed against the Golgi marker GM130. Images were scored for the number of leading-edge cells with correctly polarized Golgi, as described elsewhere (Francis et al., 2011; Mendoza-Naranjo et al., 2012).
ATP release assay
Fibroblasts were seeded at a concentration of 4 × 104/well in 24-well plates. The next day, the cells were washed once with fresh DMEM and then incubated for 30 min in DMEM or DMEM supplemented with 100 μM carbenoxolone to block connexin hemichannels or dimethyl sulfoxide as a control. Cells were washed with an ECS containing Ca2+ and Mg2+ (142 mM NaCl, 5.4 mM KCl, 1.4 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, and 25 mM d-glucose) with or without CBX. To stimulate hemichannel opening, the cells were incubated with 300 μl of DCFS, in which Ca2+ and Mg2+ were replaced with 2 mM EGTA for 15 min either with or without CBX (100 μM), as described previously (Tong et al., 2007; Sun et al., 2014). Nonstimulated cells were incubated with ECS alone. To prevent extracellular degradation of ATP, all solutions for the final incubation step contained 150 μM of the ecto-ATPase inhibitor ARL-67156 trisodium salt (Santa Cruz Biotechnology). After 15 min at 37°C, 100-μl samples were spun down in precooled Eppendorf tubes to pellet any cells or debris, and then 10 μl was removed for ATP analysis using an ATP Determination Kit (Invitrogen), according to the manufacturer’s instructions. Briefly, 90 μl of the standard reaction solution containing 1.25 μg/ml firefly luciferase was added to each sample and ATP standard in a 96-well plate and immediately imaged using the Luminescence setting on a Victor3 Multilabel Counter (PerkinElmer, Waltham, MA).
Statistics
All values are presented as mean ± SEM unless otherwise indicated. All results were analyzed using a one-way or two-way ANOVA followed by a Bonferroni, Tukey, or Dunnett post hoc test. All analyses were calculated with GraphPad Prism 4.0 statistical software. *p < 0.05, **p < 0.01, ***p < 0.001.
Acknowledgments
We thank the patients and their families for participating in this study. We also thank Jamie Simek for making the Cx43 model in Figure 1 and Ali Zhang and Kevin Tieu for help with data collection. This study was supported by Canadian Institutes of Health Research Grants 123228 and 74637 to D.W.L.
Abbreviations used:
- α-SMA
α-smooth muscle actin
- CBX
carbenoxolone
- Cx43
connexin43
- DCFS
divalent cation–free solution
- ECS
extracellular solution
- GJIC
gap junctional intercellular communication
- ODDD
oculodentodigital dysplasia
- PDGF
platelet-derived growth factor
- TGF-β
transforming growth factor-β.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0062) on May 25, 2016.
REFERENCES
- Alexander DB, Goldberg GS. Transfer of biologically important molecules between cells through gap junction channels. Curr Med Chem. 2003;10:2045–2058. doi: 10.2174/0929867033456927. [DOI] [PubMed] [Google Scholar]
- Bennett BC, Purdy MD, Baker KA, Acharya C, McIntire WE, Stevens RC, Zhang Q, Harris AL, Abagyan R, Yeager M. An electrostatic mechanism for Ca(2+)-mediated regulation of gap junction channels. Nat Commun. 2016;7:8770. doi: 10.1038/ncomms9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Kelly JJ, Lajoie P, Shao Q, Laird DW. Mutations in Cx30 that are linked to skin disease and non-syndromic hearing loss exhibit several distinct cellular pathologies. J Cell Sci. 2014;127:1751–1764. doi: 10.1242/jcs.138230. [DOI] [PubMed] [Google Scholar]
- Brueton LA, Huson SM, Farren B, Winter RM. Oculodentodigital dysplasia and type III syndactyly: separate genetic entities or disease spectrum? J Med Genet. 1990;27:169–175. doi: 10.1136/jmg.27.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Chan J, Shao Q, Laird DW. The G60S connexin43 mutant regulates hair growth and hair fiber morphology in a mouse model of human oculodentodigital dysplasia. J Invest Dermatol. 2011a;131:2197–2204. doi: 10.1038/jid.2011.183. [DOI] [PubMed] [Google Scholar]
- Churko JM, Langlois S, Pan X, Shao Q, Laird DW. The potency of the fs260 connexin43 mutant to impair keratinocyte differentiation is distinct from other disease-linked connexin43 mutants. Biochem J. 2010;429:473–483. doi: 10.1042/BJ20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Shao Q, Gong XQ, Swoboda KJ, Bai D, Sampson J, Laird DW. Human dermal fibroblasts derived from oculodentodigital dysplasia patients suggest that patients may have wound-healing defects. Hum Mutat. 2011b;32:456–466. doi: 10.1002/humu.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock M, Kerrebrouck M, Wang N, Leybaert L. Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front Pharmacol. 2013;4:120. doi: 10.3389/fphar.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hondt C, Iyyathurai J, Himpens B, Leybaert L, Bultynck G. Cx43-hemichannel function and regulation in physiology and pathophysiology: insights from the bovine corneal endothelial cell system and beyond. Front Physiol. 2014;5:348. doi: 10.3389/fphys.2014.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, Troatz C, Ghanem A, Tiemann K, Degen J, et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17:539–554. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R, Sommershof A, Willecke K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J Membr Biol. 2007;219:9–17. doi: 10.1007/s00232-007-9055-7. [DOI] [PubMed] [Google Scholar]
- Ek Vitorin JF, Pontifex TK, Burt JM. Determinants of Cx43 channel gating and permeation: the amino terminus. Biophys J. 2016;110:127–140. doi: 10.1016/j.bpj.2015.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine JL, Shao Q, Huang T, Kelly JJ, Sampson J, Laird DW. Manipulating Cx43 expression triggers gene reprogramming events in dermal fibroblasts from oculodentodigital dysplasia patients. Biochem J. 2015;472:55–69. doi: 10.1042/BJ20150652. [DOI] [PubMed] [Google Scholar]
- Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, Gong XQ, Kelsey LB, Lounsbury C, Moreno L, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132:4375–4386. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One. 2011;6:e26379. doi: 10.1371/journal.pone.0026379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong XQ, Shao Q, Langlois S, Bai D, Laird DW. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J Biol Chem. 2007;282:19190–19202. doi: 10.1074/jbc.M609653200. [DOI] [PubMed] [Google Scholar]
- Gong XQ, Shao Q, Lounsbury CS, Bai D, Laird DW. Functional characterization of a GJA1 frameshift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J Biol Chem. 2006;281:31801–31811. doi: 10.1074/jbc.M605961200. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- Guan X, Ruch RJ. Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial cells treated with DDT and lindane. Carcinogenesis. 1996;17:1791–1798. doi: 10.1093/carcin/17.9.1791. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Zackai EH, McDonald-McGinn DM, Fischbeck KH, Kamholz J. Oculodentodigital dysplasia syndrome associated with abnormal cerebral white matter. Am J Med Genet. 1991;41:18–20. doi: 10.1002/ajmg.1320410106. [DOI] [PubMed] [Google Scholar]
- Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Hirst-Jensen BJ, Sahoo P, Kieken F, Delmar M, Sorgen PL. Characterization of the pH-dependent interaction between the gap junction protein connexin43 carboxyl terminus and cytoplasmic loop domains. J Biol Chem. 2007;282:5801–5813. doi: 10.1074/jbc.M605233200. [DOI] [PubMed] [Google Scholar]
- Huang T, Shao Q, Barr K, Simek J, Fishman GI, Laird DW. Myogenic bladder defects in mouse models of human oculodentodigital dysplasia. Biochem J. 2014;457:441–449. doi: 10.1042/BJ20130810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judisch GF, Martin-Casals A, Hanson JW, Olin WH. Oculodentodigital dysplasia. Four new reports and a literature review. Arch Ophthalmol. 1979;97:878–884. doi: 10.1001/archopht.1979.01020010436007. [DOI] [PubMed] [Google Scholar]
- Kalcheva N, Qu J, Sandeep N, Garcia L, Zhang J, Wang Z, Lampe PD, Suadicani SO, Spray DC, Fishman GI. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci USA. 2007;104:20512–20516. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandouz M, Zhao J, Bier A, Di Marco S, Oviedo-Landaverde I, Gallouzi IE, Batist G. Post-transcriptional regulation of connexin43 in H-Ras-transformed cells. PLoS One. 2013;8:e58500. doi: 10.1371/journal.pone.0058500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JJ, Shao Q, Jagger DJ, Laird DW. Cx30 exhibits unique characteristics including a long half-life when assembled into gap junctions. J Cell Sci. 2015a;128:3947–3960. doi: 10.1242/jcs.174698. [DOI] [PubMed] [Google Scholar]
- Kelly JJ, Simek J, Laird DW. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015b;360:701–721. doi: 10.1007/s00441-014-2024-4. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014;588:1339–1348. doi: 10.1016/j.febslet.2013.12.022. [DOI] [PubMed] [Google Scholar]
- Levit NA, White TW. Connexin hemichannels influence genetically determined inflammatory and hyperproliferative skin diseases. Pharmacol Res. 2015;99:337–343. doi: 10.1016/j.phrs.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loddenkemper T, Grote K, Evers S, Oelerich M, Stogbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002;249:584–595. doi: 10.1007/s004150200068. [DOI] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Magdalena J, Millard TH, Machesky LM. Microtubule involvement in NIH 3T3 Golgi and MTOC polarity establishment. J Cell Sci. 2003;116:743–756. doi: 10.1242/jcs.00288. [DOI] [PubMed] [Google Scholar]
- Mendoza-Naranjo A, Cormie P, Serrano AE, Hu R, O’Neill S, Wang CM, Thrasivoulou C, Power KT, White A, Serena T, et al. Targeting Cx43 and N-cadherin, which are abnormally upregulated in venous leg ulcers, influences migration, adhesion and activation of Rho GTPases. PLoS One. 2012;7:e37374. doi: 10.1371/journal.pone.0037374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhaske PV, Levit NA, Li L, Wang HZ, Lee JR, Shuja Z, Brink PR, White TW. The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am J Physiol Cell Physiol. 2013;304:C1150–C1158. doi: 10.1152/ajpcell.00374.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molica F, Meens MJ, Morel S, Kwak BR. Mutations in cardiovascular connexin genes. Biol Cell. 2014;106:269–293. doi: 10.1111/boc.201400038. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Gap junctional intercellular communication and the regulation of connexin expression and function. Curr Opin Cell Biol. 1990;2:875–880. doi: 10.1016/0955-0674(90)90086-t. [DOI] [PubMed] [Google Scholar]
- Oyamada M, Takebe K, Oyamada Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim Biophys Acta. 2013;1828:118–133. doi: 10.1016/j.bbamem.2011.12.031. [DOI] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–733. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- Richardson R, Donnai D, Meire F, Dixon MJ. Expression of Gja1 correlates with the phenotype observed in oculodentodigital syndrome/type III syndactyly. J Med Genet. 2004;41:60–67. doi: 10.1136/jmg.2003.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, Liu Q, Lorentz R, Gong XQ, Bai D, Shaw GS, Laird DW. Structure and functional studies of N-terminal Cx43 mutants linked to oculodentodigital dysplasia. Mol Biol Cell. 2012;23:3312–3321. doi: 10.1091/mbc.E12-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RE, Griffin JW, Stine OC. Evidence for genetic anticipation in the oculodentodigital syndrome. Am J Med Genet. 1997;71:36–41. [PubMed] [Google Scholar]
- Shuja Z, Li L, Gupta S, Mese G, White TW. Connexin26 mutations causing palmoplantar keratoderma and deafness interact with connexin43, modifying gap junction and hemichannel properties. J Invest Dermatol. 2016;136:225–235. doi: 10.1038/JID.2015.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–1429. doi: 10.1016/j.febslet.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Ye ZC, Ransom BR. Functional connexin “hemichannels”: a critical appraisal. Glia. 2006;54:758–773. doi: 10.1002/glia.20429. [DOI] [PubMed] [Google Scholar]
- Stewart MK, Gong XQ, Barr KJ, Bai D, Fishman GI, Laird DW. The severity of mammary gland developmental defects is linked to the overall functional status of Cx43 as revealed by genetically modified mice. Biochem J. 2013;449:401–413. doi: 10.1042/BJ20121070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116:2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hills MD, Ye WG, Tong X, Bai D. Atrial fibrillation-linked germline GJA5/connexin40 mutants showed an increased hemichannel function. PLoS One. 2014;9:e95125. doi: 10.1371/journal.pone.0095125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong D, Li TY, Naus KE, Bai D, Kidder GM. In vivo analysis of undocked connexin43 gap junction hemichannels in ovarian granulosa cells. J Cell Sci. 2007;120:4016–4024. doi: 10.1242/jcs.011775. [DOI] [PubMed] [Google Scholar]
- Wang H, Cao X, Lin Z, Lee M, Jia X, Ren Y, Dai L, Guan L, Zhang J, Lin X, et al. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum Mol Genet. 2015;24:243–250. doi: 10.1093/hmg/ddu442. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Yang W, Lurtz MM, Ye Y, Huang Y, Lee HW, Chen Y, Louis CF, Yang JJ. Identification of the calmodulin binding domain of connexin 43. J Biol Chem. 2007;282:35005–35017. doi: 10.1074/jbc.M707728200. [DOI] [PubMed] [Google Scholar]