

A long-standing conundrum is resolved about the underlying sequence determinants and molecular mechanism responsible for the recruitment of the protein kinase Hsl1 (an indispensable component of the so-called “morphogenesis checkpoint”) exclusively to the septin collar at the bud neck.
Abstract
Passage through the eukaryotic cell cycle requires processes that are tightly regulated both spatially and temporally. Surveillance mechanisms (checkpoints) exert quality control and impose order on the timing and organization of downstream events by impeding cell cycle progression until the necessary components are available and undamaged and have acted in the proper sequence. In budding yeast, a checkpoint exists that does not allow timely execution of the G2/M transition unless and until a collar of septin filaments has properly assembled at the bud neck, which is the site where subsequent cytokinesis will occur. An essential component of this checkpoint is the large (1518-residue) protein kinase Hsl1, which localizes to the bud neck only if the septin collar has been correctly formed. Hsl1 reportedly interacts with particular septins; however, the precise molecular determinants in Hsl1 responsible for its recruitment to this cellular location during G2 have not been elucidated. We performed a comprehensive mutational dissection and accompanying image analysis to identify the sequence elements within Hsl1 responsible for its localization to the septins at the bud neck. Unexpectedly, we found that this targeting is multipartite. A segment of the central region of Hsl1 (residues 611–950), composed of two tandem, semiredundant but distinct septin-associating elements, is necessary and sufficient for binding to septin filaments both in vitro and in vivo. However, in addition to 611–950, efficient localization of Hsl1 to the septin collar in the cell obligatorily requires generalized targeting to the cytosolic face of the plasma membrane, a function normally provided by the C-terminal phosphatidylserine-binding KA1 domain (residues 1379–1518) in Hsl1 but that can be replaced by other, heterologous phosphatidylserine-binding sequences.
INTRODUCTION
In budding yeast, a filamentous superstructure is erected at the bud neck composed of five members of the septin family of GTP-binding proteins, four of which (Cdc3, Cdc10, Cdc11, and Cdc12) were originally identified in a screen for temperature-sensitive cell division cycle mutants (Hartwell, 1971; Hartwell et al., 1974). A fifth septin (Shs1) that is expressed in vegetatively growing cells was discovered by other means (Carroll et al., 1998; Mino et al., 1998). The septin superstructure at the bud neck is required for cytokinesis because it contributes to membrane deformation (Bridges and Gladfelter, 2015), serves as a scaffold to recruit the actomyosin contractile apparatus that drives membrane ingression (Wloka and Bi, 2012; Finnigan et al., 2015a), and acts as a barrier or corral to confine and locally concentrate the vesicles that deposit new membrane and the enzymatic machinery that synthesizes the chitinaceous cross wall for cell septation (Dobbelaere and Barral, 2004; Caudron and Barral, 2009). Scores of proteins localize to the bud neck in a highly orchestrated manner, concomitant with changes in the architecture of septin assembly at the bud neck, which transforms from a patch into a ring and then into an hourglass-shaped collar and finally splits into two bands at the onset of cell division (McMurray and Thorner, 2009).
Higher-order septin structures are erected from two types of linear apolar hetero-octamers: Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11 (Bertin et al., 2008; McMurray et al., 2011) and Shs1-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Shs1 (Garcia et al., 2011; Finnigan et al., 2015b). The ratio of these two types of rods and their association with membrane lipids influence the nature of the structures that are assembled from these building blocks both in vitro (Bertin et al., 2010; Garcia et al., 2011; Bridges et al., 2014) and in vivo (Bertin and Nogales, 2012; Ong et al., 2014). These forms include end-on-end polymerization into long, paired filaments and braids, lateral associations to form arcs and spirals, and more gauze- or lattice-like arrays.
The changes in the state of septin organization need to be integrated both temporally and spatially with the other processes required for cell cycle progression. Conversely, because cytokinesis cannot proceed productively unless the septin collar has been erected, yeast cells have evolved a checkpoint that delays initiation of the G2/M transition until the septin collar has been properly assembled (Shulewitz et al., 1999). This particular feedback circuitry, which is one of several that monitor and ensure correct cellular morphogenesis (Lew, 2003; Keaton and Lew, 2006; Howell and Lew, 2012), involves a network of protein kinases and additional factors. In brief, the cyclin B (Clb)-bound form of Cdk1 (Cdc28) that drives mitosis is held in check by inhibitory phosphorylation on Y19 by the yeast Wee1 orthologue (Swe1; Booher et al., 1993). The action of Swe1 is reversed, in part, by the protein-tyrosine phosphatase Mih1 (Russell et al., 1989). In addition, in conjunction with the combined actions of the protein-arginine N-methyltransferase Hsl7 (Cid et al., 2001; Sayegh and Clarke, 2008) and the protein kinase Hsl1 (Ma et al., 1996; Barral et al., 1999; Shulewitz et al., 1999), Swe1 is exported from the nucleus (Keaton et al., 2008), captured at the bud neck, and further marked there for timely cyclosome/anaphase-promoting complex protein-ubiquitin ligase (APC)–dependent ubiquitinylation and proteasome-imposed destruction by phosphorylation, mediated, in large part, by the yeast Polo orthologue (Cdc5) (Sakchaisri et al., 2004). Because Clb2-Cdc28 phosphorylation of Swe1 primes it for its Cdc5-mediated phosphorylation (Asano et al., 2005), Hsl7- and Hsl1-dependent reduction of nuclear Swe1 initiates a self-reinforcing burst of autocatalytic activation of Cdk1 and, therefore and concomitantly, hyperphosphorylation and nearly complete elimination of Swe1 (Sreenivasan and Kellogg, 1999). Of importance, Hsl1 is only active in targeting Swe1 for destruction when Hsl1 is associated with a correctly assembled septin collar at the bud neck (McMillan et al., 1999, 2002; Shulewitz et al., 1999). Hsl1 (and the checkpoint) may have additional roles besides surveillance of septin organization, such as monitoring bud size/shape and/or plasma membrane composition, that are necessary to license entry into mitosis.
Hsl1 is a large polypeptide (1518 residues; Figure 1A), with its canonical catalytic domain (residues 85–369) situated at its N-terminus. It is not clear how association with septins activates Hsl1. As judged by the two-hybrid method, it has been reported that residues 987–1100 of Hsl1 can associate with its kinase domain and, using the same method, that the same segment is able to interact with septins Cdc11 and Cdc12, suggesting a model in which binding to septins activates Hsl1 by alleviating autoinhibition (Hanrahan and Snyder, 2003). It has also been reported that Hsl1 activity requires its phosphorylation on T220 in its so-called activation loop by Elm1, yet another bud neck–localized protein kinase (Szkotnicki et al., 2008), suggesting a model in which recruitment to the septin collar activates Hsl1 by placing it in close proximity to an upstream activator. In addition, Hsl1 contains both a KEN box (residues 775–781) and a destruction box (D-box; residues 828–836) and is very efficiently degraded in an APC-dependent manner (Burton and Solomon, 2000, 2001; Simpson-Lavy et al., 2009). Perhaps binding to septins occludes the KEN and D-boxes, thereby protecting Hsl1 from destruction, suggesting a model in which “activation” simply involves elevating the number of Hsl1 molecules present (rather than increasing their specific activity per se).
FIGURE 1:
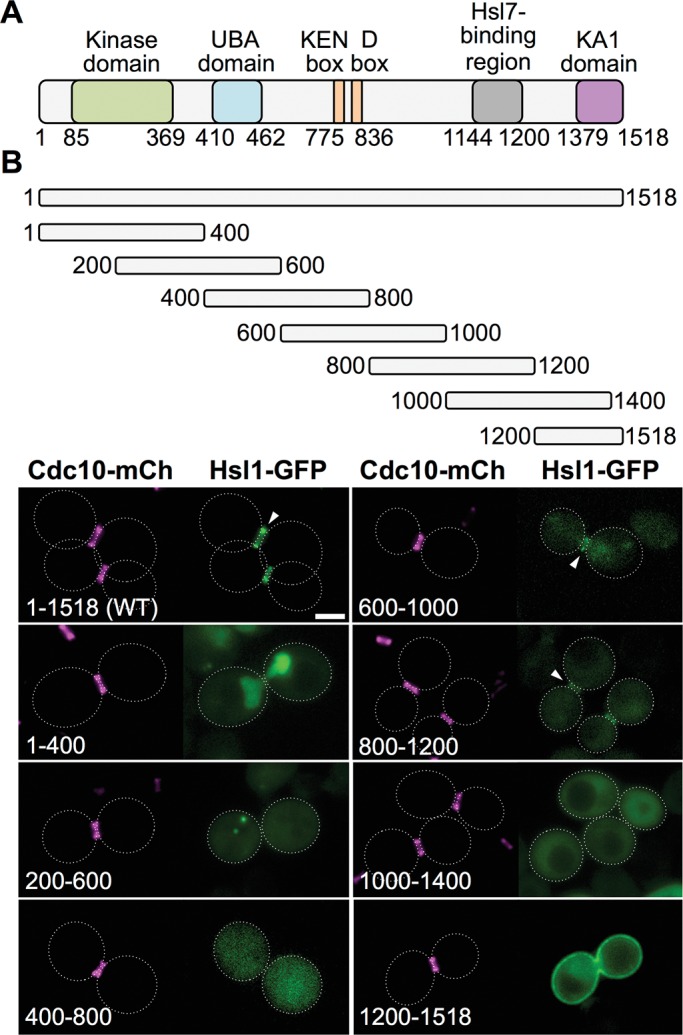
Analysis of the subcellular location of a sequential series of 400-residue overlapping fragments of protein kinase Hsl1. (A) Primary structure of Hsl1. Previously characterized sequence features include the kinase (catalytic) domain (green; Shulewitz, 2000; Szkotnicki et al., 2008); the putative ubiquitin association domain (blue; Hofmann and Bucher, 1996; Mueller and Feigon, 2002); the KEN box and D-box motifs (orange; Burton and Solomon, 2001); the Hsl7-binding element (gray; Shulewitz et al., 1999; Crutchley et al., 2009); and the KA1 domain (pink; Moravcevic et al., 2010). (B) Top, diagram depicting Hsl1 and the indicated fragments that were expressed in vivo under the control of the same promoter from the same CEN vector, each as a fusion to the N-terminus of eGFP. Bottom, yeast strain GFY-42 expressing Cdc10-mCherry from its endogenous locus transformed with plasmids expressing either full-length Hsl1 (pGF-IVL521) or each of the indicated fragments (pGF-IVL561 through pGF-IVL567) and examined by fluorescence microscopy. All images were scaled identically. Arrowheads, GFP signal at the bud neck; dotted white line, cell periphery; scale bar, 2 μm. For clarity, only one or a few representative cells are shown for each construct, and the dotted white line is omitted for constructs that exhibited significant PM fluorescence.
To devise appropriate experimental approaches to distinguish among these models, it is imperative to first pinpoint the elements in Hsl1 responsible for septin recognition and targeting to the bud neck. However, this goal has been elusive. Deletion analysis of the most highly conserved sequences in Hsl1 located several regions that, when absent, only mildly impaired its localization to the bud neck in vivo (Crutchley et al., 2009). Thus the elements in Hsl1 required for septin binding and how they contribute at the mechanistic level to Hsl1 recruitment to the bud neck remained to be determined. In the present study, we map the sequence elements in Hsl1 required for its association with the septin collar. We show that these sequences are necessary and sufficient for both Hsl1 localization to the septin collar in vivo and binding to septin filaments in vitro. Moreover, we also demonstrate that, in the cell, these septin-binding elements must work in conjunction with a phospholipid-binding fold, dubbed the kinase associated-1 (KA1) domain (Moravcevic et al., 2010), which is located at the extreme C-terminus of Hsl1.
RESULTS
Septin collar assembly is required for recruitment of Hsl1 to the bud neck
Prior work established that assembly of an intact septin collar is a prelude to localization of Hsl1 at the bud neck, that Hsl1, in turn, recruits Hsl7, and that both of these factors are necessary for efficient inactivation and degradation of Swe1 and maintenance of normal morphology (McMillan et al., 1999, 2002; Shulewitz et al., 1999; Thomas et al., 2003; Sakchaisri et al., 2004). These findings were made largely in strains of the W303 lineage or other backgrounds. For the analysis we conducted and describe here, all the constructs were derived in the now widely used BY4741 background (Brachmann et al., 1998). Reassuringly, the following observations confirmed that the prior conclusions were equally valid in BY4741 strains. First, BY4741 cells lacking Hsl1 grew on plates at a rate indistinguishable from HSL1+ cells, and the presence of an hsl1∆ mutation did not exacerbate the phenotypes of cells carrying three different types of septin mutations (Supplemental Figure S1A), consistent with septin function not requiring Hsl1 action. Second, absence of Hsl1 caused buds and especially mother cells to be somewhat more elongated than cells in the control (HSL1+) culture, indicative of a delay in the G2/M transition, consistent with prior work (Longtine et al., 2000); however, assembly of septins at the bud neck was unaffected (Supplemental Figure S1B), again consistent with septin collar formation preceding the function of Hsl1 (Cid et al., 2001; Thomas et al., 2003). Third, disruption of the septin collar (by shift of a cdc12-6ts mutant [ Johnson et al., 2015] to the restrictive temperature) ablated Hsl1-GFP localization at the bud neck (Supplemental Figure S1C), in agreement with the requirement for assembly of an intact septin superstructure for Hsl1 recruitment. Finally, by the time anaphase was executed and cytokinesis underway (as judged by splitting of the septin collar into two rings), Hsl1–green fluorescent protein (GFP) was undetectable (Supplemental Figure S1D), consistent with its known APC-dependent degradation (Burton and Solomon, 2000, 2001).
An internal fragment of Hsl1 is necessary and sufficient for its bud neck localization
Hsl1 is a relatively large protein (1518 residues) comprising a number of previously characterized functional domains and sequence elements (Figure 1A). As our initial approach to delineating the region(s) in Hsl1 required for its targeting to the septin collar, we first conducted a systematic and unbiased scan across the entire Hsl1 sequence by expressing overlapping 400-residue fragments fused at their C-terminal end to enhanced GFP (eGFP) and examined their localization pattern in vivo in cells expressing Cdc10-mCherry as a marker for the septin collar at the bud neck. Full-length Hsl1-GFP colocalized exclusively with the septin collar at the bud neck, whereas Hsl1(1–400)-GFP localized strongly in the nucleus, consistent with the presence of multiple basic residue–rich tracts in this N-terminal fragment (20AKKAAKRA27, 68KSSKRKSR75, 100KIVPKKKA116, 249KKNRRIKI256, and 379KVLRKMRK386 [N-nuclear localization signals; NLSs]), each of which resembles a classical NLS (Lange et al., 2007; Kosugi et al., 2009), and Hsl1(200–600)-GFP and Hsl1(400–800)-GFP exhibited diffuse cytosolic fluorescence (Figure 1B, left). By contrast, both Hsl1(600–1000)-GFP and Hsl1(800–1200)-GFP displayed readily detectable, albeit rather weak, colocalization with the septin collar, whereas Hsl1(1000–1400)-GFP exhibited diffuse cytosolic fluorescence, and Hsl1(1200–1518)-GFP was targeted predominantly to the plasma membrane (PM), in keeping with the presence of the phosphatidylserine (PtdSer)-binding KA1 domain (residues 1358–1518; Moravcevic et al., 2010) in this fragment (Figure 1B, right). This analysis was conducted in cells that were HSL1+ at its normal chromosomal locus, and very similar results were obtained for all of these same Hsl1-GFP constructs expressed in hsl1∆ cells (G.C.F., unpublished data).
The two fragments that displayed bud neck localization (600–1000 and 800–1200) span a 600-residue region and overlap over 200 residues. These findings suggested two possibilities: 1) the bud neck–targeting information is confined to the region of overlap (800–1000), or 2) the bud neck–targeting information is bipartite, that is, each fragment contains a partially redundant signal. We favored the later interpretation because the sequence in the region of overlap (800–1000) is poorly conserved, whereas both the upstream (600–800) and downstream (1000–1200) segments contain more highly conserved sequences (Supplemental Figure S2).
Plasma membrane association enhances recruitment to the septin collar
Native Hsl1 contains both the septin-associating region we identified (600–1200) and the C-terminal KA1 domain. To assess the contribution of the latter domain to bud neck targeting, first we prepared C-terminal truncations of Hsl1 that removed the KA1 domain but left the remainder of the protein intact and, conversely, expressed Hsl1 fragments containing only its KA1 domain (Figure 2A, top). We found that removal of the KA1 fold itself (residues 1358–1518) or an even larger deletion (residues 1245–1518) reduced but did not eliminate binding at the bud neck and concomitantly increased the cytosolic fluorescence (Figure 2A, bottom left). Quantification of the images (Supplemental Figure S3) indicated that bud neck localization was reduced by 65–70% (Figure 2B, top). Conversely, in the absence of the rest of Hsl1, its KA1 domain–containing fragments decorated the PM exclusively and did not colocalize with the septin collar (Figure 2, A, bottom right, and B, bottom). Using a similar approach with a set of internal deletions, we confirmed that residues 1144–1200 of Hsl1, which prior work demonstrated are required for its association with Hsl7 (Shulewitz et al., 1999; Shulewitz, 2000; Crutchley et al., 2009), have no role in localizing Hsl1 to the septin collar (Supplemental Figure S4).
FIGURE 2:

Residues 611–950 of Hsl1 are necessary and sufficient, in conjunction with the C-terminal KA1 domain, for optimal bud neck localization. (A) Top, diagram of Hsl1 and the indicated C-terminal truncations and C-terminal fragments that were examined. Bottom, plasmids producing full-length Hsl1 (pGF-IVL521), empty vector (pRS315), or the indicated truncations and fragments (pGF-IVL522 through pGF-IVL525), expressed and visualized as in Figure 1, A and B. Arrowheads, GFP signal at the bud neck; dotted white line, cell periphery; scale bar, 2 μm. For clarity, only one or a few representative cells are shown for each construct. and the dotted white line is omitted for constructs that exhibited significant PM fluorescence. (B) Top, relative average pixel intensity of the GFP signal at the bud neck was quantified in triplicate using ImageJ. Error bars, SEM; asterisk, statistically significant difference (p < 0.05) using an unpaired t test. Bottom, relative PM fluorescence quantified as the ratio of the maximum pixel intensity of the PM signal vs. the average cytosolic fluorescence signal, measured as described in detail in Supplemental Figure S3. (C) Top, diagram of Hsl1 and the indicated N-terminal truncations and internal deletions that were examined. Bottom, plasmids producing the indicated truncations and deletions (pGF-IVL526 through pGF-IVL533), expressed and visualized as in Figure 1, A and B. Arrowheads, structures consistent with some mitochondrial localization; scale bar, 2 μm. (D) Top, diagram of the indicated internal fragments fused, where shown, to the indicated C-terminal fragments that were examined. Bottom, plasmids producing the indicated constructs (pGF-IVL534 through pGF-IVL536, left; pGF-IVL624 through pGF-IVL626, right), expressed and visualized as in Figure 1, A and B. Scale bar, 2 μm.
These results suggested synergy or coordination between KA1 domain–mediated PM binding and the ability of the 600–1200 region to achieve efficient recruitment to the septin collar. Hence, as a second means to investigate this apparent cooperative effect, we constructed N-terminal truncations and internal deletions within otherwise full-length Hsl1 that removed all or portions of the 600–1200 region but left the KA1 domain intact (Figure 2C, top). In brief, we found that constructs containing the region 600–800 or 800–1120 or both were localized either exclusively or strongly to the bud neck, whereas removal of a portion of that region, for example, 800–1244, reduced bud neck fluorescence and increased cytoplasmic fluorescence, and removal of the entire region, for example, 620–1244, eliminated bud neck fluorescence and enhanced PM decoration and/or the nuclear signal (Figure 2C, bottom). Hsl1(620–1518)-GFP localized strongly to the bud neck but also to a number of cortical puncta, which we attribute to spurious targeting to the mitochondrion due to exposure of N-terminal Arg-, Leu-, and Ser-rich tracts (629SRYSLSRR636 and 650SLSR654; Supplemental Figure S2) that closely resemble canonical mitochondrial presequences (Allison and Schatz, 1986; Claros and Vincens, 1996; Roise, 1997). These data indicated that two separable sequence elements lying between residues 620 and 1119 are necessary for Hsl1 recruitment to the bud neck, closely matching the segment (600–1200) identified in our initial fragment scanning approach.
As a third means to assess the synergy between the PM-binding KA1 domain and the apparent septin-binding region and to ascertain the minimal sequence sufficient for septin collar targeting of Hsl1, we generated additional constructs in which fragments lying within the 600–1120 region were fused to C-terminal fragments either containing (1245–1518) or lacking (1245–1357) the KA1 domain (Figure 2D, top). Tellingly, when fused to the KA1 domain–containing C-terminal end of Hsl1 (1245–1518), a very small (81-residue) fragment, Hsl1(663–743), was sufficient to clearly decorate the bud neck yet also displayed detectable cytosolic fluorescence and weak association with the PM (Figure 2D, bottom left). This behavior mimicked that of Hsl1(800–1518)-GFP (Figure 2C), which is missing the 663–743 sequence, consistent with our initial conclusion that the 600–1120 region contains two distinct septin-associating elements in tandem. However, extending the 663–743 fragment both upstream (611–662) and downstream (744–878) weakened the bud neck signal substantially and caused significant accumulation in the nucleus. That this pattern represented localization within the nucleus was confirmed by expressing the same constructs in cells coexpressing a marker of the nuclear envelope, mCherry-tagged Nup188 (G.C.F., unpublished data).
Strikingly, extending the same 611–878 fragment an additional 73 residues (to 950) restored robust localization at the bud neck, which, as for full-length Hsl1, was completely dependent on the presence of the KA1 domain (Figure 2D, bottom). This result is consistent with the presence of a second septin-associating sequence that, in combination with the more-upstream septin-associating sequence in 663–743 and the KA1 domain, is able to override the nuclear targeting information in the 611–662 region. In addition, it was possible that hydrophobic tracts residing in the 879–950 segment that resemble the hydrophobic nuclear export signal (NES) motifs that mediate Xpo1/Crm1-based nuclear export (898ISQPVNSKVESLLQGLKF915 [NES1] and 941PVKASGVSI949 [NES2]; Güttler et al., 2010) also contributed to preventing nuclear accumulation of the Hsl1(611–950; 1245-1518)-GFP and Hsl1(611–950; 1358–1518)-GFP constructs. Indeed, deleting NES1 (Hsl1(611–925; 1245–1518)-GFP) or both NES1 and NES2 (Hsl1(611–900; 1245–1518)-GFP) from the C-terminal end of the Hsl1(611–950; 1245–1518)-GFP diminished bud neck localization and caused readily measurable nuclear accumulation, whereas co-removal of an apparent classical bipartite NLS (Robbins et al., 1991) present in this internal fragment (635RRAIHASPSTKSIHKSLSRK654 [I-NLS]) from the N-terminal end of both NES-less constructs (Hsl1(663–925; 1245–1518)-GFP and Hsl1(663–900; 1245–1518)-GFP) eliminated their nuclear accumulation (Figure 3A). (Although residues 900–950 may contain putative NES motifs, this sequence also seems to harbor information important for optimal septin targeting, as judged by the fact that, compared with Hsl1(663–950; 1245–1518), Hsl1(663–900; 1245–1518) exhibited diminished bud neck localization and some concomitant decoration of the PM [ Figure 3A, second row] and similarly for Hsl1(663–900) compared with Hsl1(663–950) [ Figure 3B, second row], even though these constructs lack the I-NLS sequence. On the other hand, we cannot rule out that removal of these 50 residues simply causes the globular KA1 [and GFP] folds to be so close as to partially occlude sterically the actual upstream septin-targeting determinants.)
FIGURE 3:

The 611–950 segment of Hsl1 contains two separable septin-association elements. (A) Right, diagram of the indicated internal fragments, each fused to the same C-terminal fragment (and eGFP), that were examined. Left, plasmids producing the indicated constructs (pGF-IVL536 and pGF-IVL601 through pGF-IVL611), expressed and visualized as in Figure 1, A and B. Scale bar, 2 μm. (B) Right, diagram of the same internal fragments as in A, each fused to eGFP only, that were examined. Left, plasmids producing the indicated constructs (pGF-IVL612 through pGF-IVL623), expressed and visualized as in Figure 1, A and B. Dotted white line, cell periphery; scale bar, 2 μm. For clarity, only one or a few representative cells are shown for each construct, and the dotted white line is omitted for constructs that exhibited significant PM fluorescence.
By fusing wild-type and mutated versions of Hsl1 fragments from the 611–950 region to a chimera (glutathione S-transferase [GST]–maltose-binding protein [MBP]-eGFP) that is otherwise excluded from the nucleus, we could independently attribute the nuclear import of Hsl1(611–878; 1245–1518)-GFP largely to exposure of the I-NLS and also found that separate segments of the 611–950 region (611–710 and 800–950) were able to target the same chimera to the bud neck in a readily detectable manner and with roughly equal efficiency (Supplemental Figure S5), again in agreement with the presence of two distinct septin-binding elements within the 611–950 region. The fact that our fragment-based analysis revealed both multiple potent NLS elements and two presumptive NES elements and yet full-length Hsl1 is found exclusively at the bud neck raised the issue of whether native Hsl1 undergoes nucleocytoplasmic shuttling but that its rate of export vastly exceeds its rate of import. However, in strains lacking the function of each of the four known yeast exportins, we did not observe any detectable nuclear accumulation whatsoever (Supplemental Figure S6). Therefore, unlike other cell cycle regulators (Keaton et al., 2008), intact Hsl1 does not appear to undergo nucleocytoplasmic shuttling.
Of most importance, successive trimming of the 611–950 segment in the Hsl1(611–950; 1245–1518)-GFP construct from its N-terminal end, its C-terminal end, or both diminished bud neck association and enhanced PM retention in a graded manner (Figure 3A), suggesting that the entire 611–950 region is required for optimal targeting to the septin collar. Moreover, when the same set of Hsl1 fragments was expressed alone (unfused to the KA1 domain–containing 1245–1518 segment), they were very unstable, regardless of whether they were in the nucleus or the cytosol (Figure 3B). This instability was due to their APC-mediated degradation, because versions of the same fragments in which we mutated the KEN box, the D-box, or both were produced at a substantially higher steady-state level (Supplemental Figure S7, left). When installed in the context of Hsl1(611–950; 1245–1518)-GFP, it was clear that mutation of the KEN box, D-box, or both, had little or no effect on the efficiency of targeting to the septin collar (Supplemental Figure S7, right).
A conserved sequence element that contributes to Hsl1 localization at the bud neck
Having shown, largely using the fragment approach, that two distinct portions of the 611–950 region were sufficient, especially in conjunction with the KA1 domain, to localize Hsl1 exclusively at the bud neck, we wanted to confirm in the context of otherwise full-length Hsl1 that these tandem elements were necessary for efficient targeting of Hsl1 to the septin collar. For this purpose, first we constructed a nested set of internal deletions (Figure 4, top). Indeed, removal of the entire 611–950 segment (340 residues), as well as of a somewhat smaller deletion (288 resides), eliminated localization at the bud neck and permitted only KA1 domain–mediated tethering to the PM, whereas two smaller and nonoverlapping deletions, 744–950 (207 residues) and 611–743 (133 residues), each exhibited robust targeting to the bud neck (Figure 4, bottom), once again consistent with the presence of two separate septin-associating elements. We noted, however, that the construct that retained the 611–743 sequence was localized exclusively at the septin collar, whereas the construct that retained the 744–950 sequence exhibited detectable fluorescence at the PM and in the cytosol, suggesting that the upstream septin-association element in the 611–950 region is somewhat more efficacious. Within the 611–743 segment, residues 643–688 are well conserved (Supplemental Figure S2). Indeed, deletion of just 24 residues (665–688) from within this region or mutation to Ala of the 19 most conserved residues in this sequence weakened localization to the bud neck, as judged by an increase in cytosolic fluorescence and the appearance of occasional PM-associated puncta (Figure 4, bottom).
FIGURE 4:
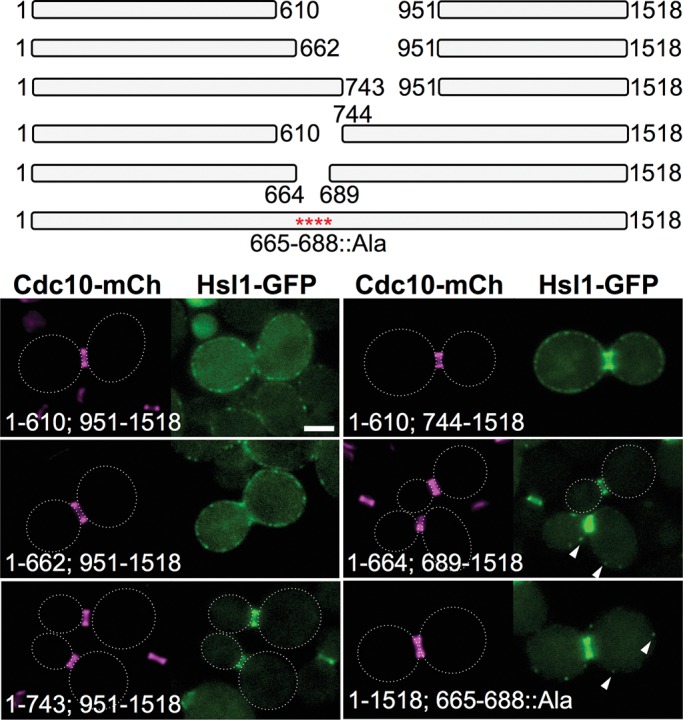
A conserved sequence element within the 611–950 segment of Hsl1 makes a major contribution to its bud neck localization. Top, diagram of the indicated internal deletions and the Hsl1(T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A) allele that were examined. Bottom, plasmids producing the indicated deletions and substitution mutant (pGF-IVL666, pGF-IVL665, pGF-IVL710, pGF-IVL709, pGF-IVL711, and pGF-IVL816), expressed and visualized as in Figure 1, A and B. Arrowheads, GFP signal in PM-associated puncta; dotted white line, cell periphery; scale bar, 2 μm. For clarity, only one or a few representative cells are shown for each construct, and the dotted white line is omitted for constructs that exhibited significant PM fluorescence.
The role of the conserved 643–688 sequence element in directing Hsl1 association with the septin collar was further confirmed using an approach in which we “sensitized” localization to perturbations by examining the behavior of two minimal fragments, Hsl1(611–900)-GFP and Hsl1(611–950)-GFP. When the basic residues in the I-NLS were mutated to Ala (Figure 5A, set 1) in either Hsl1(611–900)-GFP (lacking NES1 and NES2) or Hsl1(611–950)-GFP (containing both NES1 and NES2), nuclear import was prevented and both fragments clearly decorated the bud neck (Figure 5B, left). When just six basic residues within the conserved sequence element (residues 643–688) were mutated to Ala (Figure 5A, set 2), both fragments still decorated the bud neck but also displayed some nuclear accumulation (because the I-NLS is intact; Figure 5B, left). However, when four of the basic residues and 15 other invariant residues within the conserved sequence element were mutated to Ala (Figure 5A, set 3), bud neck localization was totally eliminated (Figure 5B, left). Similarly, when 611–900 (lacking NES1 and NES2) was fused to the KA1 domain–containing 1245–1518 fragment, the set 3 mutations also markedly reduced bud neck recruitment and caused a readily detectable increase in cytosolic and nuclear fluorescence (Figure 5B, right). Thus the conserved sequence element (residues 643–688) clearly contributes to bud neck localization of Hsl1. Tellingly, however, when 611–950 (containing NES1 and NES2) was fused to the KA1 domain–containing 1245–1518 fragment, the set 3 mutations did not detectably abrogate bud neck localization (Figure 5B, right), indicating that, when both robust export from the nucleus and KA1-mediated PM tethering occur, the second septin-associating element (possibly the conserved residues 877–910) in the 611–950 fragment is sufficient to retain Hsl1 at the bud neck.
FIGURE 5:

A cryptic bipartite NLS is separable from the conserved septin-associating sequence element in the 611–950 segment of Hsl1. (A) Positions of the residues mutated in the N-terminal portion (residues 611–710) of the 611–950 segment of S. cerevisiae Hsl1 aligned using ClustalW against the corresponding sequences of seven other yeast species (Sc, Saccharomyces cerevisiae; Cg, Candida glabrata; Kn, Kazachstania naganishii; Zr, Zygosaccharomyces rouxii; Zb, Zygosaccharomyces bailii; Nc, Naumovozyma castellii; Ka, Kazachstania africana; and Vp, Vanderwaltozyma polyspora). See also Supplemental Figure S2. White-on-black letters represent invariant residues across all eight fungal species; blue letters indicate strongly conserved residues (found in seven of the eight species). Pink, set 1 allele (R635A R636A K645A H648A K649A R653A K654A); green, set 2 allele (R663A R664A K672A R673A K683A R684A; blue, set 3 allele (T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A). (B) Right, diagram of the indicated mutated internal fragments, either free or fused to the same C-terminal fragment (and eGFP), that were examined. Bottom, plasmids producing the indicated constructs (pGF-IVL705, pGF-IVL757, pGF-IVL758, pGF-IVL760, pGF-IVL762, pGF-IVL763, pGF-IVL765, pGF-IVL766, and pGF-IVL817 through pGF-IVL820), expressed and visualized as in Figure 1, A and B. Scale bar, 2 μm.
Direct binding to septin filaments is responsible for Hsl1 localization at the bud neck
Having demonstrated, using imaging in live cells, that the 611–950 region is both necessary and sufficient for highly efficient localization of Hsl1 to the bud neck, we wanted to confirm, using biochemical methods, that the function of these sequences is to mediate direct physical association with the septin filaments that are a diagnostic hallmark and required for establishment of the bud neck constriction (Byers and Goetsch, 1976; Longtine and Bi, 2003).
For this purpose and to ensure its production as a soluble protein, we expressed and purified this segment of Hsl1 in Escherichia coli as a hexahistidine (His6)–MBP-Hsl1(611–950) fusion, as well His6-MBP alone as a control (Figure 6A). We used two independent approaches to determine the capacity of the Hsl1(611–950) sequence to bind to septin filaments. First, we devised a cosedimentation assay (Garcia, 2012), based on the fact that purified Cdc11-capped septin hetero-octamers remain unpolymerized in high-salt solution but rapidly assemble end to end to form very long, paired filaments in low-salt solution (Bertin et al., 2008; Garcia et al., 2011; Booth et al., 2015). For this reason, such septin hetero-octamers in high-salt buffer remain in the supernatant fraction upon high-speed centrifugation, whereas in low-salt buffer, they sediment into the pellet upon high-speed centrifugation (Figure 6B, lanes 1–4). In the absence of any septins, His6-MBP-Hsl1(611–950) remained in the supernatant fraction under either condition (Figure 6B, lanes 5–8); however, in the presence of purified Cdc11-capped septin hetero-octamers, a nearly stoichiometric amount of His6-MBP-Hsl1(611–950) copelleted with the filaments (Figure 6B, lanes 11 and 12). Moreover, as expected for a protein that binds solely to assembled filaments, which should shift the equilibrium toward polymerization, a detectable amount of the septins and His6-MBP-Hsl1(611–950) (which was added in only a twofold molar excess over the hetero-octamers) was able to pellet even from high-salt solution (Figure 6B, lanes 9 and 10). In marked contrast, the His6-MBP–alone control showed no capacity to copellet with the assembled septins and no ability to promote polymerization of the hetero-octamers (Figure 6B, lanes 13–20).
FIGURE 6:

The 611–950 segment of Hsl1 binds directly to assembled septin filaments in vitro. (A) Diagram of the primary structure of the His6-MBP-Hsl1(611–950) fusion protein expressed in and purified from E. coli as described in Materials and Methods. (B) Cosedimentation assay. Cdc11-capped hetero-octamers, purified as described in Materials and Methods, alone (lanes 1–4), purified His6-MBP-Hsl1(611–950) alone (lanes 5–8), a 1:2 mixture of Cdc11-capped hetero-octamers and His6-MBP-Hsl1(611–950) (lanes 9–12), purified His6-MBP alone (lanes 13–16), or a 1:2 mixture of Cdc11-capped hetero-octamers and His6-MBP (lanes 17–20) were incubated in either high-salt buffer (H) or low-salt buffer (L) and then subjected to high-speed centrifugation and separated into supernatant (s) and pellet (p) fractions, the contents of which were then resolved by SDS–PAGE and analyzed by staining with InstantBlue dye. (C) Samples of the indicated mixtures from B were applied to carbon-coated copper grids, stained, and examined by EM, and representative images were recorded. Scale bar, 100 nm.
Second, the same samples used for the cosedimentation assays were examined by transmission electron microscopy (EM) on carbon-coated copper grids after staining with uranyl formate because we previously demonstrated that, under these conditions, the stable globular fold of the MBP tag provides a reliable fiducial marker to detect the location of proteins associated with septin-based structures (Bertin et al., 2008). As expected, in low-salt buffer, purified Cdc11-capped septin hetero-octamers alone assembled end to end to form many parallel arrays of long, paired filaments (Figure 6C, left), and virtually indistinguishable images were observed for the septins in the presence of the His6-MBP control (Figure 6C, right). In marked contrast, in the presence of His6-MBP-Hsl1(611–950), Cdc11-capped septin hetero-octamers assembled into tightly cross-braced bundles in which the globular MBP domain was prominently and periodically positioned in the cross-bridges between the filaments. Thus, as judged by two independent criteria, the 611–950 segment in Hsl1 binds directly to polymerized septin filaments.
For technical reasons, we were unable to address whether either of the two apparent septin-association elements (643–688 and 877–910) lying within the 611–950 fragment is able on its own to bind to septins in vitro because, even as fusions to MBP or other tags, fragments containing these sequences were not stably expressed as soluble recombinant proteins in bacterial cells.
Plasma membrane binding is the sole role of the KA1 domain in targeting Hsl1 to the bud neck
Although the 611–950 segment of Hsl1 is necessary and sufficient for recruitment of Hsl1 to the bud neck in vivo and for binding to septin filaments in vitro, we amply demonstrated that, in the cell, the ability of this element to localize Hsl1 at the bud neck is greatly enhanced by the presence in-cis of the C-terminal KA1 domain. For example, native Hsl1 is recruited exclusively to the bud neck, whereas removal of the KA1 domain alone (residues 1358–1518) drastically reduces the amount of Hsl1 at the bud neck (Figure 2A). Conversely, attachment of the KA1 domain to the 611–950 fragment permits its highly efficient recruitment to the bud neck (Figure 2D). Hence we sought to determine whether the KA1 domain of Hsl1 had some specialized function that can only operate uniquely with the rest of the protein or whether its primary, and perhaps sole, function is to concentrate Hsl1 at the PM, thereby greatly enhancing the probability of its encounter with and binding to the septin collar at the cell cortex.
As previously shown (Moravcevic et al., 2010; Roelants et al., 2015), two Hsl1-related protein kinases, Gin4 and Kcc4, also possess C-terminal PtdSer-binding KA1 domains (although the primary sequence conservation among the three of them is modest). We first confirmed that, when each was tagged with GFP, both the KA1Gin4 and KA1Kcc4 domains decorated the PM; however, the binding of KA1Gin4 to the PM was noticeably weaker than that mediated by KA1Hsl1, and the PM binding of KA1Kcc4 was weaker still (Figure 7A). In fact, only an unrelated and evolutionarily highly divergent PtdSer-binding element, the 158-residue C2 domain of bovine lactadherin (Butler et al., 1980; Andersen et al., 2000; Shao et al., 2008), displayed a PM-binding efficacy in yeast comparable to that of KA1Hsl1 (Figure 7A). If the major role of its own KA1 domain in bud neck localization is simply to raise the effective local concentration of Hsl1 at the PM, then the KA1Gin4 and KA1Kcc4 domains and the phosphatidylserine-binding C2 domain of bovine lactadherin (C2Lact), when substituted for the KA1Hsl1 domain, should promote bud neck targeting with the same order of potency as they support PM association. Indeed, when each of these three elements was fused to the Hsl1(611–950) fragment, the weakest PM-binding domain, KA1Kcc4, allowed for only weak but detectable bud neck recruitment accompanied by prominent nuclear accumulation; the next most effective PM-binding domain, KA1Gin4, permitted prominent bud neck decoration, but still accompanied by some nuclear accumulation; and the most efficacious PM-binding domain, C2Lact, supported exclusive recruitment to the bud neck (Figure 7B), comparable to that for native Hsl1-GFP (Figures 1B and 2A).
FIGURE 7:

High-affinity PM binding is the sole role of the KA1 domain for localization of Hsl1 to the septin collar. (A) Top, diagram of the KA1 domains of the indicated protein kinases (white, Hsl1; pink, Gin4; and, blue, Kcc4) and the C2 domain of bovine (Bos taurus) lactadherin (orange), each fused to the C-terminus of eGFP, that were examined. Bottom, plasmids producing the indicated constructs (pGF-IVL181, pGF-IVL184, pGF-IVL187, and pGF-IVL708), expressed and visualized as in Figure 1, A and B. (B) Top, diagram of the fusions of the 611–950 segment of Hsl1 to the indicated heterologous membrane-targeting domains (and to eGFP) that were examined. Bottom, plasmids producing the indicated constructs (pGF-IVL639, pGF-IVL641 and pGF-IVL688), expressed and visualized as in Figure 1, A and B. (C) Top, diagram of the constructs in which the endogenous KA1 in Hsl1 was substituted with the indicated heterologous membrane-targeting domains (fused to eGFP). Bottom, plasmids producing the indicated constructs (pGF-IVL638, pGF-IVL640 and pGF-IVL687), expressed and visualized as in Figure 1, A and B. Dotted white line, cell periphery; scale bar, 2 μm. The dotted white line is omitted for constructs that exhibited significant PM fluorescence.
Quite similarly, when substituted for KAHsl1 in the context of otherwise full-length Hsl1, the KA1Kcc4 domain permitted prominent bud neck localization but with readily detectable cytosolic fluorescence, whereas the two stronger PM-binding motifs, KA1Gin4 and C2Lact, supported highly efficient and exclusive localization at the bud neck (Figure 7C). We conclude, therefore, that the primary if not sole function of the C-terminal KA1 domain is to mediate efficient association of Hsl1 with the PM, thus permitting its more efficient encounter with and binding to the septin filaments, which are themselves phosphatidylinositol-4,5-bisphosphate (PtdIns4,5P2)–binding proteins tightly associated with the PM (Rodal et al., 2005; Bertin et al., 2010, 2012; Ong et al., 2014).
In vivo competition assay confirms the role of residues 611–950 and the KA1 domain in targeting Hsl1 to the septin collar
Both Hsl1 and Mih1 oppose the action of Swe1 (Lew, 2003); the phosphatase Mih1 dephosphorylates the inhibitory P-Tyr-19 on Clb-bound Cdc28/Cdk1 that is installed by Swe1 (Russell et al., 1989), and the protein kinase Hsl1 is an integral component of the septin collar–sensing checkpoint that triggers ubiquitinylation and proteasome-mediated degradation of Swe1 (Shulewitz et al., 1999; Sakchaisri et al., 2004). Thus Mih1 and Hsl1 act in concert to release Cdk1 from Swe1-imposed inhibition, thereby permitting timely execution of the G2/M transition. For this reason, and contrary to recent evidence that other cellular phosphatases may also participate in dephosphorylating P-Tyr-19 (Kennedy et al., 2015), an mih1∆ hsl1∆ double mutant is inviable and, consistent with this phenotype arising solely from an inability to eliminate Swe1 function, an mih1∆ hsl1∆ swe1∆ triple mutant is viable (McMillan et al., 1999).
Indeed, we confirmed in strains of the BY4741 lineage that, like an hsl1∆ single mutant (Supplemental Figure S1), an mih1∆ single mutant is viable, whereas an hsl1∆ mih1∆ double mutant is inviable (Figure 8A). We reasoned that, if expressed at a sufficient level, the 611–950 fragment alone or fused to the KA1 domain might be able to displace native Hsl1 from the septin collar and compromise its function, and, if so, this outcome would be revealed as growth inhibition in the “sensitized” background of an mih1∆ mutant. To achieve high-level overexpression, we chose to drive production from a GAL promoter on a CEN vector. However, it was reported previously that overproduction of full-length Hsl1 is toxic in otherwise wild-type (HSL1+ MIH1+) cells (Sopko et al., 2006), an effect that might void our approach. Fortunately, we were able to show that this toxicity was due largely, if not exclusively, to off-target function of the N-terminal kinase domain of Hsl1 and its catalytic activity. First, when overexpressed, fragments lacking the kinase domain were not toxic (Figure 8B, left). Second, and conversely, the toxicity of overexpressed fragments containing the kinase domain were greatly ameliorated or completely eliminated by a single point mutation (D239A) that removed the Asp essential for catalysis (Figure 8B, left). By itself, the D239A mutation had no effect on localization of full-length Hsl1 to the bud neck (Figure 8C). Third, when overexpressed, the kinase domain artificially targeted to the PM via fusion to the C2Lact domain was as toxic as overexpressed full-length Hsl1, but its growth inhibitory effect was completely alleviated by the D239A mutation (Figure 8B, left).
FIGURE 8:

Cells lacking mih1∆ provide a sensitized background for assessment of Hsl1 function. (A) Simultaneous absence of Mih1 and Hsl1 is lethal. Strains of the BY4741 lineage expressing either Cdc10-mCherry (GFY-42) or Cdc11-mCherry (GFY-58) as indicated and carrying the wild-type HLS1 and/or MIH1 loci or carrying an hsl1∆ mutation (GFY-1156 and GFY-1157), an mih1∆ mutation (GFY-1881 and GFY-1882), or both (GFY-1737 and GFY-1738), and harboring a wild-type copy of the HSL1 gene on a URA3-marked CEN plasmid (pGFY-316-IVL924) were incubated for 2 d at 30˚C on medium lacking uracil (left) or containing 5-FOA (right) to select for the loss of the covering plasmid. (B) Toxicity of overexpressed Hsl1 is ameliorated by eliminating its kinase activity, revealing that the 611–950 segment combined with the KA1 domain is necessary and sufficient to compete for the function of endogenous Hsl1. Wild-type cells (BY4741, leftmost pair) or an otherwise isogenic mih1∆ derivative (GFY-1652, rightmost pair) were transformed with plasmids overexpressing from the GAL1/10 promoter eGFP alone (pGF-IVL391), full-length Hsl1 (fused to eGFP; pGF-IVL302), or the other indicated Hsl1 derivatives (pGF-IVL694, pGF-IVL689, pGF-IVL932, pGF-IVL642, pGF-IVL643, pGF-IVL929, pGF-IVL893A, pGF-IVL893B, pGF-IVL863, pGF-IVL894B, pGF-IVL895, pGF-IVL930, pGF-IVL896, pGF-IVL931, pGF-IVL644, pGF-IVL645, and pGF-IVL646), grown overnight at 30°C in liquid SD-Leu medium containing 2% raffinose and 0.2% sucrose, spotted onto agar plates containing SD-Leu (left) or SGal-Leu (right), and incubated at 30°C for 3 d. In two constructs, C2Lact domain was fused to the C-terminus of the Hsl1(1–374)-GFP or Hsl1(1–374; D239A) fragment, as indicated. Red asterisks, overexpressed fragments containing both the 611–950 segment and the KA1 domain cause an Hsl1-deficient phenotype in mih1∆ cells. (C) Hsl1 kinase-dead (D239A) allele localizes normally. Cells of strain GFY-42 expressing both Cdc10-mCherry from its endogenous locus and a full-length, catalytically inactive Hsl1 allele, Hsl1(D239A) (tagged with eGFP), from a plasmid (pGF-IVL693) were visualized by fluorescence microscopy. Scale bar, 2 μm.
Given our insight into how to circumvent the toxicity of overexpression, we were able to test whether overexpression of fragments of Hsl1 containing the 611–950 segment, the KA1 domain, or both were capable of displacing native Hsl1 from the bud neck and thereby conferring an Hsl1-deficient-like phenotype to HSL1+ mih1∆ cells. Indeed, when overexpressed, fragments containing both 611–950 and the KA1 domain, such as Hsl1(375–1518) and Hsl1(611–950; 1245–1518), efficiently prevented the growth of HSL1+ mih1∆ cells (Figure 8B, right, red asterisks). In contrast, a fragment containing only the 611–950 segment had only a modest growth-inhibitory effect, and a fragment containing only the KA1 domain had no detectable growth-inhibitory effect (Figure 8B, right).
Having demonstrated that the cytosolic level of the 611–950 fragment was reduced by both its tendency for nuclear import (Supplemental Figure S5) and its APC-mediated degradation (Supplemental Figure S7), we sought to increase the competitive advantage of this segment by eliminating its I-NLS and KEN and D-boxes by mutation. Strikingly, this maneuver revealed that, when overexpressed, the resulting mutated 611–950 segment alone was sufficient to cause a marked growth-inhibitory effect in HSL1+ mih1∆ cells (Figure 9A, right) and even to have a detectably deleterious effect on growth in HSL1+ MIH1+ cells (Figure 9A, left). Moreover, when combined with the KA1 domain and overexpressed, the stabilized 611–950 fragment potently inhibited growth in both HSL1+ mih1∆ (Figure 9A, right) and HSL1+ MIH1+ cells (Figure 9A, left). By tracking the localization of both endogenously expressed Hsl1-GFP and a septin in the cells, we could directly demonstrate that presence of the overexpressed, stabilized 611–950 segment was both necessary and sufficient to displace native Hsl1-GFP from the septin collar (Figure 9B). Revealingly, as expected for high-affinity binding of the 611–950 segment to septin filaments, overexpression of the stabilized 611–950 fragment alone often caused coalescence of the septins into a single large intracellular aggregate and, when fused to the KA1 domain, caused recruitment of septin bundles to the PM, whereas overexpression of the KA1 domain completely lacked this effect (Figure 9B). These outcomes were not specific to Cdc10-mCherry because the same phenotypes were observed in strains expressing Cdc11-mCherry and Hsl1-GFP (G.C.F., unpublished data). These data amply confirm, as observed in vitro, that the 611–950 segment is the septin-binding region of Hsl1 in vivo. Moreover, in keeping with their observed capacity to displace endogenous Hsl1 from the bud neck and/or to sequester septins and thereby disrupt septin collar organization at the bud neck, the same fragments that were growth inhibitory when overexpressed also elicited the elongated cell morphology (Figure 9C) that is the hallmark of the pronounced G2/M delay manifested by both Hsl1- and septin-deficient cells (McMillan et al., 1999; Shulewitz et al., 1999; Lew, 2003; Sakchaisri et al., 2004).
FIGURE 9:

Preventing APC-mediated degradation and blocking nuclear import of the 611–950 fragment of Hsl1 markedly enhance its ability to confer an Hsl1-deficient phenotype by displacing endogenous Hsl1 from the bud neck. (A) Wild-type cells (BY4741, leftmost pair) or an otherwise isogenic mih1∆ derivative (GFY-1652, rightmost pair) were transformed with either empty vector (pRS315) or derived plasmids overexpressing from the GAL1/10 promoter either GST alone (pGF-IVL914) or the other indicated fragments of Hsl1 fused to GST (pGF-IVL911, pGF-IVL916, pGF-IVL912, pGF-IVL917, pGF-IVL913, and pGF-IVL918), which were also tagged with a FLAG epitope at the end opposite from that fused to GST and then propagated and tested for growth as described in Figure 8B. (B) Strain GFY-1739, expressing both Cdc10-mCherry and Hsl1-GFP from their endogenous loci, was transformed with the same plasmids as in A, grown overnight to saturation in SD-Leu medium containing 2% raffinose and 0.2% sucrose, backdiluted in SD-Leu medium containing 2% galactose, incubated for 4–5 h at 30°C, and then examined by fluorescence microscopy. Arrowheads, mCherry-marked septins deposited at ectopic locations. (C) Representative cells in cultures of the same strains as in B, numbered 1–8 for clarity, that were grown overnight in SGal-Leu medium to saturation and then examined by DIC. Images were scaled identically; scale bar, 2 μm.
DISCUSSION
We demonstrated here, using multiple independent methods (localization of 400-residue fragments of Hsl1, localization of truncations and internal deletions of otherwise intact Hsl1, localization of fusions of portions of Hsl1 to a GST-MBP-eGFP reporter, localization of fusions of Hsl1 segments to its KA1 domain or alternative membrane-targeting domains, and in vivo competition assays), that coordinated action between a central region (residues 611–950) of Hsl1 and its C-terminal KA1 domain (Figure 10A) is both necessary and sufficient for recruitment of this protein kinase exclusively to the septin collar at the bud neck. Based on our collective findings, the most parsimonious model to explain this synergy is that the PtdSer-binding KA1 domain concentrates Hsl1 at the PM, permitting more efficient encounter of its 611–950 septin-binding domain with the septin filaments at the bud neck (because the septin collar is also tightly associated with the PM; Figure 10B). In addition, we also showed that within the 611–950 segment, there are two tandem yet distinct septin-association elements that contain two different conserved sequence motifs (643–688 and 877–910) and that both of these elements contribute to ensuring that Hsl1 is localized efficiently and solely to the septin collar. That these two elements act in a cooperative manner but each is sufficient for readily detectable bud neck localization presumably explains why their joint role was overlooked in a prior study (Crutchley et al., 2009) in which segments containing each of the conserved regions were deleted but not both.
FIGURE 10:
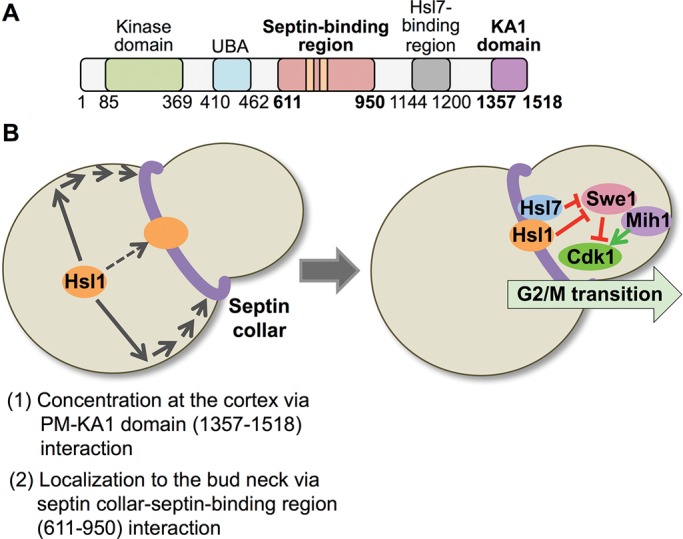
Two-step mechanism for localization of Hsl1 to the septin collar at the bud neck. (A) Diagram of Hsl1 with the locations of the septin-binding region (pink) pinpointed in this study and the KA1 domain (purple) highlighted. As described in the text, the 611–950 segment contains two distinct and separable septin-association elements, as well as the KEN and D-box APC recognition motifs (orange bars). Residues 611–950 are both necessary and sufficient for binding of Hsl1 to the septin filaments in the collar at the bud neck; however, for optimal and exclusive recruitment of Hsl1 to this location, interaction of the KA1 domain with the PM is required. (B) Two-stage model for efficient recruitment of Hsl1 to the septin collar. Left, at the bud neck, septin filaments are tightly associated with the PM via their binding to PtdIns4,5P2, a lipid enriched at the bud neck. The KA1 domain concentrates Hsl1 at the PM by binding to PtdSer, another lipid that is also enriched at the bud neck. Thus the most likely scenario to account for the observed synergy between the KA1 domain and the 611–950 septin-binding segment is that KA1-mediated concentration of Hsl1 at the PM permits more efficient encounter of its 611–950 septin-binding domain with the septin filaments. Right, subsequent Hsl1-dependent recruitment of Hsl7 to the bud neck promotes degradation of Swe1, a negative regulator of Clb-bound Cdk1/Cdc28, which, in conjunction with the action of the phosphatase Mih1, fully releases Cdk1 from inhibition, allowing for timely initiation of and passage through the G2/M transition of the cell cycle.
Here we also confirmed using biochemical methods that the recombinant 611–950 segment binds in vitro to purified polymerized septin hetero-octamers and promotes filament formation, as expected for a protein whose binding stabilizes assembled filaments. EM analysis revealed that the MBP-tagged 611–950 fragment decorates filament bundles in a regular pattern with ∼32-nm periodicity, consistent with the repeat length of a hetero-octameric rod (Bertin et al., 2008). In this regard, it is noteworthy that the two semiredundant septin-associating elements within the 611–950 segment bear little overt sequence relatedness to each other (aside from a similar spacing of basic residues) and are not clearly conserved in Gin4, Kcc4, or any other protein kinase that is also localized at the bud neck. This distinctiveness suggests that the septin-binding elements in Hsl1 have uniquely evolved to recognize some specific feature of assembled septin filaments. Therefore, in subsequent work, it will be interesting to carry out further EM analysis with tagged septin subunits (Bertin et al., 2008) and/or use other biophysical methods, such as Förster resonance energy transfer (Booth et al., 2015), to determine with which of the septin subunits the 611–950 domain makes the most intimate contact.
Strikingly, and consistent with its role in mediating direct tethering of Hsl1 to septins in vivo, overexpression of a GST-Hsl1(611–950) fusion (with or without an appended KA1 domain) in which both the cryptic I-NLS and the KEN and D-boxes have been removed (to prevent any trafficking to the nucleus and stabilize the fragment against APC-initiated destruction) physically displaced endogenously expressed Hsl1-GFP from the bud neck and also caused the formation of ectopic septin structures distinct and separate from the bud neck. The latter observation—that the 611–950 domain is able to redirect septin structures to nonnative localizations—provides additional compelling evidence that this segment of Hsl1 binds directly to septin filaments. Similarly, overexpression of the septin-binding elements in other septin-associated proteins, such as the bud site selection machinery components Bud3 (Guo et al., 2011) and Bud4 (Kang et al., 2013), are also able to redirect septins to ectopic locations in vivo.
Our findings have important implications for understanding how association with the septin collar promotes the function of Hsl1. First, using the standard two-hybrid method to assess protein–protein interaction, Hanrahan and Snyder (2003) reported that residues 987–1100 are the septin-binding region of Hsl1. Our results, however, obtained by direct visualization in vivo and direct binding assays in vitro, are completely at odds with that conclusion. In addition, that same study provided evidence, again using the two-hybrid method, that residues 987–1100 could associate with the N-terminal kinase domain of Hsl1; however, using GST pull-downs with purified proteins, we were unable to detect any such interaction (B. Gullbrand and J. Thorner, unpublished data). Moreover, that same study reported, based on Hsl1 autophosphorylation, that the presence of the putative 987–1100 inhibitory domain inactivated Hsl1 kinase activity in vitro and that addition of septins Cdc11 and Cdc12 to that mixture restored Hsl1 kinase activity in vitro; others, however, have been unable to repeat those observations (Crutchley et al., 2009; Szkotnicki et al., 2008). Similarly, in our hands, and using recombinant Shs1 as the phospho-acceptor (because it serves as an efficient exogenous substrate for Hsl1), addition of individual recombinant septins, septin hetero-octamers, or assembled septin filaments does not detectably stimulate full-length and septin-free Hsl1 purified from yeast (B. Gullbrand and J. Thorner, unpublished data). Hence it seems unlikely that Hsl1 association with the septin collar leads to its activation by alleviating intramolecular autoinhibition of this enzyme via the specific model proposed by Hanrahan and Snyder (2003).
Second, although yeast AMPK (Snf1) is strongly activated by phosphorylation on T210 in its activation loop by any of three upstream protein kinases (Sak1, Tos3, or Elm1; Elbing et al., 2006; Hedbacker and Carlson, 2008), it is not so clear that modification of the corresponding residue in members of the AMPK-like kinase group is obligatorily required for their catalytic activity (Alessi et al., 2006; Crozet et al., 2014). For example, in one study, it was reported that the bud neck–associated AMPK-like kinases Gin4 and Kcc4 could be phosphorylated and stimulated by the upstream bud neck–localized kinase Elm1 but Hsl1 could not (Asano et al., 2006). In another study, it was reported that Elm1 is capable of phosphorylating and stimulating Hsl1 but that Hsl1 has a significant basal activity and the degree of stimulation is rather modest (Szkotnicki et al., 2008). In agreement with the latter study, we find that the kinase domain of Hsl1, Hsl1(1–500), expressed and purified from Escherichia coli is intrinsically and robustly active as a protein kinase (again using purified Shs1 as the substrate) in the absence of any Elm1-mediated phosphorylation (B. Gullbrand and J. Thorner, unpublished data). For these reasons, we do not favor the model that association of Hsl1 with the septin collar leads to activation of the enzyme by placing it in close proximity to an essential upstream activator.
By contrast, because both the KEN box (residues 775–781) and D-box (828–836) that mediate APC-mediated destruction of Hsl1 lie squarely within the septin-associating region that we identified (residues 611–950; Figure 10A), we favor the view that association of Hsl1 with the septin collar promotes the function of this enzyme, at least in part, by sparing it from degradation rather than by stimulating its intrinsic catalytic activity. Overall our findings support this model. We found, first, that the level of GFP fluorescence increased for any construct that contained mutated KEN and/or D-boxes compared with the corresponding Hsl1(611–950) control. This effect was masked, however, when a KA1 domain–containing fragment was appended onto each construct: each displayed a robust signal at the septin collar regardless of the presence or absence of a mutated KEN and/or D-box. Thus Hsl1 fragments (such as 611–950) that are only weakly septin associated and largely cytosolic/nuclear are readily accessible to APC-initiated ubiquitinylation and subsequent degradation, whereas fragments (such as the 611–950; 1245–1518 fusion), once efficiently recruited and tightly bound to the septin filaments in the collar, are protected from being marked for degradation.
As noted in Results, in the course of our dissection of Hsl1, we encountered what appeared to be rather numerous cryptic NLS and NES elements in various fragments of this polypeptide, suggesting that Hsl1 may undergo nucleocytoplasmic shuttling (either constitutively or in a regulated manner). However, one hallmark of a protein that undergoes such shuttling is accumulation in the nucleus if its export is blocked. We found, however, that eliminating the function of each of the four karyopherins known to mediate the export of other proteins from the yeast nucleus did not cause any detectable accumulation of Hsl1 inside the nucleus. Of course, we cannot rule out that Hsl1 is ejected from the nucleus by either of two different exportins or that Hsl1 is exported by a karyopherin not previously implicated in export.
In many respects, Hsl1 is a remarkably modular protein (Figure 10A). In particular, we found that for targeting to the septin collar at the bud neck in vivo, the PtdSer-binding KA1 domain situated at the extreme C-terminus of Hsl1 synergizes with the septin-binding elements embedded in the middle of the protein sequence. PtdSer is a lipid that appears to be enriched at the bud neck (Fairn et al., 2011). Thus it is possible that this synergy provides a mechanism to couple information about the composition of the inner leaflet of the PM with the degree of completeness of septin collar assembly to ensure that cytokinesis is only executed when both are optimal for all the processes necessary to separate the mother and daughter cells (Howell and Lew, 2012). In the same regard, this arrangement—presence of a membrane-binding domain at the extreme C-terminus of a large protein that needs to be situated in intimate contact with the septin collar at the bud neck to carry out its function—is not confined to Hsl1. Clearly, as shown originally by Moravcevic et al. (2010) and confirmed here, both Gin4 and Kcc4 have functional KA1 domains at their extreme C-termini, and it has been shown that Gin4 contributes to regulating the lipid distribution in the inner leaflet of the PM in a way that affects the efficiency of cytokinesis (Roelants et al., 2015). Similarly, the septin-binding proteins Bud4 (Kang et al., 2013; Wu et al., 2015) and Boi1 (and its paralogue Boi2; Hallett et al., 2002), which are necessary for proper septin organization for bud emergence, have phosphoinositide-binding pleckstrin homology domains at their extreme C-termini (Yu et al., 2004), and PtdIns4,5P2 is another lipid that appears to be enriched at the bud neck (Garrenton et al., 2010). Moreover, septin filament assembly is robustly stimulated in vitro by the presence of PtdIns4,5P2 in membranes (Bertin et al., 2010; Bridges et al., 2014); in the cell, the septin filaments in the collar are tightly apposed to the PM at the bud neck, and this association is required for their function (Bezanilla et al., 2015; Finnigan et al., 2015b).
Conservation of the KA1 domain across Eukarya can be readily detected on the basis of either sequence alignments or structural homology (Moravcevic et al., 2010; Leonard and Hurley, 2011). It is still unclear, however, whether KA1 domain–mediated membrane association of Hsl1 must obligatorily precede Hsl1 binding to the septin collar (Figure 10B). Perhaps tracking newly made Hsl1 by single-molecule superresolution fluorescence microscopy could address this issue. However, our data indicate that, at least with regard to locking in Hsl1 at the septin collar, the primary role of the KA1 domain is to recruit Hsl1 to the PM. However, the KA1 domain in other protein kinases has been ascribed additional roles. In Schizosaccharomyces pombe Cdr2 (closest apparent Saccharomyces cerevisiae counterpart is Gin4), its KA1 domain is required not only for its membrane binding, but also for its apparent oligomerization/clustering at cortical septin–associated nodes that demarcate the incipient site of cell division (Rincon et al., 2014). In addition, sequences at the C-terminus of Cdr2 (perhaps involving its KA1 domain) mediate its interaction with another protein kinase, Cdr1 (closest apparent S. cerevisiae counterpart is Kcc4), a direct negative regulator of fission yeast Wee1 (Guzman-Vendrell et al., 2015). In S. cerevisiae, genetic analysis indicates that Gin4 and Kcc4 act semiredundantly with Hsl1 to down-regulate Swe1 (Barral et al., 1999), although the molecular mechanism by which they do so is rather obscure. In mammalian cells, mutations designed to disrupt the fold of the C-terminal KA1 domain in the DNA damage–activated protein kinase Chk1 (Caparelli and O’Connell, 2013) led to constitutive activity (Gong et al., 2015), consistent with an inhibitory role, perhaps mediated by interaction of the KA1 domain with the kinase domain. For MARK1/Par1c, a mammalian protein kinase involved in regulating microtubule dynamics, cell polarity, and cell migration, there is more direct, albeit preliminary, biochemical evidence that the isolated KA1 domain binds to the purified kinase domain in-trans, that this interaction inhibits the catalytic activity against a known exogenous substrate (the microtubule-binding protein Tau), and that addition of ligands that compete for binding to the KA1 domain partially alleviate the observed inhibition, consistent with an autoinhibitory mechanism (R. P. Emptage, M. A. Lemmon, and K. M. Ferguson, personal communication). Further work will be required to determine whether the C-terminal KA1 domain in Hsl1 can exert a negative regulatory effect on its catalytic activity by associating (in either cis or trans) with its N-terminal kinase domain and, if so, whether PtdSer binding to the KA1 domain, binding of septin filaments to the 611–950 domain, or both induce conformational changes that alleviate such autoinhibition and enhance the specific activity of this enzyme.
In summary, the specific advances and new insights that we obtained that were not known from any prior studies are 1) coordinated action between a central region (residues 611–950) of Hsl1 and its C-terminal KA1 domain is both necessary and sufficient for recruitment of this protein kinase exclusively to the septin collar at the bud neck, a critical synergy not previously uncovered; 2) the segment in Hsl1 (residues 987–1100) purported to mediate its recruitment to the septin collar reported by Hanrahan and Snyder (2003) was incorrect; 3) the septin-binding region of Hsl1 comprises two adjacent septin-targeting sequences, a fact previously overlooked because in a prior, more limited deletion analysis (Crutchley et al., 2009), a deletion big enough to cover both elements was not made; 4) the KEN box (residues 775–781) and D-box (828–836) lie squarely within the septin-binding region, and, therefore, association of Hsl1 with the septin collar spares Hsl1 from APC-mediated destruction; 5) with respect to promoting Hsl1 encounter and engagement with the septin collar, the sole function of the PtdSer-binding C-terminal KA1 domain in Hsl1 is to concentrate this protein kinase at the plasma membrane; 6) although Crutchley et al. (2009) reported that they were unable to detect interaction of any portion of Hsl1 with septins in vitro, we documented, using both a sedimentation assay and EM analysis, that the 611–950 segment of Hsl1 binds to septin filaments (and not to unassembled septin octamers); and 7) in agreement with their role in mediating Hsl1 binding to the septin collar, overexpression of residues 611–950 displaces endogenous Hsl1 from the bud neck.
MATERIALS AND METHODS
Yeast strains and plasmids
All budding yeast strains used in this study are listed in Table 1 (also see Supplemental Table S1). Modern molecular biology techniques were used to manipulate all DNA and yeast strains (Sambrook and Russell, 2001). Strains deleted for HSL1 were generated using the following protocol. First, chromosomal DNA was isolated from hsl1∆::KanR yeast (GFY-1541), and the HSL1 locus was PCR amplified with 500 base pairs of flanking 5′- and 3′-untranslated region (UTR) and transformed into the appropriate strains (GFY-42 to obtain GFY-1156, etc.). Proper deletions were confirmed by a series of diagnostic PCRs using oligonucleotide primers that were either upstream or downstream of the 500 base pairs of UTR used for amplification, as well as primers specific to the KanR gene cassette. For strains that also contained additional genetic modifications at other loci, diagnostic PCRs were also performed after each integration event. When appropriate, drug resistance marker cassettes (Goldstein and McCusker, 1999) were swapped to allow for use of the preferred integrating selection cassette. For creation of strains GFY-1561 to GFY-1566, the modified hsl1 alleles were first constructed on a CEN-based LEU2-marked plasmid (pRS315) using in vivo ligation and homologous recombination (Finnigan and Thorner, 2015). After plasmid construction, each Hsl1-expressing cassette was PCR amplified, including 450 base pairs of 5′-UTR, the entire open reading frame (ORF), the C-terminal triple-hemagglutinin (HA) epitope tag, 243 nucleotides of the ADH1 terminator sequence (Bennetzen and Hall, 1982), and the NatR drug cassette (Goldstein and McCusker, 1999) in two fragments: the upstream PCR product included the promoter and the front half of the HSL1 ORF, whereas the downstream fragment included the remainder of the HSL1 gene, the HA tag, terminator, and drug cassette. The two fragments contain several hundred base pairs of homology within their HSL1 sequence and, after treatment with DpnI to digest the template DNA, were cotransformed into yeast containing hsl1∆::KanR (GFY-1156). Because all of the MX4-based selection cassettes (Goldstein and McCusker, 1999) contain an identical terminator sequence following the drug resistance gene, the HSL1 PCR fragments could 1) reassemble in vivo and 2) integrate at the endogenous HSL1 locus. After subsequent selection steps to ensure the proper marker swap (and the presence of the CDC10::mCherry::SpHIS5 marker), multiple diagnostic PCRs were performed on each modified locus to ensure proper integration.
TABLE 1:
Yeast strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | MATa leu2∆ ura3∆ met15∆ his3∆ | Brachmann et al. (1998) |
| GFY-42 | BY4741 cdc10∆::CDC10::mCherry::SpHIS5 | Finnigan et al. (2015b) |
| GFY-58 | BY4741 cdc11∆::CDC11::mCherry::SpHIS5 | Finnigan et al. (2015b) |
| GFY-1156 | BY4741 cdc10∆::CDC10::mCherry::SpHIS5 hsl1∆::KanR | This study |
| GFY-1157 | BY4741 cdc11∆::CDC11::mCherry::SpHIS5 hsl1∆::KanR | This study |
| GFY-1881 | BY4741 cdc10∆::CDC10::mCherry::SpHIS5 mih1∆::KanR | This study |
| GFY-1882 | BY4741 cdc11∆::CDC11::mCherry::SpHIS5 mih1∆::KanR | This study |
| GFY-1737a | BY4741 cdc10∆::CDC10::mCherry::SpHIS5 hsl1∆::NatR mih1∆::KanR + pGF-316-IVL924 | This study |
| GFY-1738a | BY4741 cdc11∆::CDC11::mCherry::SpHIS5 hsl1∆::NatR mih1∆::KanR + pGF-316-IVL924 | This study |
| GFY-1652b | BY4742 mih1∆::KanR | Life Technologies |
| GFY-1739c | BY4741 cdc10∆::CDC10::mCherry::SpHIS5 hsl1∆::HSL1::GFP::KanR | This study |
aStrains 1737 and 1738 were constructed by first switching the KanR cassette of strains GFY-1156 and GFY-1157, respectively, to NatR, followed by transformation of the pRS316-based covering vector expressing WT HSL1 (pGF-316-IVL924) and, finally, deletion of MIH1 using the knockout cassette from GFY-1652.
bAn isogenic clone was tested by multiple diagnostic PCRs from the haploid yeast genome deletion collection (Brachmann et al., 1998).
cThis strain was constructed by integrating prHSL1::HSL1(WT)::GFP::KanR (amplified from pGF-IVL560) at the native HSL1 locus in GFY-1656.
Plasmids used in this study are listed in Table 2 (also see Supplemental Table S2). Unless otherwise indicated, the same in vivo ligation protocol for vector generation was used (Finnigan and Thorner, 2015). Briefly, all modified Hsl1-expressing plasmids used a parent vector that contained 500 base pairs of 5′-UTR to the CDC11 gene, a unique NotI restriction site, followed by the ADH1(t)-KanR cassette (pGF-IVL520). PCR amplification of one or more fragments of HSL1 was performed to generate the appropriate gene fusions or domain deletions, followed by cotransformation of linearized parent vector and selection in yeast on rich medium containing G418 disulfide (Life Technologies, Carlsbad, CA). After rescue of the plasmid out of yeast (Finnigan and Thorner, 2015), vector DNA was transformed into chemically competent TOP10 E. coli (Life Technologies). Single clonal isolates were extracted from bacteria and tested using diagnostic PCRs to ensure that the resulting vectors contained all of the required PCR fragments. Finally, the entire ORF (including junctions to both the promoter and terminator) was confirmed using DNA sequencing (University of California, Berkeley, DNA Sequencing Facility). In general, the DNA template for PCR amplification for Hsl1-expressing vectors was either full-length wild-type (WT) HSL1 (from pGF-IVL521) or a small fragment (residues 611–950; pGF-V672) that was subcloned into a TOPO II bacterial vector (Life Technologies). When appropriate, other Hsl1-containing vectors were used as DNA template for the creation of additional vectors; DpnI digestion after PCR amplification was used in these cases.
TABLE 2:
Plasmids used in this study. Continued
| Plasmid | Description | Reference |
|---|---|---|
| pGF-IVL521a,b | pRS315; prCDC11::HSL1(1-1518)::eGFP::KanR | This study |
| pRS315 | CEN, LEU2 | Sikorski and Hieter (1989) |
| pGF-IVL561 | pRS315; prCDC11::hsl1(1-400)::eGFP::KanR | This study |
| pGF-IVL562c | pRS315; prCDC11::hsl1(200-600)::eGFP::KanR | This study |
| pGF-IVL563 | pRS315; prCDC11::hsl1(400-800)::eGFP::KanR | This study |
| pGF-IVL564 | pRS315; prCDC11::hsl1(600-1000)::eGFP::KanR | This study |
| pGF-IVL565 | pRS315; prCDC11::hsl1(800-1200)::eGFP::KanR | This study |
| pGF-IVL566 | pRS315; prCDC11::hsl1(1000-1400)::eGFP::KanR | This study |
| pGF-IVL567 | pRS315; prCDC11::hsl1(1200-1518)::eGFP::KanR | This study |
| pGF-IVL522 | pRS315; prCDC11::hsl1(1-1244)::eGFP::KanR | This study |
| pGF-IVL523 | pRS315; prCDC11::hsl1(1-1357)::eGFP::KanR | This study |
| pGF-IVL524 | pRS315; prCDC11::hsl1(1358-1518)::eGFP::KanR | This study |
| pGF-IVL525 | pRS315; prCDC11::hsl1(1245-1518)::eGFP::KanR | This study |
| pGF-IVL526 | pRS315; prCDC11::hsl1(1120-1518)::eGFP::KanR | This study |
| pGF-IVL527 | pRS315; prCDC11::hsl1(800-1518)::eGFP::KanR | This study |
| pGF-IVL528 | pRS315; prCDC11::hsl1(620-1518)::eGFP::KanR | This study |
| pGF-IVL529 | pRS315; prCDC11::hsl1(375-1518)::eGFP::KanR | This study |
| pGF-IVL530 | pRS315; prCDC11::hsl1(1-1119; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL531 | pRS315; prCDC11::hsl1(1-799; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL532 | pRS315; prCDC11::hsl1(1-619; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL533 | pRS315; prCDC11::hsl1(1-374; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL534 | pRS315; prCDC11::hsl1(663-743; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL535 | pRS315; prCDC11::hsl1(611-878; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL536 | pRS315; prCDC11::hsl1(611-950; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL624 | pRS315; prCDC11::hsl1(611-950; 1245-1357)::eGFP::KanR | This study |
| pGF-IVL625 | pRS315; prCDC11::hsl1(611-950; 1358-1518)::eGFP::KanR | This study |
| pGF-IVL626 | pRS315; prCDC11::hsl1(1245-1357)::eGFP::KanR | This study |
| pGF-IVL601 | pRS315; prCDC11::hsl1(663-950; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL602 | pRS315; prCDC11::hsl1(711-950; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL603 | pRS315; prCDC11::hsl1(744-950; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL604 | pRS315; prCDC11::hsl1(611-925; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL605 | pRS315; prCDC11::hsl1(663-925; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL606 | pRS315; prCDC11::hsl1(711-925; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL607 | pRS315; prCDC11::hsl1(744-925; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL608 | pRS315; prCDC11::hsl1(611-900; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL609 | pRS315; prCDC11::hsl1(663-900; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL610 | pRS315; prCDC11::hsl1(711-900; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL611 | pRS315; prCDC11::hsl1(744-900; 1245-1518)::eGFP::KanR | This study |
| pGF-IVL612 | pRS315; prCDC11::hsl1(611-950)::eGFP::KanR | This study |
| pGF-IVL613 | pRS315; prCDC11::hsl1(663-950)::eGFP::KanR | This study |
| pGF-IVL614 | pRS315; prCDC11::hsl1(711-950)::eGFP::KanR | This study |
| pGF-IVL615 | pRS315; prCDC11::hsl1(744-950)::eGFP::KanR | This study |
| pGF-IVL616 | pRS315; prCDC11::hsl1(611-925)::eGFP::KanR | This study |
| pGF-IVL617 | pRS315; prCDC11::hsl1(663-925)::eGFP::KanR | This study |
| pGF-IVL618 | pRS315; prCDC11::hsl1(711-925)::eGFP::KanR | This study |
| pGF-IVL619 | pRS315; prCDC11::hsl1(744-925)::eGFP::KanR | This study |
| pGF-IVL620 | pRS315; prCDC11::hsl1(611-900)::eGFP::KanR | This study |
| pGF-IVL621 | pRS315; prCDC11::hsl1(663-900)::eGFP::KanR | This study |
| pGF-IVL622 | pRS315; prCDC11::hsl1(711-900)::eGFP::KanR | This study |
| pGF-IVL623 | pRS315; prCDC11::hsl1(744-900)::eGFP::KanR | This study |
| pGF-IVL665 | pRS315; prCDC11::hsl1(1-662; 951-1518)::eGFP::KanR | This study |
| pGF-IVL666 | pRS315; prCDC11::hsl1(1-610; 951-1518)::eGFP::KanR | This study |
| pGF-IVL709 | pRS315; prCDC11::hsl1(1-610; 744-1518)::eGFP::KanR | This study |
| pGF-IVL710 | pRS315; prCDC11::hsl1(1-743; 951-1518)::eGFP::KanR | This study |
| pGF-IVL711 | pRS315; prCDC11::hsl1(1-664; 689-1518)::eGFP::KanR | This study |
| pGF-IVL816d | pRS315; prCDC11::hsl1(1-1518 T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A)::eGFP::KanR | This study |
| pGF-IVL 765d | pRS315; prCDC11::hsl1(611-900 R635A R636A K645A H648A K649A R653A K654A)::eGFP::KanR | This study |
| pGF-IVL 766d | pRS315; prCDC11::hsl1(611-900 R663A R664A K672A R673A K683A R684A)::eGFP::KanR | This study |
| pGF-IVL818d | pRS315; prCDC11::hsl1(611-900 T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A)::eGFP::KanR | This study |
| pGF-IVL762d | pRS315; prCDC11::hsl1(611-950 R635A R636A K645A H648A K649A R653A K654A)::eGFP::KanR | This study |
| pGF-IVL763d | pRS315; prCDC11::hsl1(611-950 R663A R664A K672A R673A K683A R684A)::eGFP::KanR | This study |
| pGF-IVL817d | pRS315; prCDC11::hsl1(611-950 T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A)::eGFP::KanR | This study |
| pGF-IVL705d | pRS315; prCDC11::hsl1(611-900; 1245-1518 R635A R636A K645A H648A K649A R653A K654A)::eGFP::KanR | This study |
| pGF-IVL760d | pRS315; prCDC11::hsl1(611-900; 1245-1518 R663A R664A K672A R673A K683A R684A)::eGFP::KanR | This study |
| pGF-IVL820d | pRS315; prCDC11::hsl1(611-900; 1245-1518 T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A)::eGFP::KanR | This study |
| pGF-IVL757d | pRS315; prCDC11::hsl1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A)::eGFP::KanR | This study |
| pGF-IVL758d | pRS315; prCDC11::hsl1(611-950; 1245-1518 R663A R664A K672A R673A K683A R684A)::eGFP::KanR | This study |
| pGF-IVL819d | pRS315; prCDC11::hsl1(611-950; 1245-1518 T665A L666A N668A S669A S671A K672A R673A S674A L675A Y676A S677A S680A I681A S682A K683A R684A S685A N687A L688A)::eGFP::KanR | This study |
| pGF-IVL638e | pRS315; prCDC11::hsl1(1-1357)::gin4(1003-1142)::eGFP::KanR | This study |
| pGF-IVL639e | pRS315; prCDC11::hsl1(611-950)::gin4(1003-1142)::eGFP::KanR | This study |
| pGF-IVL640e | pRS315; prCDC11::hsl1(1-1357)::kcc4(877-1037)::eGFP::KanR | This study |
| pGF-IVL641e | pRS315; prCDC11::hsl1(611-950)::kcc4(877-1037)::eGFP::KanR | This study |
| pGF-IVL181e | pRS315; prCDC11::eGFP::kcc4(877-1037)::KanR | This study |
| pGF-IVL184e | pRS315; prCDC11::eGFP::gin4(1003-1142)::KanR | This study |
| pGF-IVL187 | pRS315; prCDC11::eGFP::hsl1(1358-1518)::KanR | This study |
| pGF-IVL687f | pRS315; prCDC11::hsl1(1-1357)::eGFP::Lact-C2(1-158)::KanR | This study |
| pGF-IVL688f | pRS315; prCDC11::hsl1(611-950)::eGFP::Lact-C2(1-158)::KanR | This study |
| pGF-IVL708f | pRS315; prCDC11::eGFP::Lact-C2(1-158)::KanR | This study |
| pGFY-316-IVL924g | pRS316; prHSL1::HSL1(1-1518)::SHS1(3′UTR) | This study |
| pGF-IVL693 | pRS315; prCDC11::hsl1(1-1518 D239A)::eGFP::KanR | This study |
| pGF-IVL391 | pRS315; prGAL1/10::eGFP::KanR | This study |
| pGF-IVL302h | pRS315; prGAL1/10::HSL1(1-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL694i | pRS315; prGAL1/10::hsl1(1-1518 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL689 | pRS315; prGAL1/10::hsl1(1-374)::eGFP::SpHIS5 | This study |
| pGF-IVL932 | pRS315; prGAL1/10::hsl1(1-374 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL642 | pRS315; prGAL1/10::hsl1(375-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL643 | pRS315; prGAL1/10::hsl1(1-1244)::eGFP::SpHIS5 | This study |
| pGF-IVL929 | pRS315; prGAL1/10::hsl1(1-1244 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL893A | pRS315; prGAL1/10::hsl1(1-610; 951-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL893B | pRS315; prGAL1/10::hsl1(1-610; 951-1518 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL863 | pRS315; prGAL1/10::hsl1(1-662; 951-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL894B | pRS315; prGAL1/10::hsl1(1-662; 951-1518 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL895 | pRS315; prGAL1/10::hsl1(1-374; 1358-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL930 | pRS315; prGAL1/10::hsl1(1-374; 1358-1518 D239A)::eGFP::SpHIS5 | This study |
| pGF-IVL896f | pRS315; prGAL1/10::hsl1(1-374)::eGFP::Lact-C2::SpHIS5 | This study |
| pGF-IVL931f | pRS315; prGAL1/10::hsl1(1-374 D239A)::eGFP::Lact-C2::SpHIS5 | This study |
| pGF-IVL645 | pRS315; prGAL1/10::hsl1(611-950)::eGFP::SpHIS5 | This study |
| pGF-IVL644 | pRS315; prGAL1/10::hsl1(611-950; 1245-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL646 | pRS315; prGAL1/10::hsl1(1245-1518)::eGFP::SpHIS5 | This study |
| pGF-IVL914j | pRS315; prGAL1/10::GST::KanR | This study |
| pGF-IVL911k,l | pRS315; prGAL1/10::GST::Hsl1(611-950 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::FLAG::KanR | This study |
| pGF-IVL916 | pRS315; prGAL1/10::FLAG::Hsl1(611-950 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::GST::KanR | This study |
| pGF-IVL912 | pRS315; prGAL1/10::GST::Hsl1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::FLAG::KanR | This study |
| pGF-IVL917 | pRS315; prGAL1/10::FLAG::Hsl1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::GST::KanR | This study |
| pGF-IVL913 | pRS315; prGAL1/10::GST::Hsl1(1245-1518)::FLAG::KanR | This study |
| pGF-IVL918 | pRS315; prGAL1/10::FLAG::Hsl1(1245-1518)::GST::KanR | This study |
| p42m | 2E; CDC3 CDC10 CDC11 6xHis-CDC12 | This study |
| pXSMS001n | pHM3C-LIC; 6xHis::MBP::PreScission cleavage::Hsl1(611-950) KanR | This study |
| pXSMS008n | pHM3C-LIC; 6xHis::MBP::PreScission cleavage KanR | This study |
aFor clarity, rather than present the deleted residues, the numbering system for alleles of HSL1 presents the amino acid resides still present within the final protein sequence.
bFor variants of Hsl1 that include the CDC11 promoter and are C-terminally tagged with eGFP::KanR, the following construction strategy was used to aid in plasmid generation. First, a parent vector (pGF-IVL520) was generated that contained prCDC11::eGFP::KanR with a unique NotI restriction site between the promoter sequence and the first codon of eGFP. In vivo ligation and homologous transformation was used to gap repair the linearized parent vector to insert the fragment(s) of HSL1 sequence. In addition, due to the large size of the HSL1 gene, multiple PCRs were often generated containing a significant amount (several hundred bases) of homology to link adjacent fragments.
cFor Hsl1 constructs that did not include the native N-terminal sequence of Hsl1 (such as pGF-IVL562), a start codon was included within the parent vector (pGF-IVL520); the numbering scheme excludes this initiator Met residue.
dSuccessive rounds of a modified QuikChange PCR mutagenesis protocol (Zheng et al., 2004) were performed on a fragment of Hsl1(611-950) subcloned into a TOPO-II vector (pGF-V672; Life Technologies) to mutate the desired residues to alanine before creation of the final yeast vector using in vivo ligation and homologous recombination.
eThe KA1 domains Gin4(1003-1142) and Kcc4(877-1037) were defined as previously described (Moravcevic et al., 2010).
fThe Bos taurus lactadherin C2 (Lact-C2) domain (Shao et al., 2008) was fused to each construct on the C-terminus of eGFP. The numbering scheme for Lact-C2 refers to the specific domain and not the full-length lactadherin protein. The template DNA used to amplify the Lact-C2 domain is plasmid 22853 (Addgene, Cambridge, MA).
gThe URA3-based covering vector expressing WT HSL1 was constructed by first constructing prHSL1::HSL1(1-1518)::SHS1(3′UTR) in a pRS315 vector containing a unique NotI restriction site downstream of the SHS1 3′ UTR sequence and the NatR cassette (pGF-IVL924). The HSL1 cassette was then subcloned to pRS316 (NotI/SpeI sites).
hFor plasmids expressing Hsl1 under control of the GAL1/10 promoter and C-terminally tagged with eGFP::SpHIS5, a parental vector was first constructed (pGF-IVL300), which included the GAL1/10 promoter linked to eGFP::SpHIS5 on pRS315 with a unique NotI restriction site. In vivo ligation and homologous recombination were used to insert the desired HSL1 fragment(s) into this vector.
iFor alleles of Hsl1 that included point mutation(s), a modified QuikChange protocol (Zheng et al., 2004) was used to generate the appropriate substitutions on fragments of Hsl1 subcloned into a TOPO-II vector (Life Technologies) before PCR amplification for plasmid construction. Multiple substitutions, such as in pGF-IVL821, were performed by successive rounds of QuikChange PCR.
jThe GST tag was amplified from vector pJT4649 in constructs pGF-IVL911 to pGF-IVL914 and pGF-IVL916 to pGF-IVL918 and ends with the amino acid sequence PGIHRDGG in each of the aforementioned vectors.
kThe single FLAG epitope tag has the sequence DYKDDDDK in vectors pGF-IVL911 to pGF-IVL913 and pGF-IVL916 to pGF-IVL918 and was inserted using in vivo ligation.
lThe Hsl1 vectors pGF-IVL911, pGF-IVL912, pGF-IVL916, and pGF-IVL917, which contain the fragment from residue 611–950, also contain the mutations to disrupt the putative NLS signal (R635A R636A K645A H648A K649A R653A K654A), as well as both the KEN and D-box motifs (K775A E776A N777A R828A L831A). The constructs were assembled using in vivo ligation and used pGF-IVL762 and pGF-IVL654 as templates to combine all of the necessary mutations and fragments.
mAddgene vector 29775, provided by Scott Gradia at the QB3 Macrolab, University of California, Berkeley. This vector includes a kanamycin resistance gene.
nBoth vectors contain the kanamycin resistance gene. In addition, the PreScission cleavage site included after the MBP sequence is from the human rhinovirus 3C protease cleavage site with the sequence LEVLFQGP. For plasmid pXSMS008, a STOP codon was mutated within the first codon of the Hsl1 sequence.
Culture conditions
Yeast were grown in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) or in synthetic drop-out medium containing the appropriate amino acids and either dextrose (2%) or a raffinose (2%) and sucrose (0.2%) mixture. Serial dilution (fivefold) growth assays were performed by spotting ∼5 μl of an overnight yeast culture diluted to ∼1 A600 nm unit of cells with water onto agar plates. For strains harboring temperature-sensitive mutations or strains grown at varying temperatures, the following protocol was used. First, yeast were selected twice at room temperature on medium containing 5-fluoroorotic acid (5-FOA; Oakwood Products, West Columbia, SC; final concentration of 0.5 mg/ml; heated for 30 min to at least 70°C and filter sterilized, not autoclaved) in order to counterselect any URA3-based covering vectors expressing WT copies of essential septin subunits (e.g., CDC11 in pJT1520). Next, strains were grown overnight in YPD at 22–25°C before spotting and incubating for 2–3 d at the indicated temperatures before being imaged. For the temperature-sensitive strains expressing either xpo1-1 or cse1-1, two clonal isolates were struck onto YPD plates and incubated for 4 d (25, 30, and 37°C) or 14 d (11°C). For growth assays on medium containing galactose, strains were grown overnight at 30°C in synthetic drop-out cultures containing raffinose and sucrose before being directly spotted onto plates containing (2%) galactose and lacking leucine and incubated for 3 d before imaging. For growth tests of strains deleted for both HSL1 and MIH1, cultures were grown overnight in SD-URA (to maintain the URA3-based covering plasmid expressing WT HSL1) and spotted onto synthetic complete medium with or without 5-FOA and incubated for 3 d before imaging.
Fluorescence microscopy
For all microscopy imaging, yeast were grown to exponential phase (A600 nm ≈ 0.7–1.0 U) in YPD cultures at 30°C unless otherwise noted, harvested, washed with water, and prepared on standard microscope slides with a coverslip. Samples were imaged within 5 min of slide preparation on an Olympus BH-2 upright fluorescence microscope (Olympus, Tokyo, Japan) with a 100× objective lens and illuminated with a SOLA light source (Lumencore, Beaverton, OR). A CoolSNAP MYO charge-coupled device camera (Photometrics, Tucson, AZ), Micro-Manager software (Edelstein et al., 2010), and ImageJ (National Institutes of Health, Bethesda, MD) software were used to obtain and analyze all images. All images were treated identically and rescaled together. The cell perimeter was determined using an overexposed fluorescence image. Representative images were chosen for each genotype, and all experiments were performed in triplicate or quadruplicate. All images were taken using identical exposures for both fluorescence channels. To quantify GFP fluorescence at the bud neck, a box tool was used in ImageJ to outline the bud neck region in cells that also had a septin ring (marked with mCherry) that had not yet split into two rings. The average pixel intensity was measured in triplicate across multiple images from separate trials, and error represents the SD of the mean value. For quantification of the fluorescent signal present on the cell periphery, the following strategy was used (Supplemental Figure S3). First, the maximum pixel intensity was measured for four lines (ImageJ line tool) bisecting the plasma membrane (PM) of a given cell. Next, four lines measured the average pixel intensity of randomly chosen sections of the cytosol for the same cell that did not extend across the cell periphery. Finally, the average PMmax measurement was divided by the cytosolavg measurement to yield the arbitrary ratio of PM:cytosol. At least 35–50 cells for each genotype were measured in triplicate, and the error represents the SD of the mean. For strains expressing the cdc12–6::mCherry temperature-sensitive allele, yeast were grown in SD-Leu cultures overnight at 25°C, backdiluted to A600 nm = 0.30, and grown for 5 h in fresh SD-Leu. Samples were shifted to a 37°C water bath for 1 h, harvested, and imaged within 5 min. For yeast expressing exportin ts alleles, cultures of SD-Ura (harboring vector pGF-IVL774) were grown overnight at 30°C, backdiluted to 22°C for 5 h, and then shifted to a 12°C water bath (for cse1-1) or to 37°C (for xpo1-1) for 6 h before imaging. For yeast strains containing vectors under control of the GAL1/10 promoter, cultures were grown overnight in a raffinose and sucrose mixture, backdiluted into synthetic medium containing 2% galactose, and grown at 30°C for 4–5 h before imaging. For differential interference contrast (DIC) microscopy (Figure 8C), cultures were grown overnight to saturation in synthetic medium containing galactose at 30°C, washed with water, and directly imaged.
Expression and purification of septin complexes
Cdc11-capped octamers were expressed from a single vector constructed via ligation-independent cloning (Aslanidis and de Jong, 1990) such that CDC3, CDC10, CDC11, and CDC12 were incorporated into a bicistronic DUET (Life Technologies) vector. The vector contained compatible origins of replication that were previously used for septin complex expression (Versele et al., 2004; Bertin et al., 2008). Plasmids were transformed into E. coli NiCo21 (DE3; New England Biolabs, Ipswich, MA) to allow for removal of chitin-modified endogenous E. coli gene products (Robichon et al., 2011; Booth et al., 2015). Bacterial cultures (1 l) were grown at 37°C to A600 nm = 0.8–1.0 and induced with isopropyl-β-d-thiogalactoside at a final concentration of 0.5 mM. After 16–20 h of induction at 16°C, cells were harvested by centrifugation and resuspended in lysis buffer at 4°C (300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], 2 mM MgCl2, 20 mM imidazole, 12% glycerol, 0.5% Tween-20, protease inhibitor mix [Complete, EDTA-free; Roche, Indianapolis, IN], 40 μM GDP, 0.1% 1-thioglycerol, and 0.2 mg/ml lysozyme). Cells were frozen and stored at −80°C until purification.
Frozen cell pellets were thawed and agitated for 30 min in 4°C water. Cells were physically broken via sonication at 4°C for six 30-s pulses with 2-min cooling intervals via a Misonix 3000 sonicator. The cell lysate was clarified by centrifugation at 25,000 × g for 45 min at 4°C. Immobilized metal affinity chromatography (IMAC)–contaminating proteins tagged with a chitin-binding domain were first removed by incubating clarified lysate with chitin-agarose resin for 30 min. The flowthrough containing the septin complexes and remaining endogenous E. coli gene products was subjected to IMAC on Ni2+–nitriloacetic acid (NTA) agarose beads (GE Healthcare, Pittsburgh, PA), leveraging an N-terminal His6 tag on Cdc12. After incubation, the beads were washed (wash buffer: 300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], 25 mM imidazole, and 0.1% 1-thioglycerol), and products were eluted in 0.5-ml fractions (elution buffer: 300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], 300 mM imidazole, and 0.1% 1-thioglycerol). Fractions containing the highest concentration of protein (visual via Thermo Scientific Pierce Coomassie Plus [Bradford] Protein Assay and separate A280nm measurement with a NanoDrop 8000 [Thermo Scientific, Wilmington, DE]) were pooled and filtered with a 0.2 μm polyvinylidene fluoride membrane. A 5-ml amount of the filtered product was loaded on a Superdex 200 HiLoad 16/60 column (GE Healthcare) via an AKTA FPLC system (GE Healthcare) for size-exclusion chromatography in high-salt buffer (300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], and 0.1% 1-thioglycerol). The 1-ml fractions were collected, analyzed by SDS–PAGE, and stained with colloidal Coomassie G-250 (InstantBlue; Expedeon, San Diego, CA). Fractions with the highest concentration (A280nm measurement with a NanoDrop 8000) of stoichiometric septin complexes were pooled, flash frozen (liquid nitrogen), and stored at −80°C until use.
Expression and purification of MBP-Hsl1(611–950) and MBP
Using the In-Fusion HD Cloning Plus kit (Takara Clontech, Mountain View, CA), we inserted Hsl1(611–950) into the pHM3C-LIC vector at the Sma1 site. The Hsl1(611–950) sequence was derived from pGF1966+IVL678. Standard site-directed mutagenesis (Wang and Malcolm, 1999) was used to construct the MBP expression vector from the MBP-Hsl1(611–950) vector by substitution of a single nucleotide creating a stop codon. Expression of each construct followed the procedure detailed for the septin complexes. The purification process for each protein followed the details of the septin complex purification with the addition of an anion exchange chromatography step after the IMAC. Specifically, for MBP-Hsl1(611–950), Ni2+-NTA fractions with the highest concentration were pooled and diluted to 82 mM KCl, 20 mM Tris-HCl (pH 8.0 at 4°C), 82 mM imidazole and 0.1% 1-thioglycerol for loading on a 1-ml HiTrap Q HP column (GE Healthcare) via an AKTA FPLC system (10-ml sample loop). For MBP, a single Ni2+-NTA fraction was diluted to 22.5 mM KCl, 20 mM Tris-HCl (pH 8.0 at 4°C), 22.5 mM imidazole, and 0.1% 1-thioglycerol for loading on a 1-ml HiTrap Q HP column via an AKTA FPLC system (5-ml sample loop). Protein was eluted via a salt gradient from the starting salt concentration listed for each protein to 1 M KCl, 20 mM Tris-HCl (pH 8.0 at 4°C), and 0.1% 1-thioglycerol. Fractions were analyzed via SDS–PAGE and visualized with InstantBlue, and the peak fractions were pooled, filtered, and subjected to size-exclusion chromatography as described for the septin complexes. Proteins were flash frozen (liquid nitrogen), and stored at −80°C until use.
Cosedimentation assay
All proteins were thawed on ice, and protein concentration was determined via A280nm measurement (NanoDrop 8000). Septin complexes were diluted alone or combined with MBP-Hsl1(611–950) or MBP in a 1:2 M ratio (Cdc11-capped octamers: MBP-Hsl1(611–950) or MBP alone) in both high-salt (300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], and 0.1% 1-thioglycerol) and low-salt conditions (85 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], and 0.1% 1-thioglycerol). As controls, MBP-Hsl1(611-950) and MBP were also subjected to the same dilution in high- and low-salt buffers. The diluted samples were incubated at room temperature for 30 min, followed by centrifugation at 313,000 × g for 15 min at room temperature (TLA-100 rotor, Optima Max UltraCentrifuge; Beckman Coulter, Brea, CA). The supernatant fraction was removed immediately and the corresponding pellet resuspended in an equivalent volume of high-salt buffer (300 mM KCl, 50 mM Tris-HCl [pH 8.0 at 4°C], and 0.1% 1-thioglycerol). The resulting samples were resolved by SDS–PAGE and stained with InstantBlue, and the gel was imaged using a Gel Doc EZ system (Bio-Rad, Hercules, CA).
EM and image collection
Protein samples were diluted and combined in the same ratio and for the same incubation period as in the cosedimentation assay. Continuous carbon grids (carbon supported by nitrocellulose on a copper grid, 400 mesh) were plasma cleaned (Solarus, Gatan, Pleasanton, CA), and the samples were adsorbed on the surface for 45 s, followed by staining with 2% uranyl formate and air drying. Electron micrographs were collected in a Tecnai T12 electron microscope (FEI, Hillsboro, OR) operated at 120 kV and equipped with a CMOS camera (TVIPS TemCam F416; Gauting, Germany). Micrographs were taken at 49,000× magnification using an ~−1-μm defocus.
Supplementary Material
Acknowledgments
We thank Anne Paoletti (Institut Curie, Paris, France) and Kate Ferguson and Mark Lemmon (Yale University, New Haven, CT) for helpful discussions and/or communication of unpublished results, the QB3 MacroLab (University of California, Berkeley), supported by funds from the W. M. Keck Foundation, for assistance with construction of plasmids for recombinant septin expression, and Libby Booth (Thorner lab) and Avinash Patel (Nogales lab) for assistance with construction of plasmids for expression of MBP and MBP-Hsl1(611–950). This work was supported by a postdoctoral fellowship from the Adolph C. and Mary Sprague Miller Institute for Basic Research in Science (to G.C.F.), a National Science Foundation Predoctoral Fellowship (to G.G.), funds from the Howard Hughes Medical Institute (to E.N.), and National Institutes of Health R01 Research Grants GM101314 (to E.N. and J.T.) and GM21841 (to J.T.).
Abbreviations used:
- APC
cyclosome/anaphase-promoting complex protein-ubiquitin ligase
- C2Lact
phosphatidylserine-binding C2 domain of bovine lactadherin
- D-box
destruction box
- DIC
differential interference microscopy
- eGFP
enhanced green fluorescent protein
- EM
electron microscopy
- 5-FOA
5-fluoro-orotic acid
- GST
glutathione S-transferase
- KA1
kinase associated-1
- MBP
maltose-binding protein
- NES
nuclear export signal
- NLS
nuclear localization signal
- PM
plasma membrane
- PtdIns4,5P2
phosphatidylinositol-4,5-bisphosphate
- PtdSer
phosphatidylserine.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0177) on May 18, 2016.
REFERENCES
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Allison DS, Schatz G. Artificial mitochondrial presequences. Proc Natl Acad Sci USA. 1986;83:9011–9015. doi: 10.1073/pnas.83.23.9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry. 2000;39:6200–6206. doi: 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Park JE, Yu LR, Zhou M, Sakchaisri K, Park CJ, Kang YH, Thorner J, Veenstra TD, Lee KS. Direct phosphorylation and activation of a Nim1- related kinase Gin4 by Elm1 in budding yeast. J Biol Chem. 2006;281:27090–27098. doi: 10.1074/jbc.M601483200. [DOI] [PubMed] [Google Scholar]
- Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD. The primary structure of the Saccharomyces cerevisiae gene for alcohol dehydrogenase. J Biol Chem. 1982;257:3018–3025. [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Grob P, Park SS, Garcia G, 3rd, Patanwala I, Ng HL, Alber T, Thorner J, Nogales E. ). Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA. 2008;105:8274–8279. doi: 10.1073/pnas.0803330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Pierson J, Thai L, McDonald KL, Zehr EA, Garcia G, 3rd, Peters P, Thorner J, Nogales E. Three-dimensional ultrastructure of the septin filament network in Saccharomyces cerevisiae. Mol Biol Cell. 2012;23:423–432. doi: 10.1091/mbc.E11-10-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Thai L, Garcia G, 3rd, Votin V, Grob P, Allyn T, Thorner J, Nogales E. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J Mol Biol. 2010;404:711–731. doi: 10.1016/j.jmb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, Nogales E. Septin filament organization in Saccharomyces cerevisiae. Commun Integr Biol. 2012;5:503–505. doi: 10.4161/cib.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla M, Gladfelter AS, Kovar DR, Lee WL. Cytoskeletal dynamics: a view from the membrane. J Cell Biol. 2015;209:329–337. doi: 10.1083/jcb.201502062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth EA, Vane EW, Dovala D, Thorner J. A Forster resonance energy transfer (FRET)-based system provides insight into the ordered assembly of yeast septin hetero-octamers. J Biol Chem. 2015;290:28388–28401. doi: 10.1074/jbc.M115.683128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Bridges AA, Gladfelter AS. Septin form and function at the cell cortex. J Biol Chem. 2015;290:17173–17180. doi: 10.1074/jbc.R114.634444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci USA. 2014;111:2146–2151. doi: 10.1073/pnas.1314138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Hsl1p, a Swe1p inhibitor, is degraded via the anaphase- promoting complex. Mol Cell Biol. 2000;20:4614–4625. doi: 10.1128/mcb.20.13.4614-4625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. D-box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 2001;15:2381–2395. doi: 10.1101/gad.917901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JE, Pringnitz DJ, Martens CL, Crouch N. Bovine-associated mucoprotein: I. Distribution among adult and fetal bovine tissues and body fluids. Differentiation. 1980;17:31–40. doi: 10.1111/j.1432-0436.1980.tb01079.x. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. A highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol. 1976;69:717–721. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparelli ML, O’Connell MJ. Regulatory motifs in Chk1. Cell Cycle. 2013;12:916–922. doi: 10.4161/cc.23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143:709–717. doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudron F, Barral Y. Septins and the lateral compartmentalization of eukaryotic membranes. Dev Cell. 2009;16:493–506. doi: 10.1016/j.devcel.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Cid VJ, Shulewitz MJ, McDonald KL, Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol Biol Cell. 2001;12:1645- 1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros MG, Vincens P. Computational method to predict mitochondrially-mported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- Crozet P, Margalha L, Confraria A, Rodrigues A, Martinho C, Adamo M, Elias CA, Baena-González E. Mechanisms of regulation of SNF1/AMPK/SnRK1 protein kinases. Front Plant Sci. 2014;5:190.1–190.17. doi: 10.3389/fpls.2014.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley J, King KM, Keaton MA, Szkotnicki L, Orlando DA, Zyla TR, Bardes ES, Lew DJ. Molecular dissection of the checkpoint kinase Hsl1p. Mol Biol Cell. 2009;20:1926–1936. doi: 10.1091/mbc.E08-08-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere J, Barral Y. Spatial coordination of cytokinetic events by compartmentalization of the cell cortex. Science. 2004;305:393–396. doi: 10.1126/science.1099892. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol. 2010;14 doi: 10.1002/0471142727.mb1420s92. Unit 14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K, McCartney RR, Schmidt MC. Purification and characterization of the three Snf1-activating kinases of Saccharomyces cerevisiae. Biochem J. 2006;393:797–805. doi: 10.1042/BJ20051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, Hermansson M, Somerharju P, Grinstein S. Phosphatidylserine is polarized and required for proper Cdc42 localization and for development of cell polarity. Nat Cell Biol. 2011;13:1424–1430. doi: 10.1038/ncb2351. [DOI] [PubMed] [Google Scholar]
- Finnigan GC, Booth EA, Duvalyan A, Liao EN, Thorner J. The carboxy-terminal tails of septins Cdc11 and Shs1 recruit myosin-II binding factor Bni5 to the bud neck in Saccharomyces cerevisiae. Genetics. 2015a;200:841–861. doi: 10.1534/genetics.115.176503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Takagi J, Cho C, Thorner J. Comprehensive genetic analysis of paralogous terminal septin subunits Shs1 and Cdc11 in Saccharomyces cerevisiae. Genetics. 2015b;200:821–840. doi: 10.1534/genetics.115.176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Thorner J. Complex in vivo ligation using homologous recombination and high-efficiency plasmid rescue from Saccharomyces cerevisiae. Bio Protoc. 2015;5:e1521. doi: 10.21769/bioprotoc.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia GI. Septin Self-Assembly: Plasticity and Protein Scaffolding. PhD Thesis. Berkeley: University of California; 2012. [Google Scholar]
- Garcia G, 3rd, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol. 2011;195:993–1004. doi: 10.1083/jcb.201107123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrenton LS, Stefan CJ, McMurray MA, Emr SD, Thorner J. Pheromone- induced anisotropy in yeast plasma membrane phosphatidylinositol-4,5-bisphosphate distribution is required for MAPK signaling. Proc Natl Acad Sci USA. 2010;107:11805–11810. doi: 10.1073/pnas.1005817107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Gong EY, Smits VA, Fumagallo F, Piscitello D, Morrice N, Freire R, Gillespie DA. KA1-targeted regulatory domain mutations activate Chk1 in the absence of DNA damage. Sci Rep. 2015;5:10856. doi: 10.1038/srep10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Gong T, Gao XD. Identification of an amphipathic helix important for the formation of ectopic septin spirals and axial budding in yeast axial landmark protein Bud3p. PLoS One. 2011;6:e16744. doi: 10.1371/journal.pone.0016744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Görlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- Guzman-Vendrell M, Rincon SA, Dingli F, Loew D, Paoletti A. Molecular control of the Wee1 regulatory pathway by the SAD kinase Cdr2. J Cell Sci. 2015;128:2842–2853. doi: 10.1242/jcs.173146. [DOI] [PubMed] [Google Scholar]
- Hallett MA, Lo HS, Bender A. Probing the importance and potential roles of the binding of the PH-domain protein Boi1 to acidic phospholipids. BMC Cell Biol. 2002;3:16. doi: 10.1186/1471-2121-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan J, Snyder M. Cytoskeletal activation of a checkpoint kinase. Mol Cell. 2003;12:663–673. doi: 10.1016/j.molcel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, Pringle JR, Reid BJ. Genetic control of the cell division cycle in yeast. Science. 1974;183:46–51. doi: 10.1126/science.183.4120.46. [DOI] [PubMed] [Google Scholar]
- Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front Biosci. 2008;13:2408–2420. doi: 10.2741/2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P. The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem Sci. 1996;21:172–173. [PubMed] [Google Scholar]
- Howell AS, Lew DJ. Morphogenesis and the cell cycle. Genetics. 2012;190:51–77. doi: 10.1534/genetics.111.128314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CR, Weems AD, Brewer JM, Thorner J, McMurray MA. Cytosolic chaperones mediate quality control of higher-order septin assembly in budding yeast. Mol Biol Cell. 2015;26:1323–1344. doi: 10.1091/mbc.E14-11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang PJ, Hood-DeGrenier JK, Park HO. Coupling of septins to the axial landmark by Bud4 in budding yeast. J Cell Sci. 2013;126:1218–1226. doi: 10.1242/jcs.118521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaton MA, Lew DJ. Eavesdropping on the cytoskeleton: progress and controversy in the yeast morphogenesis checkpoint. Curr Opin Microbiol. 2006;9:540–546. doi: 10.1016/j.mib.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Keaton MA, Szkotnicki L, Marquitz AR, Harrison J, Zyla TR, Lew DJ. Nucleocytoplasmic trafficking of G2/M regulators in yeast. Mol Biol Cell. 2008;19:4006–4018. doi: 10.1091/mbc.E08-03-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EK, Dysart M, Lianga N, Williams EC, Pilon S, Doré C, Deneault JS, Rudner AD. Redundant regulation of Cdk1 tyrosine dephosphorylation in Saccharomyces cerevisiae. Genetics. 2015;202:903–910. doi: 10.1534/genetics.115.182469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem. 2007;282:5101–5105. doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard TA, Hurley JH. Regulation of protein kinases by lipids. Curr Opin Struct Biol. 2011;21:785–791. doi: 10.1016/j.sbi.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Bi E. Regulation of septin organization and function in yeast. Trends Cell Biol. 2003;13:403–409. doi: 10.1016/s0962-8924(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Lu Q, Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- McMillan JN, Longtine MS, Sia RA, Theesfeld CL, Bardes ES, Pringle JR, Lew DJ. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol Cell Biol. 1999;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Theesfeld CL, Harrison JC, Bardes ES, Lew DJ. Determinants of Swe1p degradation in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:3560- 3575. doi: 10.1091/mbc.E02-05-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Bertin A, Garcia G, 3rd, Lam L, Nogales E, Thorner J. Septin filament formation is essential in budding yeast. Dev Cell. 2011;20:540–549. doi: 10.1016/j.devcel.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Div. 2009;4:18.1–18.40. doi: 10.1186/1747-1028-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino A, Tanaka K, Kamei T, Umikawa M, Fujiwara T, Takai Y. Shs1p: a novel member of septin that interacts with spa2p, involved in polarized growth in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1998;251:732–736. doi: 10.1006/bbrc.1998.9541. [DOI] [PubMed] [Google Scholar]
- Moravcevic K, Mendrola JM, Schmitz KR, Wang YH, Slochower D, Janmey PA, Lemmon MA. Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell. 2010;143:966–977. doi: 10.1016/j.cell.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller TD, Feigon J. Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J Mol Biol. 2002;319:1243–1255. doi: 10.1016/S0022-2836(02)00302-9. [DOI] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat Commun. 2014;5:5698. doi: 10.1038/ncomms6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon SA, Bhatia P, Bicho C, Guzman-Vendrell M, Fraisier V, Borek WE, Alves Fde L, Dingli F, Loew D, Rappsilber J, et al. Pom1 regulates the assembly of Cdr2-Mid1 cortical nodes for robust spatial control of cytokinesis. J Cell Biol. 2014;206:61–77. doi: 10.1083/jcb.201311097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, Dilworth SM, Laskey RA, Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–623. doi: 10.1016/0092-8674(91)90245-t. [DOI] [PubMed] [Google Scholar]
- Robichon C, Luo J, Causey TB, Benner JS, Samuelson JC. Engineering Escherichia coli BL21(DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Appl Environ Microbiol. 2011;77:4634–4646. doi: 10.1128/AEM.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Kozubowski L, Goode BL, Drubin DG, Hartwig JH. Actin and septin ultrastructures at the budding yeast cell cortex. Mol Biol Cell. 2005;16:372–384. doi: 10.1091/mbc.E04-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants FM, Su BM, von Wulffen J, Ramachandran S, Sartorel E, Trott AE, Thorner J. Protein kinase Gin4 negatively regulates flippase function and controls plasma membrane asymmetry. J Cell Biol. 2015;208:299–311. doi: 10.1083/jcb.201410076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roise D. Recognition and binding of mitochondrial presequences during the import of proteins into mitochondria. J Bioenerg Biomembr. 1997;29:19–27. doi: 10.1023/a:1022403604273. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Sakchaisri K, Asano S, Yu LR, Shulewitz MJ, Park CJ, Park JE, Cho YW, Veenstra TD, Thorner J, Lee KS. Coupling morphogenesis to mitotic entry. Proc Natl Acad Sci USA. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sayegh J, Clarke SG. Hsl7 is a substrate-specific type II protein arginine methyltransferase in yeast. Biochem Biophys Res Commun. 2008;372:811–815. doi: 10.1016/j.bbrc.2008.05.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Novakovic VA, Head JF, Seaton BA, Gilbert GE. Crystal structure of lactadherin C2 domain at 1.7A resolution with mutational and computational analyses of its membrane-binding motif. J Biol Chem. 2008;283:7230–7241. doi: 10.1074/jbc.M705195200. [DOI] [PubMed] [Google Scholar]
- Shulewitz MJ. Septin Assembly Regulates Cell Cycle Progression Through Activation of a Protein Kinase Signaling Pathway. PhD Thesis. Berkeley: University of California; 2000. [Google Scholar]
- Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson-Lavy KJ, Sajman J, Zenvirth D, Brandeis M. APC/CCdh1 specific degradation of Hsl1 and Clb2 is required for proper stress responses of S. cerevisiae. Cell Cycle. 2009;8:3003–3009. [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Sreenivasan A, Kellogg D. The Elm1 kinase functions in a mitotic signaling network in budding yeast. Mol Cell Biol. 1999;19:7983–7994. doi: 10.1128/mcb.19.12.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkotnicki L, Crutchley JM, TR Z, Bardes ES, Lew DJ. The checkpoint kinase Hsl1p is activated by Elm1p-dependent phosphorylation. Mol Biol Cell. 2008;19:4675–4686. doi: 10.1091/mbc.E08-06-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CL, Blacketer MJ, Edgington NP, Myers AM. Assembly interdependence among the S. cerevisiae bud neck ring proteins Elm1p, Hsl1p and Cdc12p. Yeast. 2003;20:813–826. doi: 10.1002/yea.1003. [DOI] [PubMed] [Google Scholar]
- Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, Barth P, Alber T, Thorner J. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:4568–4583. doi: 10.1091/mbc.E04-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- Wloka C, Bi E. Mechanisms of cytokinesis in budding yeast. Cytoskeleton. 2012;69:710–726. doi: 10.1002/cm.21046. [DOI] [PubMed] [Google Scholar]
- Wu H, Guo J, Zhou YT, Gao XD. The anillin-related region of Bud4 is the major functional determinant for Bud4’s function in septin organization during bud growth and axial bud site selection in budding yeast. Eukaryot Cell. 2015;14:241–251. doi: 10.1128/EC.00268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, Murray D, Emr SD, Lemmon MA. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell. 2004;13:677–688. doi: 10.1016/s1097-2765(04)00083-8. [DOI] [PubMed] [Google Scholar]
- Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site- saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.