

Atypical PKC, Par6, and Par3 constitute a conserved complex signaling cell asymmetry. In contrast to its role in other tissues, atypical PKC inhibits NF-κB activation in epithelia and may function in maintaining low levels of inflammation in addition to establishing apicobasal polarity.
Abstract
The conserved proteins of the polarity complex made up of atypical PKC (aPKC, isoforms ι and ζ), Par6, and Par3 determine asymmetry in several cell types, from Caenorhabditis elegans oocytes to vertebrate epithelia and neurons. We previously showed that aPKC is down-regulated in intestinal epithelia under inflammatory stimulation. Further, expression of constitutively active PKCι decreases NF-κB activity in an epithelial cell line, the opposite of the effect reported in other cells. Here we tested the hypothesis that aPKC has a dual function in epithelia, inhibiting the NF-κB pathway in addition to having a role in apicobasal polarity. We achieved full aPKC down-regulation in small intestine villi and colon surface epithelium using a conditional epithelium-specific knockout mouse. The results show that aPKC is dispensable for polarity after cell differentiation, except for known targets, including ROCK and ezrin, claudin-4 expression, and barrier permeability. The aPKC defect resulted in increased NF-κB activity, which could be rescued by IKK and ROCK inhibitors. It also increased expression of proinflammatory cytokines. In contrast, expression of anti-inflammatory IL-10 decreased. We conclude that epithelial aPKC acts upstream of multiple mechanisms that participate in the inflammatory response in the intestine, including, but not restricted to, NF-κB.
INTRODUCTION
Partition-deficient (PAR) mutant genes encoding PAR proteins and PCK-3 (the orthologue atypical PKC [aPKC]) were identified in Caenorhabditis elegans as essential components of cell polarity mechanisms (Guo and Kemphues, 1996). These proteins are highly conserved in metazoan evolution and participate in polarization of various cell types, including epithelial apicobasal polarity (Suzuki and Ohno, 2006; Pieczynski and Margolis, 2011; Chen and Zhang, 2013). Typically, the aPKC-Par6-Par3 polarity complex is highly polarized itself (Chen and Zhang, 2013). However, most of the evidence supporting a role of aPKC in epithelial apicobasal polarity in vertebrates was obtained in polarized tissue culture epithelial cell lines, such as Madin–Darby canine kidney (MDCK) and Caco-2 (intestine) cells (Suzuki et al., 2004; Liu et al., 2007; Zihni et al., 2014). Global mouse knockout models for both aPKC isoforms (PKCι and ζ) were not informative regarding the role of aPKC in polarity in vivo. The PKCι knockout is embryonic lethal (Bandyopadhyay et al., 2004), whereas the PKCζ knockout model displays a mild phenotype with defective immune response and compromised NF-κB activation in B-lymphocytes (Martin et al., 2002)
Two other groups independently reported the use of conditional PKCι-deficient mice for studies of epithelia (Imai et al., 2006; Calcagno et al., 2011). In one case, defects of adherens junctions in neuroepithelial cells were found (Imai et al., 2006), whereas in the other, the status of apicobasal polarity was not reported. Calcagno et al. (2011), however, reported an intriguing hypersensitivity of PKCι-knockout mice to chemical colitis, compatible with an anti-inflammatory role of this kinase, but evidence for a specific role in modulating the NF-κB pathway was lacking. In contrast, the role of aPKC in other cell types has been extensively studied, especially in relationship to its role in glucose metabolism (Farese and Sajan, 2010; Farese et al., 2014) and activation of NF-κB (Diaz-Meco and Moscat, 2012). In those functions, aPKC typically partners with p62 via the N-terminal PB1 domain (Moscat et al., 2006) rather than Par6 in the PAR polarity complex (Chen and Zhang, 2013).
Our interest in the possible role of aPKC in inflammatory pathways was sparked when we observed that aPKC is deeply down-regulated in intestinal epithelia under proinflammatory stimuli in Caco-2 cells, a mouse model of colitis (Mashukova et al., 2011), and samples from human inflammatory bowel disease (IBD) patients (Wald et al., 2011). We demonstrated that aPKC down-regulation is posttranslational, due to destabilization, and under the control of NF-κB activation (Mashukova et al., 2011). It was natural to ask what consequences, if any, result from loss of aPKC in inflammation. Overexpressing wild-type and constitutively active PKCι in Caco-2 cells, we found inhibition of basal NF-κB activity. Furthermore, there was no detectable effect on apicobasal polarity by a constitutively active mutant defective in the PB1 domain and, thus, depolarized. These results were intriguing because they are not consistent with prevalent views of aPKC activating NF-κB. We hypothesized that different effects of aPKC on NF-κB might be tissue specific or perhaps due to the transformed status of Caco-2 cells.
This work was undertaken to test the hypothesis that PKCι down-regulation affects both apical polarity and inflammation in vivo. To that end, we used a PKCιflox/flox mouse developed in our facility to achieve a conditional knockout in intestinal epithelia. The results showed a modest role of aPKC in the maintenance of apical polarity and a critical control of NF-κB activation.
RESULTS
Effects of the conditional PKCι knockout
Exon 4 of the gene encoding PKCι (Prkci) (Figure 1A) was floxed by breeding Prkciflox/flox animals with transgenic mice expressing CRE under a 12.4-kb villin promoter. In these animals, CRE is expressed in the intestinal epithelium (Madison et al., 2002), active in all lineages, and possibly expressed in stem cells (Koo et al., 2009). To analyze the phenotype, we performed studies with an antibody that recognizes the phosphorylated turn domain in both aPKC isoforms, ι and ζ (T555 and T560, respectively). Because the turn domain is autophosphorylated, it reports that the kinase is in the active conformation. Therefore this strategy enabled us to identify cells lacking both aPKC isoforms so that the results would not be confused by isoform redundancy. The characteristic signal of aPKC in the tight junction region in villus enterocytes (Figure 1B, arrows) and colonocytes (Figure 1E, arrows) was missing in the Prkciflox/flox Vil-CRE+ (hereafter referred to as KO). However, we found aPKC signal in the intestinal glands (“crypts”; Figure 1, C, arrow in E). Using an isoform-specific PKCζ antibody, we found that this isoform was up-regulated in the KO crypts (Figure 1D) but not in the surface epithelium (colon) or in the villus (small intestine), except for isolated patches of cells at the tip of villi (unpublished data). The subcellular apical localization of PKCζ in KO crypts was identical to that of total aPKC in control animals (Figure 1, C and D). This is important because the predicted protein encoded by Prkci exons 1–3, if stable, would comprise the PB1 domain, which is responsible for binding to Par6 and, consequently, aPKC apical localization and activation (Graybill et al., 2012). Competition for PB1 domain partners by the possible product of the floxed Prkci gene is therefore unlikely. For the interpretation of the results in the following sections, it is necessary to bear in mind that the intestinal epithelial cells display a very fast turnover. The stem cells are located at or near the bottom of the crypts, and proliferating cells move along the crypt. Therefore up-regulation of PKCζ must follow loss of PKCι, acting as a redundant aPKC only in the crypts.
FIGURE 1:

Effect of conditional Prkciflox/flox Vil-CRE± (KO) on aPKC protein expression. (A) Structure of the targeted flox/flox insertion in the Prkci locus after homologous recombination. Numbers in gray represent Prkci exons. Boxes represent the selection genes. Triangles represent the targets for Flp (FRP) and CRE (loxP) recombinases. Arrows represent orientation of genotyping primers. (B) Villus enterocytes or (C) crypts in small intestine frozen sections were stained with anti–phosphorylated aPKC turn domain antibody, which recognizes the active conformation of PKCι/λ and ζ. (D) Crypts in the small intestine mucosa stained with an isoform-specific antibody against PKCζ that does not recognize the ι isoform. (E) Large intestine mucosa sections were processed with anti–p-PKCι/λ antibody as in B and C. Arrows point at aPKC signal in the apical domain. Bars, 10 μm (B), 25 μm (C, E), 20 μm (D).
Villus enterocytes are differentiated and postmitotic (Umar, 2010). The lifespan of these cells as they move from the base of the villus to desquamation at the tip is ∼2 d (Eastwood, 1977). This is the maximum time villus enterocytes lack aPKC activity after spontaneous down-regulation of PKCζ in the KO villus. In summary, these conditional PKCι-deficient mice lack all aPKC activity (PKCι + ζ) in the villus (small intestine) or surface epithelia (colon) but retain aPKC activity in the crypts because of compensatory expression of redundant PKCζ.
The KO animals developed normally, the proportion of KO pups born alive was Mendelian, and weight gain was indistinguishable from that of the controls (Figure 2A). Previous studies showed that the conditional PKCι defect in enterocytes does not result in changes in proliferation or apoptosis in the absence of chemical injury (Calcagno et al., 2011). The proportion of goblet cells was found to be normal as well (Calcagno et al., 2011). Here we found no changes in the number or localization of Paneth cells (Figure 2B, arrows), suggesting that the loss of PKCι does not affect lineage determination, at least in the presence of compensatory PKCζ. The intestinal barrier permeability, measured by diffusion of 3-kDa fluorescent dextran into the blood, showed a significant threefold increase in the KO animals (Figure 2C). Such an increase was abolished by prior treatment with oral sulfasalazine (SS), an inhibitor of NF-κB, which interferes with IKKα and β (Wahl et al., 1998; Weber et al., 2000). Sulfasalazine is approved by the U.S. Food and Drug Administration for use in IBD patients (Nielsen and Munck, 2007) and is also safe for use in rodents. These results led us to ask about the extent to which, if any, the total loss of aPKC in villus enterocytes would affect apicobasal polarity or junction structure.
FIGURE 2:

Functional phenotype of Prkciflox/flox Vil-CRE± (KO) mice. (A) Weight gain after birth for male and female KO (blue diamond) or control littermates (red squares, Prkciflox/flox Vil-CRE- or Prkciflox/− Vil-CRE+; lm). (B) Hematoxylin and eosin images showing the bottom of the crypts displaying Paneth cells (arrow) in control littermates and KO mice. Bar, 20 μm. (C) Female and male control lm and KO mice from 3 to 5 mo old were stomach gavaged with 3-kDa fluorescent dextran and killed 3 h later; fluorescence in plasma was measured. Some animals received NF-κB inhibitor SS in the water before gavage. Values are relative to one control animal in each group. Horizontal lines represent average values. *Kruskal–Wallis test, p < 0.05.
aPKC is dispensable for maintenance of epithelial polarity in differentiated enterocytes but necessary for correct localization of direct targets
The question of how loss of aPKC affects apicobasal polarity is relevant because these kinases are deeply down-regulated in intestinal inflammation (Wald et al., 2011). We tested polarized proteins and tight junction (TJ) markers. Several markers remained well polarized in the villus enterocytes despite the lack of aPKC. Brush border actin, NHE3, and occludin (Figure 3), as well as alkaline phosphatase, Par3, and ZO-1 (Figure 4A), were normally localized in the KO. An increase in the basolateral F-actin signal was noted in the KO villus enterocytes (Figure 3, arrow). The ultrastructure of brush borders (Figure 4, B and C) and apical junctional complexes (Figure 4, D and E) was also indistinguishable (Figure 4, D and E). Claudin-4, which is not confined to TJs in intestinal epithelia and is expressed almost exclusively in villi (Rahner et al., 2001), was deeply down-regulated in the KO (Figure 3). Finally, as expected, the homologue of Drosophila lethal giant larvae, LLGL2, a known aPKC target (Kjaer et al., 2013), changed its distribution from diffuse cytoplasmic signal in control enterocytes to cortical, including the apical pole in KO (Figure 3).
FIGURE 3:

Apicobasal polarity and junction markers in Prkciflox/flox Vil-CRE± (KO) mice. Frozen sections of villus enterocytes from KO and control littermates (lm) were processed with fluorescent phalloidin or the following antibodies: Na+/H+-exchanger 3 (NHE3), claudin-4, occludin, and the human homologue of Drosophila lethal giant larvae protein 2 (LLGL2). Arrow points at increased lateral F-actin signal. Bars, 10 μm, except claudin-4, 25 μm.
FIGURE 4:

Apical and junction markers and intestinal epithelium ultrastructure in Prkciflox/flox Vil-CRE± (KO) mice. (A) Frozen sections of villus enterocytes from KO and control littermates (lm) were processed with the following antibodies: alkaline phosphatase (iAP), active (pT567) ezrin, ezrin, ZO-1, and human homologue of Par3. Ezrin inset, cross sections of the crypts displaying normal apical ezrin levels. Bars, 10 μm (iAP, pT567ezrin), 45 μm (ezrin), 25 μm (ZO-1, Par3). (B–E) Electron microscopy images of villus enterocytes from control (B, D) or KO (C, E) animals. Bars, 10 μm (B, C), 0.5 μm (D, E).
Both total ezrin and p-T567 ezrin signals were significantly decreased in the KO villus enterocytes, with the exception of positive patches (Figure 4A). However, ezrin was normally localized to the apical domain in the crypts (Figure 4A, inset). This is consistent with our finding that PKCι phosphorylates ezrin in cultured intestinal Caco-2 cells (Wald et al., 2008). Because this result seems to contradict the normal ultrastructure of brush borders in KO enterocytes (Figure 4C), we further quantified apical and cytoplasmic ezrin signals in confocal images. There was a 15-fold decrease in average pixel intensity for the brush border (Figure 5, A and B, black bars; note the difference in the pixel-value scales) in the KO enterocytes. However, despite the fact that it is difficult to see when the images are acquired at the same gain, there was a significant remnant of apical ezrin, well above the background level in the KO (Figure 5B). We conclude that a substantial fraction of T567 ezrin phosphorylation depends on aPKC activity.
FIGURE 5:
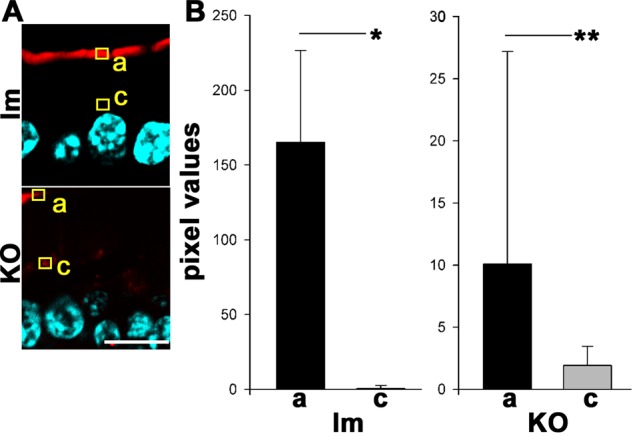
Quantification of ezrin distribution in control littermates (lm) or Prkciflox/flox Vil-CRE + (KO) villus enterocytes. (A) Pixels in the red channel of images of frozen sections of villus enterocytes showing ezrin distribution as in Figure 4 were quantified in ROIs randomly located at the apical edge of cells (a) or in the supranuclear cytoplasm (c). Enterocytes were randomly selected in the blue channel at the distal half of the villus but not at the tip. Bar, 10 μm. (B) Quantification of pixel intensities for pT567 ezrin signal in the apical pole (a; black bars) or supranuclear cytoplasm (c; gray bars) ROIs in enterocytes at the distal half of villi. Control, n = 20; KO, n = 56; t test, *p < 0.0001, **p < 0.05.
With regard to basolateral markers, except for LLGL2 (Figure 3), we did not find any mistargeting to the apical domain. Na+-K+ATPase images showed variability (Figure 6A) but no apical localization. We also analyzed another basolateral marker, E-cadherin (Figure 6B), and reached a similar conclusion. Finally, we determined pixel values in the basolateral domain for both markers (after background subtraction; Figure 6C). The results indicate that there were no significant differences in the E-cadherin signal and a small but significant increase in Na+-K+ATPase signal in the KO mice. In summary, the phenotype of basolateral markers in Prkciflox/flox Vil-CRE+ villus enterocytes is complex. There are differences in the steady-state levels of expression of different markers: claudin-4 decreases considerably, E-cadherin does not change, and Na+-K+ATPase slightly increases in the KO. Therefore it is difficult to conceive that a single mechanism can account for all these observations. In addition, considering the normal images of the junctions (Figure 4, B–E), we conclude that there was no global disruption of the lateral domain in the KO enterocytes, although there were changes in the expression of some specific basolateral proteins.
FIGURE 6:

Expression of basolateral markers in control littermates (lm) or Prkciflox/flox Vil-CRE± (KO) villus enterocytes. Frozen sections of villus enterocytes were processed with (A) anti–Na+-K+ATPase antibody or (B) anti–E-cadherin antibodies. Examples of typical ROIs used for quantification of pixel values for basolateral signal (b) or cytoplasmic background (c) are shown. Bars, 20 μm. (C) Pixel values per cell (ROIs b and c) in littermates (black bars) or KO (gray bars) for Na+-K+ATPase or E-cadherin. In each case, the results are from three animals; Na+-K+ATPase, n = 41; E-cadherin, n = 30; * p < 0.05.
aPKC is an inhibitor of NF-κB activity in vivo
Because PKCι overexpression inhibits NF-κB activity in the Caco-2 (intestinal epithelia) cell line (Forteza et al., 2013), we hypothesized that the conditional PKCι KO would display intestinal NF-κB activation and, possibly, inflammation. Up to ∼30% of the animals developed anal prolapse after 5 mo of age, which suggested the existence of a mild colitis. A role of PKCι in colitis has been shown (Calcagno et al., 2011). Therefore we wanted to determine whether PKCι-dependent pathways inhibit NF-κB in the absence of or with minimal external proinflammatory stimuli. Accordingly, we focused our attention on the small intestine, where there are small microbial loads and fewer infiltrating myeloid cells than in the colon and local inflammation levels are generally low. Studies in the large intestine and ileum are more common and clinically relevant due to the localization of lesions in IBD. However, the complexity of the interactions among microbiota, epithelia, and immune cells in the colon obscures mechanistic conclusions. Specifically, we analyzed villus enterocytes in which the aPKC defect was complete (Figure 1).
To test the hypothesis, we used various independent criteria of NF-κB activation in conditional PKCι-knockout mouse enterocytes. First, we examined NF-κB nuclear translocation. In control (lm) duodenum villus enterocytes, relA(p65) signal was found excluded from the nuclei. Conversely, Prkciflox/flox Vil-CRE+ (KO) enterocytes showed nuclear relA (Figure 7A). The average ratio of nuclear/cytoplasmic relA signal doubled in KO enterocytes, and the difference was significant (Figure 7B). Second, we semiquantitatively measured pSer536 relA signal in epithelial and lamina propria connective tissue (examples of regions of interest [ROIs] in Figure 7C, yellow squares). The overall epithelial signal was lower than the lamina propria signal in control cells (lm) but showed similar intensities in KO (Figure 7, C and D). In other words, the levels of phospho-relA signal in the lamina propria were indistinguishable between control and KO, but the levels in epithelial cells significantly increased threefold in the KO enterocytes. As a negative control, we treated the animals with SS and observed minimal levels of phospho-relA signal (Figure 7, C and D).
FIGURE 7:

Activation of NF-κB in of Prkciflox/flox Vil-CRE± (KO) villus enterocytes. (A) Frozen sections of villus enterocytes from untreated KO or littermate control (Prkciflox/flox Vil-CRE- or Prkciflox/− Vil-CRE+; lm) animals processed with anti-relA(p65) (red channel) and DAPI (blue channel). (B) Quantification of the data in A. Average pixel values were measured in ROIs in the cytoplasm or the nucleus. Several cells were measured in four to seven random sections. Each dot represents the average of all of the measured cells from one animal. Horizontal bars show averages of each group. *Kruskal–Wallis test, H0 = control and KO populations are equal, p < 0.003. (C) Frozen sections of intestinal villi were processed with anti-pSer536 relA (red channel) antibody. Animals were untreated controls (lm) or KO, as well as KO mice treated with ROCK inhibitor Y27628 or SS. Yellow squares show examples of randomly located ROIs in epithelia or lamina propria cells used for measurements in D. (D) Quantification of pixel values in images like those in C using ROIs in epithelial cells (black bars) or in lamina propria cells (gray bars) for control littermate (lm) or KO animals or KO animals treated with Y27628 or SS. Data shown as average and SD. *t test for difference between average in untreated KO epithelial cells (n = 22) and corresponding controls (n = 28) or between untreated KO epithelial cells and KO treated with Y27628 (n = 29) or between untreated lamina propria cells in KO compared with SS-treated cells, p < 0.001. (E) The sections were processed with anti-ROCK1/2 antibody. Bars, 10 μm.
Atypical PKC phosphorylates Rho-associated protein kinase (ROCK) in S1333, T1334, T1337, and S1341. These residues comprise the ROCK Rho-PH binding domain. Phosphorylation renders ROCK inactive and detached from the membrane (Ishiuchi and Takeichi, 2011). In turn, it was shown that ROCK activates IKK (Guo et al., 2012; Kim et al., 2014). To test the hypothesis that aPKC acts on NF-κB activation pathway through ROCK, we determined the subcellular distribution of ROCK1/2. As expected from evidence in MDCK cells (Ishiuchi and Takeichi, 2011), ROCK cortical and supranuclear distribution increased dramatically in KO enterocytes, but no changes were noted in lamina propria cells (Figure 7E). It is of note that the increase in ROCK cortical localization was more prominent in the apical pole but was also observed in the basolateral domain (Figure 7E). Consistent with this result, the ROCK inhibitor Y27632 significantly rescued >50% of the increase in phospho-Ser536 relA in the KO enterocytes, with little effect on the same signal in lamina propria cells (Figure 7, C and D). These results suggest that ROCK1/2 may be involved in NF-κB activation in aPKC-deficient enterocytes. We speculate that the widespread subcellular distribution of potentially active ROCK may multiply the effect of highly localized aPKC.
The third criterion to assess NF-κB activity was nonmuscle myosin II light chain phosphorylation (pMLC). NF-κB increases myosin light chain kinase (MLCK) transcription (He et al., 2012), which phosphorylates myosin II regulatory light chain (MLC), enabling assembly of functional nonmuscle myosin II (Graham et al., 2006). Phosphorylation of MLC by MLCK in enterocytes is a hallmark of intestinal inflammation and inflammation-associated carcinogenesis (Suzuki et al., 2014). pMLC signal was consistently higher in all KO animals studied (Figure 8). Treatment of KO animals with SS abrogated pMLC signal increase. These results independently suggest NF-κB activation in Prkciflox/flox Vil-CRE+ enterocytes. It is important to note that activation of myosin II is considered a major cause of TJ opening and barrier permeabilization (Al-Sadi et al., 2010), which is consistent with the results in Figure 2C.
FIGURE 8:
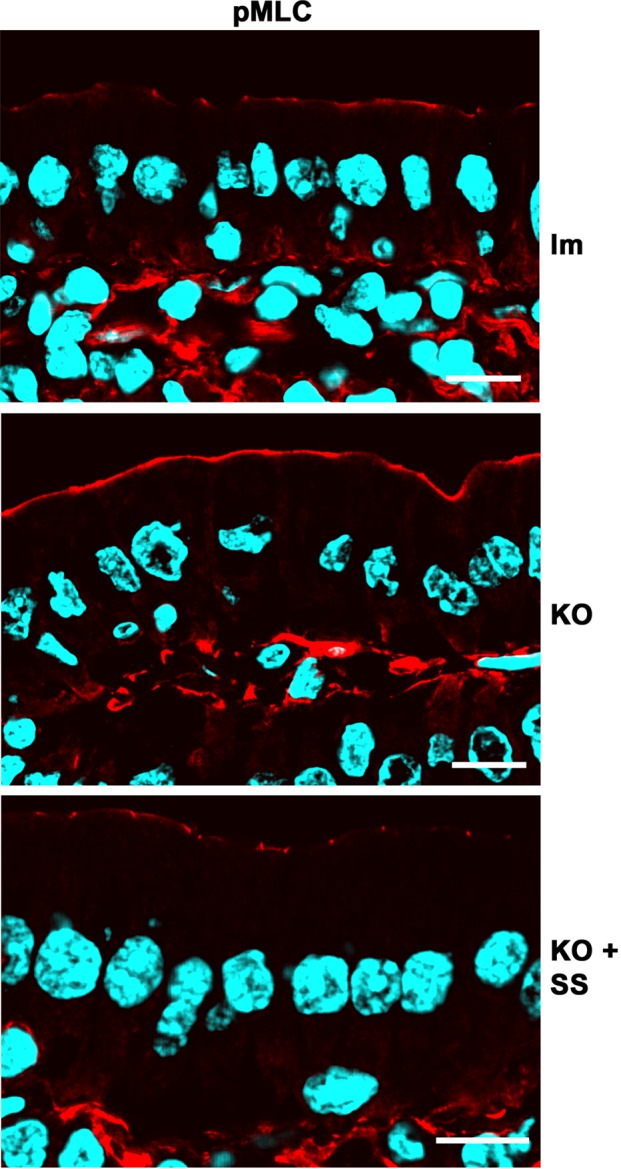
Effects of NF-κB activation in Prkciflox/flox Vil-CRE± (KO) villus enterocytes. Frozen sections of villi from control (lm), KO, or KO mice treated with sulfasalazine SS were processed with anti–phospho Ser-21 nonmuscle myosin II light chain (pMLC, red channel) antibody. Note that changes in signal were observed in epithelia and not in lamina propria cells. Bar, 10 μm.
Finally, to assess directly NF-κB transcriptional activity, we measured CXCL-1 (GRO-1) and CXCL-2 (GRO-2) mRNAs, broadly accepted as NF-κB target genes (El-Guendy and Sinai, 2008; Patterson et al., 2013). Reverse transcription-PCR (RT-PCR) was performed on RNA extracts from purified enterocytes. Both cytokines were found to be significantly increased (ninefold and fivefold, respectively) in KO enterocytes. However, KO animals treated with SS or Y27628 did not show increased CXCL-1 mRNA levels (Figure 9). These data provide additional evidence for NF-κB activation in the absence of PKCι, likely through the canonical IKK pathway via ROCK.
FIGURE 9:

Cytokine mRNA expression from isolated small intestine enterocytes from Prkciflox/flox Vil-CRE± (KO). Each data set comprises mRNAs from two different groups of animals for control littermates (lm) and KO, except for CXCL-1, which also comprises animals treated with oral SS or intraperitoneal Y27628. For each group, values were normalized to one control animal (lm). Horizontal lines represent average. Vimentin and IL-1β mRNA were measured in preparations of isolated enterocytes (E) or spleen (S) from the same animals to assess connective tissue contamination of the epithelium preparations. Kruskal–Wallis test, H0 = control lm and KO populations are equal, p < 0.005 (CXCL-1), p < 0.05 (CXCL-2), p < 0.05 (IL-1α), SS or Y27628 groups, not significant. For vimentin and IL-1b, H0 = spleen data are equal to enterocytes, p < 0.001.
Two other cytokines have been found to be essential in the development of intestinal inflammation: interleukin 1α (IL-1α) and IL-10. The former is proinflammatory, and the latter is anti-inflammatory (Kole and Maloy, 2014; Scarpa et al., 2015). These cytokines have been commonly studied in myeloid cells but are known to be secreted by the intestinal epithelium as well (Bersudsky et al., 2014; Olszak et al., 2014; Scarpa et al., 2015). IL-1α mRNA was significantly increased in the KO. Conversely, IL-10 in the KO was found to be ∼25% the levels of the control animals, a significant reduction (Figure 9). It is worth highlighting that these two cytokines are not known to be directly under NF-κB transcriptional control. The possibility that these results may be due to a contamination with cells of mesenchymal lineage, including myeloid cells, was controlled by measuring mRNAs that are not expressed in epithelial cells. Spleen parenchymal cells (S) were used as a positive control of mesenchymal/myeloid lineage cells and were compared with purified enterocytes (E) from the same animal (Figure 9). Two mRNAs not expressed in epithelial cells were used. Vimentin mRNA showed a 200-fold enrichment of epithelial over mesenchymal RNAs in our preparations. IL-1β mRNA is transcribed at lower levels than cytoskeletal proteins mRNAs. By in situ hybridization, it has been shown that IL-1β mRNA is expressed in lamina propria but not in epithelial cells in the intestine (Woywodt et al., 1999). Because IL-1β expression was 400-fold higher in a spleen cells compared with enterocytes, we further concluded that the contamination of our purified enterocyte mRNA with RNAs from other cells was minimal. These data support the conclusion that basal (unstimulated) cytokine expression in Prkciflox/flox Vil-CRE+ enterocytes is proinflammatory via NF-κB and possibly other, unidentified pathways as well.
DISCUSSION
This work focused on innate immunity activity in epithelial cells under minimal exogenous proinflammatory signaling: The goal was to identify epithelium-specific functions of polarity complex aPKC. The evidence leads to two conclusions. First, aPKC is dispensable for the maintenance of apicobasal polarity in the short term, that is, the 2-d lifespan of postmitotic villus enterocytes. Second, aPKC is indeed a negative regulator of NF-κB activity in enterocytes. The latter is a tissue-specific effect of aPKC and indicates that results from other cell types, in which aPKC activates NF-κB, cannot be generalized to epithelia. Furthermore, although there are commonalities between epithelia in the small and large intestine, the conclusions from this work should not be generalized to colon epithelium.
Apical polarity persists in PKCι KO enterocytes after PKCζ is spontaneously down-regulated at the crypt/villus boundary for the short lifespan of the villus enterocytes. The Prkciflox/flox Vil-CRE animals showed normal weight gains, in stark contrast with the phenotype of mutations disturbing apical membrane traffic, such as Rab8 (Sato et al., 2007) and Myo5b (Carton-Garcia et al., 2015) KO models. Even defects in single apical transporters, such as the GLUT8 transporter, have been shown to affect body weight (DeBosch et al., 2012). Overall, therefore, nutrient absorption in PKCι KO animals must be normal. On the other hand, changes in the distribution of direct aPKC targets such as LLGL2 and ROCK were observed, confirming the loss of aPKC activity.
The loss of ezrin phosphorylation in the PKCι KO supports our previous observation in Caco-2 cells that PKCι phosphorylates ezrin (Wald et al., 2008). The rapid turnover of the ezrin activation phosphosite (T567; Viswanatha et al., 2012) is consistent with dephosphorylation immediately after PKCζ is down-regulated in the PKCι KO enterocytes. This issue has been controversial: Mst4 was also shown to phosphorylate ezrin in intestinal epithelia (ten Klooster et al., 2009). Both splice variants of LKB1 are activated by aPKC (Zhu et al., 2013). Therefore the results shown here demonstrate that PKCι is key for ezrin phosphorylation, either directly or indirectly via the LKB1-Mst4 pathway, and are consistent with observations in other systems (Liu et al., 2013).
It may be counterintuitive that KO enterocytes still show a normal brush border (Figure 4C). In ezrin-knockout mice, enterocytes lacking ezrin throughout their differentiation still show microvilli, albeit ones that are shorter and more disorganized (Saotome et al., 2004). PKCι KO enterocytes, on the other hand, show a 15-fold decrease in apical ezrin but still display a sizable apical accumulation of ezrin. Considering that the ezrin decrease is short-lived due to the limited lifespan of the cells, it is not surprising that the brush border structure is not affected. In summary, the data suggest that persistence of a polarized phenotype may be more related to the temporal stability of the structure. In other words, the PKCι-defective enterocytes would display a phenotype only if the dephosphorylation of aPKC targets and its effects appear within a 2-d period. Conversely, Par3 localizes in an aPKC-independent manner through the NTD and PDZ domains (Chen and Zhang, 2013). In fact, Par3 localization to the adherens junctions drives aPKC-Par6 localization to the TJ (Horikoshi et al., 2009). Therefore, predictably, Par3 remained polarized in the absence of aPKC activity.
Despite the normal appearance of ZO-1 and occludin, the decreased expression of claudin-4 may partially explain the increased solute permeability in PKCι-deficient enterocytes: in lung cells, claudin-4 knockout increases solute permeability with no changes in ion permeability (Kage et al., 2014). In addition, claudin-4 is down-regulated in ulcerative colitis (Oshima et al., 2008). On the other hand, innate immunity mechanisms increase TJ permeability by multiple mechanisms, including NF-κB–dependent assembly of myosin II via MLCK activation (Pasparakis, 2012) and interferon γ–dependent pathways (Boivin et al., 2009). In addition, PKCι has been shown to modulate claudin-2 as well (Lu et al., 2015). Finally, there is extensive evidence that ROCK activation mediates opening of TJs (Ivanov et al., 2009; Clark et al., 2015) by inhibiting MLC dephosphorylation (Kaneko-Kawano et al., 2012). The results from the PKCι KO mouse presented here strongly suggest that, in addition to “local” regulation of nonmuscle myosin II via ROCK at the junctional level, aPKC exerts control over the canonical NF-κB activation pathway and, accordingly, is involved in transcriptional control. All of these non–mutually exclusive mechanisms can explain the increase in TJ permeability.
Of importance, although a threefold increase in solute permeability seems modest, it mimics the levels of increased permeability in IBD patients in remission or in asymptomatic relatives (Buhner et al., 2006), including the barrier permeability in the small intestine (Buning et al., 2012). Accordingly, we speculate that the conditional PKCι-null mouse model might be clinically relevant to mimic increased barrier permeability close to the threshold that triggers colitis.
aPKC defect increases NF-κB activation
The evidence presented here strongly supports activation of NF-κB in epithelial cells within an environment with low levels of inflammatory stimuli as compared with the colon. Bearing in mind that in other cell types, aPKC activates NF-κB (Diaz-Meco and Moscat, 2012), the identity of the pathway(s) connecting a highly localized aPKC to inhibition of NF-κB activation is important. Two pieces of evidence suggest that the effect must be exerted on the canonical NF-κB activation pathway. First, expression of constitutively active PKCι results in IkB accumulation (Forteza et al., 2013). Second, PKCι deficiency increased pSer 536 relA signal (Figure 7, C and D), an effect rescued by two different small molecules (SS and Y27632), which both inhibit IKKs directly or indirectly via ROCK. On the other hand, the transcriptional effects of an aPKC defect on IL-1α and IL-10 suggest that additional pathways might be involved. Evidence supports the possibility that aPKC might signal through STAT3 (Guyer and Macara, 2015), which in turn activates IL-10. Moreover, aPKC also signals through CREB-binding protein (CBP) by direct phosphorylation in S436 (He et al., 2009). In turn, CBP is involved in the control of NF-κB and IL-10 transcription (Alvarez et al., 2009; Mukherjee et al., 2013). Dissecting all of these possible mechanisms is beyond the scope of this work.
Evidence from other laboratories has been contradictory regarding the effects of the Rho pathway in NF-κB activation. However, there is support for the notion that RhoA potentiates NF-κB activation (Perez-Moreno et al., 2006; Bist et al., 2015). On the basis of the poor effect of Y27632 on phospho-relA in lamina propria cells, we speculate that this pathway of aPKC inhibition of ROCK, which in turn activates IKK (Anwar et al., 2004; Kim et al., 2014), might provide clues for the tissue specificity of the effect of PKCι on NF-κB. Additional studies are necessary to further test this hypothesis.
Recently Guyer and Macara (2015) confirmed our original observation that Par3 is also an inhibitor of NF-κB (Forteza et al., 2013) in carcinoma cells. The data presented here, however, challenge the notion that loss of Par3 activates NF-κB through the activation of aPKC. The results in this work, conversely, support our interpretation that the anti–NF-κB activity of Par3 is exerted independently of aPKC (Forteza et al., 2013). It is possible that the differences arise from the use of different cell types in culture in the cited work and the analysis in vivo in this work.
In summary, PKCι expression in differentiated enterocytes is critical to control transcription factors that mediate effects on MLC and barrier permeability (NF-κB), proinflammatory cytokine transcription inhibition (CXCL-1 and -2, IL-1α), and anti-inflammatory cytokine expression (IL-10, possibly through STAT3 or CBP). All of these mechanisms seem to be additional to apicobasal polarity functions and sufficient to predispose the intestinal mucosa to inflammation. It seems reasonable to speculate that this function might be general to polarized epithelia under conditions of low inflammatory stimulation. Whether this mechanism interacts with, for example, activated TLR4 or TNFR pathways remains to be determined. However, the down-regulation of aPKC during intestinal inflammation is likely to contribute to the intensity or persistence of the inflammatory response. The data presented here add to a growing body of evidence (Bersudsky et al., 2014; Olszak et al., 2014; Scarpa et al., 2015) that intestinal epithelial cells are important regulators of inflammation, not only as a passive barrier, but also through the secretion of cytokines.
MATERIALS AND METHODS
Animal model
All studies in animals were conducted according to guidelines in the Public Health Service Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the local Institutional Animal Care and Use Committee. Embryonic stem cells carrying the recombinant insertions in the Prkci gene shown in Figure 1A were obtained from the International Mouse Knockout Consortium (www.mousephenotype.org/; Project 71671). The cells were selected, cloned, and injected into mouse blastocysts in the C57BL/6 genomic background. For genotyping, we used the following primers (Figure 1A, GenBank accession JN952602.1):
14729: CAGGCCCTCTGCAGAGATAGTTC
21131: GGCTGCATACGCTTGATCCG
22182: CCAACAAAAGTCCCTCTCCCCT
Genotyping PCR was performed as follows: primers 2 and 3 were used for germline transmission, amplimer 1051 base pairs; primers 1–3 yielded a 350–base pair amplimer in WT and a 550–base pair amplimer in the flox insertion. After germline transmission was verified, the animals were bred with B6(C3)-Tg(Pgk1-FLPo)10Sykr/J mice (Jackson Laboratory) to remove the FRT insert. After removal of the Neo cassette, animals carrying the flox insertions flanking exon 4 were backcrossed with WT C57BL/6 mice for five generations. Finally, these animals were further bred with B6.Cg-Tg(Vil-cre)997Gum/J (Jackson Laboratory). Prkciflox/− Vil-CRE+ or Prkciflox/flox littermates were used as controls. These animals were cohoused with the Prkciflox/flox Vil-CRE+ animals. Expression of PKCι was verified independently by immunofluorescence in each animal.
Reagents
Reagents used were as follows: Texas red–dextran (3000 Da; Invitrogen, Carlsbad, CA), Alexa Fluor 546–phalloidin (Invitrogen), SS (Fluka, Sigma-Aldrich, St. Louis, MO), and Y27632 (Tocris, R&D Systems, Minneapolis, MN). The antibodies used in this work were PKCζ (directed against a synthetic peptide comprising amino acids 174–203 of the human orthologue; human–mouse similarity in this region, 100%; identity, 90%; ThermoFisher, Waltham, MA), PKCι (5282; Abcam, Cambridge, MA), pT555 PKCζ (GeneTex, Irvine, CA), CD68 (Bio-Rad, Hercules, CA), ezrin (Abcam), pT567 ezrin (Cell Signaling, Danvers, MA), intestinal alkaline phosphatase (iAP; Accurate), ZO-1 (Invitrogen), Par3 (Millipore, Billerica, MA), NF-κB p65 relA (Cell Signaling), occludin (ThermoFisher), NHE3 (Abcam), claudin-4 (Invitrogen), LLGL2 (Bioss, Woburn, MA), Na+-K+ ATPase (Accurate, Westbury, NY), pSer21-MLC (Abcam), pSer 536 relA (Invitrogen), and ROCK1/2 (Assay Biotech, Sunnyvale, CA).
Intestinal permeability
The animals were food starved overnight but allowed free access to water. Then they received 100 μl of 4 mg/ml 3-kDa Texas red–dextran via stomach gavage. After 3 h, the animals were killed by deep anesthesia and exsanguination. In all cases, a Texas red–dextran bolus was macroscopically localized to the small intestine after death. Samples of the blood were anticoagulated with 2 mM EDTA, and plasma was separated by centrifugation. Texas red fluorescence was measured with a SpectraMax Gemini EM fluorometer (Molecular Devices) after dilution. Background in plasma from animals not receiving fluorescent dextran was subtracted. The results from different groups of animals are expressed as fluorescence relative to one control animal in the same group.
Frozen sections, immunofluorescence, and electron microscopy
Methods for these techniques have been published (Ameen et al., 2001; Mashukova et al., 2012). Immunofluorescence using phosphoepitope antibodies was performed after fixation in trichloroacetic acid (Hayashi et al., 1999). All immunofluorescence images were collected with a Leica SP5 confocal microscope, and semiquantitative image analysis was performed with Leica software as described later. Electron microscope samples were prepared by standard techniques, and images were obtained with a Jeol JEM-1400 instrument, using Gatan software.
For fluorescence image analysis, pixel quantification was performed as described previously (Wald et al., 2011). Briefly, images were collected, avoiding pixel saturation in the channel to be measured. ROIs matching the size of positive signal images were defined and used throughout the sampling. Examples of ROIs are shown in Figures 5A, 6A, and 7C (yellow squares). The analyses were conducted blindly, and ROIs were positioned on the desired region of cells (e.g., apical edge, cytoplasm, or nucleus) in the 4′,6-diamidino-2-phenylindole (DAPI) channel to randomize the sampling. One average of pixel values in the ROI per cell was collected and used for statistics, which comprised several cells from different animals.
RT-quantitative PCR
Immediately after euthanasia, small intestine epithelial cells were purified by extraction in EDTA-dithiothreitol as described (McNicholas et al., 1994), with five to eight cycles of centrifugation and resuspension to wash mucus and separate nonepithelial cells. Spleen parenchymal cells were obtained by cutting the capsule with a razor blade in phosphate-buffered saline and shaking the organ with tweezers to separate the capsule. The resulting cell suspension was pipetted several times to separate debris. RNA was purified using mechanical homogenization by passing samples through needles 18G11/2 and 20G11/2 several times and TRIzol Reagent (Ambion Thermo Fisher Scientific). cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR amplification was performed using TaqMan gene expression assays (Life Technologies), including gene-specific primer and probe sets specifically designed against our molecules of interest (Mm04207460_m1 for CXCL-1, Mm00436450_m1 for CXCL-2, Mm00434151_m1 for IL-10, Mm00439620_m1 for IL-1α, Mm01333430-m1 for vimentin, and Mm00439620-m1 for IL-1β). The relative quantification method (ΔΔCt) was used for analysis of mRNA fold change using endogenous control glyceraldehyde-3-phosphate dehydrogenase for normalization.
Sulfasalazine and Y27632 treatments
Sulfasalazine was dissolved at 37°C in tap water supplemented with 50 mM sucrose (for taste). The pH was titered to 7.0 with NaOH. Accordingly, the SS vehicle for controls was tap water supplemented with 5 mM NaCl and 50 mM sucrose. The animals were allowed to drink ad libitum.
The Y27632 treatment was slightly modified from published protocols. The dose was at the low range of standard doses (2 mg/kg; Liao et al., 2013) and repeated twice a day. The rationale for this dosing was based on published bioavailability studies showing Y27632 half-life of ∼90 min after intraperitoneal administration (Li et al., 2009). We aimed at peak plasma concentrations of ∼1 μM. In addition, Y27632 doses were calculated to be above the EC50 of 0.14 μM for ROCK (Routhier et al., 2010) for much of the day, thus justifying two injections per day. At concentrations >20 μM, Y27632 blocks other kinases (Routhier et al., 2010), so higher doses were avoided. In summary, Y27628 was injected intraperitoneally, at 2 mg/kg twice a day.
Statistics
For normal distribution values, statistical significance was determined by Student’s t test. For data not conforming to normal distribution, the nonparametric Kruskal–Wallis test was used.
Acknowledgments
We thank Yingcai Wang, Transgenic Animal Core Facility, for embryonic stem cell handling and blastocyst injections and Margaret Bates (Miami Project to Cure Paralysis) for skillful handling of electron microscopy specimens. We are especially thankful to Robert Warren for critically reading the manuscript. This work was supported by National Institutes of Health Grant R01-DK076652 to P.J.S. and a Nova Southeastern University Health Professions Research Grant to A.M. R.F. was a recipient of National Institutes of Health Ruth L. Kirschstein National Research Service Award Fellowship F32-DK095503.
Abbreviations used:
- aPKC
atypical protein kinase C
- CBP
CREB-binding protein
- IBD
inflammatory bowel disease
- IL
interleukin
- LLGL2
homologue of Drosophila lethal giant larvae
- MLC
myosin II regulatory light chain
- NF-kB
nuclear factor kappa beta
- NHE3
sodium-proton exchanger 3
- Par
homologues of partition-deficient mutations in C. elegans
- Prkci
murine gene encoding PKC lambda
- ROCK
Rho-associated protein kinase
- SS
sulfasalazine
- TJ
tight junction
- TLR4
Toll-like receptor 4
- TNFR
tumor necrosis factor receptor
- Vil
villin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0086) on May 25, 2016.
REFERENCES
- Al-Sadi R, Ye D, Said HM, Ma TY. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am J Pathol. 2010;177:2310–2322. doi: 10.2353/ajpath.2010.100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y, Municio C, Alonso S, Sanchez Crespo M, Fernandez N. The induction of IL-10 by zymosan in dendritic cells depends on CREB activation by the coactivators CREB-binding protein and TORC2 and autocrine PGE2. J Immunol. 2009;183:1471–1479. doi: 10.4049/jimmunol.0900312. [DOI] [PubMed] [Google Scholar]
- Ameen NA, Figueroa Y, Salas PJ. Anomalous apical plasma membrane phenotype in CK8-deficient mice indicates a novel role for intermediate filaments in the polarization of simple epithelia. J Cell Sci. 2001;114:563–575. doi: 10.1242/jcs.114.3.563. [DOI] [PubMed] [Google Scholar]
- Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol. 2004;173:6965–6972. doi: 10.4049/jimmunol.173.11.6965. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay G, Standaert ML, Sajan MP, Kanoh Y, Miura A, Braun U, Kruse F, Leitges M, Farese RV. Protein kinase C-lambda knockout in embryonic stem cells and adipocytes impairs insulin-stimulated glucose transport. Mol Endocrinol. 2004;18:373–383. doi: 10.1210/me.2003-0087. [DOI] [PubMed] [Google Scholar]
- Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I, Aychek T, Dinarello CA, et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut. 2014;63:598–609. doi: 10.1136/gutjnl-2012-303329. [DOI] [PubMed] [Google Scholar]
- Bist P, Phua QH, Shu S, Yi Y, Anbalagan D, Lee LH, Sethi G, Low BC, Lim LH. Annexin-A1 controls an ERK-RhoA-NFkappaB activation loop in breast cancer cells. Biochem Biophys Res Commun. 2015;461:47–53. doi: 10.1016/j.bbrc.2015.03.166. [DOI] [PubMed] [Google Scholar]
- Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J Interferon Cytokine Res. 2009;29:45–54. doi: 10.1089/jir.2008.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, Kuechler I, Krueger S, Schmidt HH, Lochs H. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation. Gut. 2006;55:342–347. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buning C, Geissler N, Prager M, Sturm A, Baumgart DC, Buttner J, Buhner S, Haas V, Lochs H. Increased small intestinal permeability in ulcerative colitis: rather genetic than environmental and a risk factor for extensive disease. Inflam Bowel Dis. 2012;18:1932–1939. doi: 10.1002/ibd.22909. [DOI] [PubMed] [Google Scholar]
- Calcagno SR, Li S, Shahid MW, Wallace MB, Leitges M, Fields AP, Murray NR. Protein kinase C iota in the intestinal epithelium protects against dextran sodium sulfate-induced colitis. Inflam Bowel Dis. 2011;17:1685–1697. doi: 10.1002/ibd.21547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carton-Garcia F, Overeem AW, Nieto R, Bazzocco S, Dopeso H, Macaya I, Bilic J, Landolfi S, Hernandez-Losa J, Schwartz S, et al. Myo5b knockout mice as a model of microvillus inclusion disease. Sci Rep. 2015;5:12312. doi: 10.1038/srep12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang M. The Par3/Par6/aPKC complex and epithelial cell polarity. Exp Cell Res. 2013;319:1357–1364. doi: 10.1016/j.yexcr.2013.03.021. [DOI] [PubMed] [Google Scholar]
- Clark PR, Kim RK, Pober JS, Kluger MS. Tumor necrosis factor disrupts claudin-5 endothelial tight junction barriers in two distinct NF-kappaB-dependent phases. PLoS One. 2015;10:e0120075. doi: 10.1371/journal.pone.0120075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch BJ, Chi M, Moley KH. Glucose transporter 8 (GLUT8) regulates enterocyte fructose transport and global mammalian fructose utilization. Endocrinology. 2012;153:4181–4191. doi: 10.1210/en.2012-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Meco MT, Moscat J. The atypical PKCs in inflammation: NF-kappaB and beyond. Immunol Rev. 2012;246:154–167. doi: 10.1111/j.1600-065X.2012.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood GL. Gastrointestinal epithelial renewal. Gastroenterology. 1977;72:962–975. [PubMed] [Google Scholar]
- El-Guendy N, Sinai AP. Potential problems inherent in cell-based stable NF-kappaB-GFP reporter systems. Mol Cell Biochem. 2008;312:147–155. doi: 10.1007/s11010-008-9730-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Lee MC, Sajan MP. Hepatic atypical protein kinase C: an inherited survival-longevity gene that now fuels insulin-resistant syndromes of obesity, the metabolic syndrome and type 2 diabetes mellitus. J Clin Med. 2014;3:724–740. doi: 10.3390/jcm3030724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Sajan MP. Metabolic functions of atypical protein kinase C: “good” and “bad” as defined by nutritional status. Am J Physiol Endocrinol Metab. 2010;298:E385–E394. doi: 10.1152/ajpendo.00608.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forteza R, Wald FA, Mashukova A, Kozhekbaeva Z, Salas PJ. Par-complex aPKC and Par3 cross-talk with innate immunity NF-kappaB pathway in epithelial cells. Biol Open. 2013;2:1264–1269. doi: 10.1242/bio.20135918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, Lin A, Turner JR. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281:26205–26215. doi: 10.1074/jbc.M602164200. [DOI] [PubMed] [Google Scholar]
- Graybill C, Wee B, Atwood SX, Prehoda KE. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J Biol Chem. 2012;287:21003–21011. doi: 10.1074/jbc.M112.360495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ. Molecular genetics of asymmetric cleavage in the early Caenorhabditis elegans embryo. Curr Opin Genet Dev. 1996;6:408–415. doi: 10.1016/s0959-437x(96)80061-x. [DOI] [PubMed] [Google Scholar]
- Guo F, Zhou Z, Dou Y, Tang J, Gao C, Huan J. GEF-H1/RhoA signalling pathway mediates lipopolysaccharide-induced intercellular adhesion molecular-1 expression in endothelial cells via activation of p38 and NF-kappaB. Cytokine. 2012;57:417–428. doi: 10.1016/j.cyto.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Guyer RA, Macara IG. Loss of the polarity protein PAR3 activates STAT3 signaling via an atypical protein kinase C (aPKC)/NF-kappaB/interleukin-6 (IL-6) axis in mouse mammary cells. J Biol Chem. 2015;290:8457–8468. doi: 10.1074/jbc.M114.621011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yonemura S, Matsui T, Tsukita S. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J Cell Sci. 1999;112:1149–1158. doi: 10.1242/jcs.112.8.1149. [DOI] [PubMed] [Google Scholar]
- He F, Peng J, Deng XL, Yang LF, Camara AD, Omran A, Wang GL, Wu LW, Zhang CL, Yin F. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. 2012;59:264–272. doi: 10.1016/j.cyto.2012.04.008. [DOI] [PubMed] [Google Scholar]
- He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, Radovick S, Wondisford FE. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Suzuki A, Yamanaka T, Sasaki K, Mizuno K, Sawada H, Yonemura S, Ohno S. Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J Cell Sci. 2009;122:1595–1606. doi: 10.1242/jcs.043174. [DOI] [PubMed] [Google Scholar]
- Imai F, Hirai S, Akimoto K, Koyama H, Miyata T, Ogawa M, Noguchi S, Sasaoka T, Noda T, Ohno S. Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development. 2006;133:1735–1744. doi: 10.1242/dev.02330. [DOI] [PubMed] [Google Scholar]
- Ishiuchi T, Takeichi M. Willin and Par3 cooperatively regulate epithelial apical constriction through aPKC-mediated ROCK phosphorylation. Nat Cell Biol. 2011;13:860–866. doi: 10.1038/ncb2274. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Samarin SN, Bachar M, Parkos CA, Nusrat A. Protein kinase C activation disrupts epithelial apical junctions via ROCK-II dependent stimulation of actomyosin contractility. BMC Cell Biol. 2009;10:36. doi: 10.1186/1471-2121-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage H, Flodby P, Gao D, Kim YH, Marconett CN, DeMaio L, Kim KJ, Crandall ED, Borok Z. Claudin 4 knockout mice: normal physiological phenotype with increased susceptibility to lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L524–L536. doi: 10.1152/ajplung.00077.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko-Kawano T, Takasu F, Naoki H, Sakumura Y, Ishii S, Ueba T, Eiyama A, Okada A, Kawano Y, Suzuki K. Dynamic regulation of myosin light chain phosphorylation by Rho-kinase. PLoS One. 2012;7:e39269. doi: 10.1371/journal.pone.0039269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim JG, Moon MY, Park SH, Park JB. IkappaB kinase gamma/nuclear factor-kappaB-essential modulator (IKKgamma/NEMO) facilitates RhoA GTPase activation, which, in turn, activates Rho-associated KINASE (ROCK) to phosphorylate IKKbeta in response to transforming growth factor (TGF)-beta1. J Biol Chem. 2014;289:1429–1440. doi: 10.1074/jbc.M113.520130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaer S, Linch M, Purkiss A, Kostelecky B, Knowles PP, Rosse C, Riou P, Soudy C, Kaye S, Patel B, et al. Adenosine-binding motif mimicry and cellular effects of a thieno[2,3-d]pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem J. 2013;451:329–342. doi: 10.1042/BJ20121871. [DOI] [PubMed] [Google Scholar]
- Kole A, Maloy KJ. Control of intestinal inflammation by interleukin-10. Curr Top Microbiol Immunol. 2014;380:19–38. doi: 10.1007/978-3-662-43492-5_2. [DOI] [PubMed] [Google Scholar]
- Koo BK, Lim HS, Chang HJ, Yoon MJ, Choi Y, Kong MP, Kim CH, Kim JM, Park JG, Kong YY. Notch signaling promotes the generation of EphrinB1-positive intestinal epithelial cells. Gastroenterology. 2009;137:145–155. doi: 10.1053/j.gastro.2009.03.046. [DOI] [PubMed] [Google Scholar]
- Li M, Huang Y, Ma AA, Lin E, Diamond MI. Y-27632 improves rotarod performance and reduces huntingtin levels in R6/2 mice. Neurobiol Dis. 2009;36:413–420. doi: 10.1016/j.nbd.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Liao MH, Shih CC, Tsao CM, Chen SJ, Wu CC. RhoA/Rho-kinase and nitric oxide in vascular reactivity in rats with endotoxaemia. PLoS One. 2013;8:e56331. doi: 10.1371/journal.pone.0056331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wu Z, Shi X, Li W, Liu C, Wang D, Ye X, Liu L, Na J, Cheng H, et al. Atypical PKC, regulated by Rho GTPases and Mek/Erk, phosphorylates Ezrin during eight-cell embryocompaction. Dev Biol. 2013;375:13–22. doi: 10.1016/j.ydbio.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Liu KD, Datta A, Yu W, Brakeman PR, Jou TS, Matthay MA, Mostov KE. Rac1 is required for reorientation of polarity and lumen formation through a PI 3-kinase-dependent pathway. Am J Physiol Renal Physiol. 2007;293:F1633–F1640. doi: 10.1152/ajprenal.00053.2007. [DOI] [PubMed] [Google Scholar]
- Lu R, Dalgalan D, Mandell EK, Parker SS, Ghosh S, Wilson JM. PKCiota interacts with Rab14 and modulates epithelial barrier function through regulation of claudin-2 levels. Mol Biol Cell. 2015;26:1523–1531. doi: 10.1091/mbc.E14-12-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Martin P, Duran A, Minguet S, Gaspar ML, Diaz-Meco MT, Rennert P, Leitges M, Moscat J. Role of zeta PKC in B-cell signaling and function. EMBO J. 2002;21:4049–4057. doi: 10.1093/emboj/cdf407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashukova A, Forteza R, Wald FA, Salas PJ. PDK1 in apical signaling endosomes participates in the rescue of the polarity complex atypical PKC by intermediate filaments in intestinal epithelia. Mol Biol Cell. 2012;23:1664–1674. doi: 10.1091/mbc.E11-12-0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashukova A, Wald FA, Salas PJ. Tumor necrosis factor alpha and inflammation disrupt the polarity complex in intestinal epithelial cells by a posttranslational mechanism. Mol Cell Biol. 2011;31:756–765. doi: 10.1128/MCB.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas CM, Brown CD, Turnberg LA. Na-K-Cl cotransport in villus and crypt cells from rat duodenum. Am J Physiol. 1994;267:G1004–G1011. doi: 10.1152/ajpgi.1994.267.6.G1004. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Albert A, Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol Cell. 2006;23:631–640. doi: 10.1016/j.molcel.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013;11:e1001647. doi: 10.1371/journal.pbio.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen OH, Munck LK. Drug insight: aminosalicylates for the treatment of IBD. Nat Clin Pract Gastroenterol Hepatol. 2007;4:160–170. doi: 10.1038/ncpgasthep0696. [DOI] [PubMed] [Google Scholar]
- Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, Lin CS, Jobin C, Brand S, Sotlar K, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509:497–502. doi: 10.1038/nature13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, Miwa H, Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol. 2008;23(Suppl 2):S146–S150. doi: 10.1111/j.1440-1746.2008.05405.x. [DOI] [PubMed] [Google Scholar]
- Pasparakis M. Role of NF-kappaB in epithelial biology. Imm Rev. 2012;246:346–358. doi: 10.1111/j.1600-065X.2012.01109.x. [DOI] [PubMed] [Google Scholar]
- Patterson EK, Yao LJ, Ramic N, Lewis JF, Cepinskas G, McCaig L, Veldhuizen RA, Yamashita CM. Lung-derived mediators induce cytokine production in downstream organs via an NF-kappaB-dependent mechanism. Mediators Inflamm. 2013;2013:586895. doi: 10.1155/2013/586895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieczynski J, Margolis B. Protein complexes that control renal epithelial polarity. Am J Physiol Renal Physiol. 2011;300:F589–F601. doi: 10.1152/ajprenal.00615.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahner C, Mitic LL, Anderson JM. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology. 2001;120:411–422. doi: 10.1053/gast.2001.21736. [DOI] [PubMed] [Google Scholar]
- Routhier A, Astuccio M, Lahey D, Monfredo N, Johnson A, Callahan W, Partington A, Fellows K, Ouellette L, Zhidro S, et al. Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol Rep. 2010;23:861–867. [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell. 2004;6:855–864. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Sato T, Mushiake S, Kato Y, Sato K, Sato M, Takeda N, Ozono K, Miki K, Kubo Y, Tsuji A, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature. 2007;448:366–369. doi: 10.1038/nature05929. [DOI] [PubMed] [Google Scholar]
- Scarpa M, Kessler S, Sadler T, West G, Homer C, McDonald C, de la Motte C, Fiocchi C, Stylianou E. The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol. 2015;185:1624–1637. doi: 10.1016/j.ajpath.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Hirata M, Kamimura K, Maniwa R, Yamanaka T, Mizuno K, Kishikawa M, Hirose H, Amano Y, Izumi N, et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr Biol. 2004;14:1425–1435. doi: 10.1016/j.cub.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Nagaishi T, Yamazaki M, Onizawa M, Watabe T, Sakamaki Y, Ichinose S, Totsuka M, Oshima S, Okamoto R, et al. Myosin light chain kinase expression induced via tumor necrosis factor receptor 2 signaling in the epithelial cells regulates the development of colitis-associated carcinogenesis. PLoS One. 2014;9:e88369. doi: 10.1371/journal.pone.0088369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, Di Giacomo V, Colland F, de Koning J, Maurice MM, Hornbeck P, et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell. 2009;16:551–562. doi: 10.1016/j.devcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Umar S. Intestinal stem cells. Curr Gastroenterol Rep. 2010;12:340–348. doi: 10.1007/s11894-010-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanatha R, Ohouo PY, Smolka MB, Bretscher A. Local phosphocycling mediated by LOK/SLK restricts ezrin function to the apical aspect of epithelial cells. J Cell Biol. 2012;199:969–984. doi: 10.1083/jcb.201207047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl C, Liptay S, Adler G, Schmid RM. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Invest. 1998;101:1163–1174. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald FA, Forteza R, Diwadkar-Watkins R, Mashukova A, Duncan R, Abreu MT, Salas PJ. Aberrant expression of the polarity complex atypical PKC and non-muscle myosin IIA in active and inactive inflammatory bowel disease. Virchows Arch. 2011;459:331–338. doi: 10.1007/s00428-011-1102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald FA, Oriolo AS, Mashukova A, Fregien NL, Langshaw AH, Salas PJ. Atypical protein kinase C (iota) activates ezrin in the apical domain of intestinal epithelial cells. J Cell Sci. 2008;121:644–654. doi: 10.1242/jcs.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CK, Liptay S, Wirth T, Adler G, Schmid RM. Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology. 2000;119:1209–1218. doi: 10.1053/gast.2000.19458. [DOI] [PubMed] [Google Scholar]
- Woywodt A, Ludwig D, Neustock P, Kruse A, Schwarting K, Jantschek G, Kirchner H, Stange EF. Mucosal cytokine expression, cellular markers and adhesion molecules in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1999;11:267–276. doi: 10.1097/00042737-199903000-00010. [DOI] [PubMed] [Google Scholar]
- Zhu H, Moriasi CM, Zhang M, Zhao Y, Zou MH. Phosphorylation of serine 399 in LKB1 protein short form by protein kinase Czeta is required for its nucleocytoplasmic transport and consequent AMP-activated protein kinase (AMPK) activation. J Biol Chem. 2013:16495–16505. doi: 10.1074/jbc.M112.443580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zihni C, Munro PM, Elbediwy A, Keep NH, Terry SJ, Harris J, Balda MS, Matter K. Dbl3 drives Cdc42 signaling at the apical margin to regulate junction position and apical differentiation. J Cell Biol. 2014;204:111–127. doi: 10.1083/jcb.201304064. [DOI] [PMC free article] [PubMed] [Google Scholar]