

Assembly of the microtubule-nucleating γ-tubulin ring complex (γTuRC) requires higher-order oligomerization of Spc110p, which connects γTuRC to the yeast spindle pole body (SPB). Because Spc110p is highly concentrated at the SPB, spatial regulation of microtubule nucleation is thus achieved by exclusive assembly of γTuRCs proximal to the SPB.
Abstract
The microtubule (MT) cytoskeleton plays important roles in many cellular processes. In vivo, MT nucleation is controlled by the γ-tubulin ring complex (γTuRC), a 2.1-MDa complex composed of γ-tubulin small complex (γTuSC) subunits. The mechanisms underlying the assembly of γTuRC are largely unknown. In yeast, the conserved protein Spc110p both stimulates the assembly of the γTuRC and anchors the γTuRC to the spindle pole body. Using a quantitative in vitro FRET assay, we show that γTuRC assembly is critically dependent on the oligomerization state of Spc110p, with higher-order oligomers dramatically enhancing the stability of assembled γTuRCs. Our in vitro findings were confirmed with a novel in vivo γTuSC recruitment assay. We conclude that precise spatial control over MT nucleation is achieved by coupling localization and higher-order oligomerization of the receptor for γTuRC.
INTRODUCTION
The microtubule (MT) cytoskeleton plays important roles in many cellular processes, including signaling, intracellular transport, polarization, motility, and cell division. Although the pathways controlling these processes are complex, in general they impinge upon the cytoskeleton to regulate MT nucleation, elongation, and catastrophe. Whereas elongation and catastrophe are largely controlled by soluble proteins, nucleation is regulated by factors anchored in microtubule-organizing centers (MTOCs), such as the budding yeast spindle pole body (SPB) and the metazoan centrosome (Kollman et al., 2011). Within the MTOC, the γ-tubulin ring complex (γTuRC) nucleates MTs by forming a ring-shaped template from which MTs grow. In all eukaryotes, γTuRCs are built from multiple copies of the conserved core complex known as the γ-tubulin small complex (γTuSC), which in turn is composed of two γ-tubulins bound at the top of a Y-shaped complex formed by Spc97p and Spc98p (Kollman et al., 2008). In budding yeast, seven γTuSCs associate laterally to form a one-start helix with one-half γTuSC overlap after one turn, yielding 13 γ-tubulins presented as MT template (Kollman et al., 2010, 2015). In metazoans, canonical γTuSCs are mixed with γTuSC-like structures composed of γ-tubulin complexed with homologues of Spc97p and Spc98p, known as the γTuRC-specific components GCP4, -5, and -6, to form γTuRCs (Guillet et al., 2011; Kollman et al., 2011).
In metazoans, γTuRCs exist predominantly as soluble complexes that are recruited to sites of MT nucleation by a variety of attachment factors (Moudjou et al., 1996; Kollman et al., 2011). In contrast, yeast γTuSC alone does not form γTuRC-like structures (Kollman et al., 2008). However, coexpression of γTuSC with the N-terminal domain of Spc110p, which anchors γTuSCs to the nuclear face of the SPB (Knop and Schiebel, 1997, 1998; Kollman et al., 2010), leads to formation of larger ring-shaped and filamentous assemblies (Kollman et al., 2010). Coexpression with larger Spc110p fragments precludes filament formation (Kollman et al., 2015). Previous reports implicated Spc110p phosphorylation in stimulating γTuRC assembly (Lin et al., 2014). In addition, a Schizosaccharomyces pombe protein homologous to budding yeast Spc72p, the counterpart of Spc110p on the cytoplasmic face of the SPB, has been shown to oligomerize, which potentially stabilizes the γTuRC (Lynch et al., 2014). While suggestive, a detailed analysis of the processes underlying Spc110p-dependent γTuRC assembly is lacking. We therefore aimed to describe the γTuRC assembly process using a quantitative biochemical, biophysical, and cell biological approach that would allow assessment of the relative contribution of the various regulatory mechanisms affecting Spc110p and γTuSC.
Using budding yeast γTuSC and a novel fluorescence resonance energy transfer (FRET) assay, we reconstituted γTuRC assembly in vitro and dissected the features of Spc110p required for assembly. We find that higher-order oligomerization of Spc110p is the principal driver of γTuRC assembly, with oligomerization state affecting γTuRC assembly much more dramatically than a phosphomimetic mutation previously reported to enhance γTuRC assembly (Lin et al., 2014). In N-terminal deletion studies, deleting up to, but not through, the conserved centrosome motif 1 (CM1) domain of Spc110p preserves the ability to assemble γTuRCs. We use a novel in vivo γTuSC recruitment assay to confirm the importance of Spc110p oligomerization in the cellular context. Our results suggest a molecular mechanism by which higher-order Spc110p oligomerization can restrict γTuRC assembly and hence MT-nucleating ability to the SPB, ensuring precise spatiotemporal regulation of the MT cytoskeleton.
RESULTS
γTuSC assembly reconstituted in vitro
Spurred by the observation that coexpression of GST-Spc110p1-220 with γTuSC in baculovirus-infected insect cells led to formation of rings and larger filamentous assemblies (Kollman et al., 2010), we developed a FRET assay to monitor the Spc110p-dependent γTuSC ring assembly process in vitro (Figure 1A; see also Materials and Methods and Supplemental Figure S1; for clarity, we will refer to the process of γTuSC oligomerization as “γTuSC assembly” and reserve γTuRC for fully formed rings). The FRET assay is highly quantitative, allowing different forms of Spc110p to be accurately compared, and, of importance, it is sensitive enough to allow measurements to be made at physiologically relevant γTuSC concentrations. The FRET analysis measures both apparent affinity (from the concentration of Spc110p, yielding half-maximal signal) and the extent of γTuSC assembly (from the plateau value).
FIGURE 1:

Reconstitution of γTuSC assembly in vitro. (A) Schematic of FRET assay for γTuRC assembly. Spc110p-induced γTuRC assembly increases CFP-YFP FRET. (B) γTuRC assembly measured by FRET in the presence of 75 nM γTuSC. Dissociation constants are E. coli GST-Spc110p1-220: 370 nM; Sf9 GST-Spc110p1-220: 1290 nM; E. coli Spc110p1-220: 1700 nM. (C) Diagram of Spc110p residues 1–220. Regions with high coiled-coil probability, calculated with MARCOIL (Delorenzini and Speed, 2002), are indicated with darker shades of gray. The residues mutated in the 5D phosphomimetic mutant are highlighted. (D) SEC-MALS analysis of untagged E. coli Spc110p1-220 at concentrations ranging from 3.9 μM (light gray differential refractive index [dRI] trace) to 195 μM (dark gray dRI trace) calculated on a monomer basis. The molecular weights calculated are between the predicted monomer (26 kDa) and predicted dimer (52 kDa), indicating that untagged Spc110p1-220 is in monomer–dimer equilibrium. (E) SEC-MALS analysis of E. coli GST‑Spc110p1-220 at concentrations ranging from 2.3 μM (light gray dRI trace) to 136 μM (dark gray dRI trace) calculated on a monomer basis. GST-Spc110p1-220 is at least dimeric (predicted molecular weight 104 kDa) at all concentrations, with small amounts of tetramers present.
We expressed and purified γTuSC labeled with cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) FRET probes on the C-termini of Spc98p and Spc97p, respectively, from baculovirus-infected insect cells (Choy et al., 2009). To decrease sample heterogeneity, we purified γTuSC based on its ability to interact with Spc110p, thereby discarding any protein lacking posttranslational modifications or other features required for Spc110p interaction. We estimate that ∼50% of γTuSC in insect cells is capable of interacting with Spc110p (Supplemental Figure S2). When combined with glutathione S-transferase (GST)–Spc110p1-220, which we previously showed induces γTuSC self-assembly (Kollman et al., 2010), γTuSC oligomers form with an apparent Kd of 1290 nM (Figure 1B). Although this confirms the ability of Spc110p1-220 to stimulate γTuSC assembly, the Kd is surprisingly high, given that the in vivo concentration of Spc110p dimer is estimated to be ∼80 nM (Ghaemmaghami et al., 2003).
Because Spc110p is phosphorylated at numerous positions within residues 1–220 both in yeast and when expressed in insect cells (Friedman et al., 2001; Keck et al., 2011; Lin et al., 2014; Figure 1C), we considered that phosphorylation of Spc110p might affect the efficiency of the γTuSC assembly process. We therefore compared baculovirus-expressed Spc110p with that expressed in Escherichia coli, which lacks the relevant cell-cycle regulatory kinases. To our surprise, E. coli–expressed Spc110p induced assembly approximately threefold more efficiently, with an apparent Kd of 370 nM (Figure 1B). This suggests that phosphorylation sites targeted by insect cell kinases have a mild inhibitory effect on γTuSC assembly.
We next considered that the GST fusion protein might perturb the assembly process, as GST itself forms dimers and might shift the oligomeric state of Spc110p. Untagged Spc110p1-220 was produced via proteolytic cleavage of the hexahistidine (6His) purification tag and was nearly fivefold less efficient at γTuSC assembly (Kd = 1700 nM; Figure 1B). To understand this difference, we analyzed the oligomeric state of Spc110p using size exclusion chromatography coupled with multiangle light scattering (SEC-MALS) over a range of concentrations (Figure 1, D and E). Untagged Spc110p1-220 had a calculated molar mass intermediate between that of a monomer and a dimer (Figure 1D). Because molar masses calculated by SEC-MALS are a weighted average of the species present, this indicates that untagged Spc110p1-220 exists in a monomer–dimer equilibrium whose species are not resolved by the size exclusion column. In contrast, GST-Spc110p1-220 was dimeric under all concentration conditions tested, with a small fraction of tetrameric species (Figure 1E). Thus the oligomerization state of Spc110p1-220 was strongly perturbed by the GST tag, and the oligomerization state of the Spc110p constructs correlates with the apparent Kd for γTuSC assembly.
Higher-order Spc110p oligomerization is necessary for γTuSC assembly at physiological concentrations
To avoid the confounding effects of the GST tag and phosphorylation state, we addressed the role of Spc110p oligomerization in a well-defined system, using a protein engineering approach with bacterially expressed protein. Spc110p1-220, which is sufficient for assembly of γTuSC filaments (Kollman et al., 2010) but has only a weak tendency to dimerize on its own (Figure 1D), was fused with either the dimeric coiled-coil domain of the transcription factor GCN4 or a mutant version that preferentially forms tetramers (Harbury et al., 1993; Figure 2A). Analysis of oligomeric state by blue native PAGE (Figure 2B) and molar mass determination by SEC-MALS (Figure 2C) confirmed that engineered Spc110p derivatives formed the expected oligomers.
FIGURE 2:

Spc110p oligomerization is required for γTuRC assembly at physiological concentrations. (A) Diagram of Spc110p1-220-GCN4 coiled-coil domain fusion construct. The predicted coiled-coil register of Spc110p1-220 is fused in-frame with the register of GCN4. (B) Analysis of Spc110p1-220-dimer and -tetramer oligomerization state by blue native PAGE. (C) Molecular weights of Spc110p1-220 dimer and tetramer measured by SEC-MALS are consistent with dimers or tetramers of ∼29.5-kDa monomers. Samples in HB250 were separated on a Shodex KW-804 column. (D) Spc110p1-220 dimer has no activity in the presence of 5 nM γTuSC, whereas Spc110p1-220 tetramer stimulates robust assembly. The wild-type binding curve was fitted to a tight-binding model, whereas the 5D phosphomimetic was fitted to a simple binding model. (E) Spc110p1-220 dimer stimulates γTuRC assembly with reduced affinity in the presence of 50 nM γTuSC. (F) Stoichiometry analysis of Spc110p:γTuSC complex. At 100 nM γTuSC, far greater than the apparent Kd, the assembly curve saturates at an Spc110p1-220-tetramer concentration of ∼50 nM, indicating a stoichiometry of one Spc110p1-220 tetramer to two γTuSC, equivalent to one Spc110p dimer per γTuSC.
Engineered Spc110p dimers failed to induce any detectable γTuSC assembly at a γTuSC concentration of 5 nM (Figure 2D). In contrast, under these conditions, tetramers promoted robust assembly, with an apparent dissociation constant of 4 nM, determined using a tight- binding formalism (Pollard, 2010; Figure 2D). At the higher γTuSC concentration of 50 nM, Spc110p1‑220 dimer was able to induce assembly but with a substantially reduced affinity of 170 nM (Figure 2E). This dependence on γTuSC concentration indicates that γTuSC–γTuSC self-association contributes to γTuSC assembly but must be stabilized by interactions with Spc110p oligomers. The striking difference in assembly efficiency between dimeric and tetrameric Spc110p was also evident in negative-stain electron micrographs, in which γTuSC assemblies were much less prevalent in the presence of Spc110p1-220 dimer than with the tetramer (Figure 3, A–F). This confirms that higher-order oligomerization of Spc110p is necessary for γTuSC assembly under physiological concentration regimes, estimated at ∼80 nM Spc110p dimer (Ghaemmaghami et al., 2003).
FIGURE 3:

Structural and functional analysis of γTuSC-Spc110p1-220-dimer and -tetramer complexes. Representative negative-stain micrograph and class averages of 75 nM γTuSC (A, B), γTuSC + Spc110p1-220-dimer (C, D), and γTuSC + Spc110p1-220-tetramer (E, F). Red arrows indicate complexes containing two γTuSCs; green arrows indicate complexes with larger numbers of γTuSC (A, C, E). Numbers indicate the number of particles represented in each class (B, D, F). γTuSC-only class averages (B) are shown padded to the same box size as γTuSC-Spc110p classes. (G) Engineered Spc110p1-220 oligomers form MT nucleation–competent complexes with γTuSC. γTuSC was incubated with a fourfold excess of Spc110p1-220 dimer or tetramer, calculated on dimer basis, and 1.5 μΜ S. cerevisiae tubulin. Spc110p1-220 tetramer in the absence of γTuSC was included as a negative control. Representative epifluorescence images. (H) Quantification of G. Mean (two technical replicates) microtubule count from 10 randomly chosen fields.
We next compared the relative importance of oligomerization and phosphorylation using phosphomimetic aspartic acid mutations to residues S36, S91 (Cdk1 targets), S60, T64, and T68 (Mps1 targets). This mutation, denoted 5D (Figure 1C), was shown to enhance assembly of γTuSCs by dimeric GST-Spc110p1-220 (Lin et al., 2014). However, we found that the 5D phosphomimetic mutation had only mild effects and actually weakened the apparent Kd to 15 nM for the tetrameric Spc110p and from 170 to 310 nM for Spc110p1-220 dimer. The 5D mutation did not affect Spc110p oligomerization state as analyzed by SEC-MALS (Supplemental Figure S3A). The effects of the 5D phosphomimetic mutation were similar when analyzed with a size exclusion chromatography assay for γTuSC assembly similar to that used by Lin et al. (2014; Supplemental Figure S3B). We also compared their Tris pH 7 buffer to our 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5 buffer and found that the 5D phosphomimetic was inhibitory in both cases, with the Tris pH 7 buffer promoting a higher level of Spc110-independent γTuSC assembly (Supplemental Figure S3C). The enhancement of γTuSC assembly upon higher-order oligomerization is thus much more robust than this previously examined set of phosphomimetic mutations.
The more-homogeneous E. coli Spc110p1-220-tetramer preparation allowed the Spc110p:γTuSC stoichiometry required for assembly to be determined. When γTuSC is present at 100 nM, a concentration much greater than the apparent Kd, the FRET signal saturates at ∼50 nM Spc110p tetramer. This indicates a stoichiometry of one Spc110p tetramer per γTuSC dimer or 2:1 Spc110p monomer:γTuSC (Figure 2F), consistent with previous observations (Erlemann et al., 2012; Kollman et al., 2015). Taken together, these data indicate that a dimer of dimers is the minimal Spc110p species sufficient to allow γTuSC assembly, with each γTuSC being bound by a single Spc110p dimer.
To ensure that the engineered Spc110p1-220 tetramer assembles γTuSCs into native-like conformations, we assessed their ability to nucleate MTs in vitro using Saccharomyces cerevisiae αβ-tubulin, which is much more active for yeast γTuRCs than the typically used porcine brain tubulin (Figure 3, G and H; Kollman et al., 2015). Although complete γTuRCs were not observed by negative-stain electron microscopy (EM) due to the relatively low protein concentration used (Figure 3, C–F), γTuSC incubated with Spc110p1‑220 tetramer nucleates twofold to threefold more MTs than does Spc110p1-220 dimer. This confirms that increased FRET elicited by Spc110p1-220 tetramer is due to authentic and functional γTuSC assembly interactions.
The γTuSC assembly process quantified by computational simulation
Although Spc110p-dependent γTuSC assembly data can be adequately fitted by the simple, two-component, single-site binding model (Figures 1B and 2, D and E), several observations suggest that the assembly process is more complex. We observed large differences in binding curves at varying γTuSC concentration and a reproducible fall-off in FRET with 5 nM γTuSC at high Spc110p1-220-tetramer concentration (Figure 4A). In addition, the formation of microtubules by Spc1101-220 tetramer and the presence of larger γTuSC assemblies observed by negative-stain EM (Figure 3E) indicate that complexes containing more than two γTuSCs can form, potentially mediated by higher-order oligomerization of Spc110p. To account for these observations, a more comprehensive assembly model is needed in which γTuSC monomers are bound sequentially by an Spc110p tetramer to form a dimer or γTuSC dimers are captured by an Spc110p tetramer (Figure 4B). The ability to form γTuSC tetramers was included in the model to account for higher-order assemblies.
FIGURE 4:

Quantifying interaction affinities underlying Spc110p-dependent γTuRC assembly. (A) Spc110p1-220-tetramer-induced γTuRC assembly behavior at varying γTuSC concentrations, with fits derived from computational simulation. Best-fit γTuSC concentrations are indicated. (B) Schematic of γTuSC-Spc110p1-220-tetramer interactions, with interactions giving rise to FRET indicated with a red star. Dissociation constants derived from computational simulation and fitting are indicated.
To estimate the resulting five dissociation constants, we modeled the γTuSC assembly process as a system of ordinary differential equations (ODEs) according to the scheme shown in Figure 4B. For a given set of initial species concentrations and rate constants, equilibrium concentrations are obtained by numerical integration of the ODE system. Experimental FRET data were then fitted to the model by assuming a fixed on-rate for each reaction and numerically optimizing the off-rates, yielding a dissociation constant for each reaction. Because fits using the nominal initial γTuSC concentrations were unsatisfactory, the lowest initial γTuSC concentration was included as an additional free parameter in the fit, with the higher γTuSC concentrations scaled relative to this based on the mean total fluorescence at each concentration (see Methods and Methods and Supplemental Figure S4A). This approach led to robust binding constant estimates for almost every parameter (Supplemental Figure S4, C–I).
Although the Kd1 for γTuSC dimerization in the absence of Spc110p is not well constrained due to limitations on the maximum practical γTuSC concentration (Supplemental Figure S4C), the model fits suggest a dissociation constant for γTuSC dimerization (Kd1) of ∼1.7 μM. The affinity of Spc110p tetramer for a single γTuSC is substantially stronger, with Kd2 = 43 nM. Binding of a second γTuSC to this complex is stabilized by both Spc110p and the γTuSC self-interaction, with a substantial local concentration effect yielding a very strong Kd3 = 3 nM. The formation of γTuSC tetramers from two preassembled γTuSC:Spc110p complexes occurs with Kd5 = 420 nM, which is stronger than the γTuSC self-interaction and indicates that larger γTuSC assemblies are also stabilized through additional Spc110p-mediated interactions. The dissociation constants account for the lack of full ring assemblies in electron micrographs (Figure 3, C–F), as the 75 nM γTuSC and Spc110p concentrations would limit abundance of large assemblies, given Kd5 = 420 nM. Taking the results together, our model provides a detailed mechanism underlying the observed requirement for Spc110p in γTuRC assembly: in the absence of stabilization by Spc110p oligomers, the γTuSC self-interaction affinity is simply too weak to support assembly.
Higher-order Spc110 oligomerization is required for γTuSC assembly in vivo
Our results indicate that Spc110p must oligomerize beyond a dimer in order for γTuSCs to assemble into a MT nucleation–competent state. To assess the relevance of Spc110p oligomerization in vivo, we designed an approach that would allow interrogation of γTuSC-Spc110p binding in live cells. Chimeric Spc110p1-220 constructs bearing the GCN4 dimerization or tetramerization domains along with green fluorescent protein (GFP) and the lacI DNA-binding domain (Figure 5A) were artificially targeted to a chromosomally integrated lacO repeat array (Figure 5B). We then measured colocalization between mCherry-labeled Spc97p and GFP-labeled Spc110p chimeras (Figure 5, B and C). In asynchronous cells, no colocalization to either Spc110p dimer or tetramer was observed. However, in cells arrested at the spindle assembly checkpoint by nocodazole treatment, 46 ± 1.8% of Spc110p-tetramer foci were colocalized with γTuSC, whereas only 6.7 ± 1.1% colocalization was observed with the Spc110p dimer. Consistent with the results of the in vitro FRET assay, introducing the 5D phosphomimetic mutation into the Spc110p-tetramer mildly reduced γTuSC colocalization to 30 ± 7%. Although these results suggest that nocodazole arrest is required to establish a state permitting γTuSC recruitment, it also confirms that an Spc110p tetramer is the minimal species required for cooperative assembly of γTuSC oligomers in vivo.
FIGURE 5:

Spc110p oligomerization is required for γTuSC recruitment in vivo. (A) Diagram of Spc110p1-220-GCN4-GFP-lacI chimeric fusion protein. Width of bars is proportional to molecular weight. (B) Schematic of in vivo recruitment assay. Recruitment of γTuSC by the Spc110p1-220 variants was measured by colocalization of Spc97-mCherry to the GFP puncta formed by the Spc110 chimeras at lacO arrays positioned on the right arm of chromosome 12, at 80 kb from the centromere. Cells were arrested in mitosis with nocodazole. (C) Representative epifluorescence images and quantification of the colocalization. Top, colocalization between Spc97p-mCherry and the tetrameric form of the Spc110p1-220 chimera. Middle, predominant lack of colocalization between Spc97-mCherry and dimeric Spc110p chimera. Bottom, partial colocalization of the 5D mutant of the tetrameric Spc110p chimera. Note that besides the reduction in the percentage of colocalization, cells expressing the 5D mutant show reduced mCherry signal at the lacO array. Colocalization values represent the average of two independent experiments, with approximately equal numbers of puncta examined in each experiment.
The conserved centrosomin motif 1 of Spc110p is required for γTuSC binding
Previous studies characterized the N-terminal 150 residues of Spc110p as the minimal domain required for interaction with γTuSC in vivo (Knop and Schiebel, 1998; Nguyen et al., 1998). However, the manner in which these residues contribute to γTuRC function is unclear. To interrogate their role more quantitatively, we constructed fusion proteins of N-terminally truncated Spc110p and the GCN4 tetramerization domain and measured their ability to stimulate γTuSC assembly. The resulting curves were fitted as described (Figure 4). However, Kd1 and Kd5 were fixed at the values determined in Figure 4, as these parameters were poorly constrained (Figure 6, A–C, and Supplemental Figure S5). Spc110p lacking the first 34 residues, which includes the Spc110/Pcp1 motif (Lin et al., 2014), supported robust assembly in vitro, with Kd2 decreased approximately fourfold but Kd3 approximately the same as wild type. This Δ34 mutant was viable in vivo as assessed by a plasmid shuffle assay (Figure 6, B and C, and Supplemental Figure S6B). Surprisingly, Spc110p lacking the first 111 residues supported assembly in vitro, albeit with reduced Kd2 and Kd3. Because this truncation eliminates a nuclear localization sequence (NLS) at residues 24–59 (Adams and Kilmartin, 1999), we assessed viability with an exogenous NLS fused to the N-terminus. Even with the added NLS, the Δ111 mutant was unviable (Supplemental Figure S6, C and D), suggesting the γTuSC interaction affinity is too low to support γTuRC assembly in vivo or that a domain essential for interacting with other factors was removed. A further truncation removing the predicted α-helix from residues 112–147 abolished assembly in vitro, indicating that the minimal γTuSC interaction domain includes at least this region. This region contains the CM1 motif, which is found in γTuRC-binding proteins from diverse organisms (Figure 6D; Sawin et al., 2004), suggesting that the core γTuSC-binding determinants are broadly conserved.
FIGURE 6:

The centrosomin motif 1 domain is required for Spc110p-dependent γTuRC assembly. (A) Domain diagrams of Spc110p1-220-tetramer N-terminal truncation constructs. Predicted α-helices and loops are shown as colored boxes and lines, respectively, and 4-mer indicates the GCN4 tetramerization domain. (B) γTuRC assembly in the presence of ∼20 nM γTuSC and Spc110p1-220-tetramer N-terminal truncation mutants. Data were fitted using the computational simulation approach described in Figure 4, with Kd1 and Kd5 as fixed parameters (Supplemental Figure S5). (C) Dissociation constants derived from fitting by computational simulation. Viability was assessed by red/white plasmid shuffle assay (Supplemental Figure S6). n.d., not determined. (D) Amino acid sequence conservation of CM1-containing proteins with Spc110p CM1, located within residues 112–146.
DISCUSSION
γTuSC assembly in budding yeast requires higher-order oligomerization of Spc110p
We demonstrated that γTuSC assembly in budding yeast is cooperative and strictly dependent on Spc110p, as the weak γTuSC self-interaction is dramatically enhanced by additional interactions with Spc110p (Figure 4). Further, we showed that higher-order Spc110p oligomerization is a fundamental requirement for promoting γTuSC assembly both in vitro and, using stable γTuSC recruitment as a proxy, in vivo. These observations provide a mechanistic explanation for how MT nucleation is restricted to the SPB in budding yeast. In vivo, the weak self-interaction of γTuSCs prevents assembly of γTuRCs elsewhere in the cell, whereas the high local concentration of Spc110p at the SPB, where ∼300 Spc110p molecules are bound (Erlemann et al., 2012), provides a favorable environment for Spc110p oligomerization, which in turn stabilizes assembly of γTuRCs. Thus γTuRC formation represents the first regulatory step in MT nucleation, after which the processes of ring closure (Kollman et al., 2015) and potentially allosteric conformational activation of γ-tubulin act in a multilevel regulatory cascade (Figure 7).
FIGURE 7:

Model for Spc110p-dependent γTuSC assembly. Spc110p coiled-coil dimers assemble into higher-order oligomers, possibly directed by the hexagonal Spc42p lattice (Bullitt et al., 1997). The weak self-interaction of γTuSCs prevents assembly of γTuRCs away from the SPB, whereas dramatically stronger interactions with Spc110p higher-order oligomers drive γTuRCs to assemble exclusively at SPBs. Spc110p phosphorylation by Cdk1 and Mps1 occurs after the onset of S phase, after γΤuRCs have assembled. Once γTuRCs are assembled, they are activated by conformational changes, including ring closure and potentially allosteric activation of γ-tubulin, to allow MT nucleation.
In vivo, Spc110p assembles at the SPB via interactions with Spc29p, calmodulin, and Spc42p, which forms a two-dimensional, crystal-like lattice at the central plaque (Bullitt et al., 1997; Elliott et al., 1999; Muller et al., 2005). We propose that Spc110p oligomer assembly is organized by interactions with the Spc42p lattice and is further driven by coiled-coil self-interactions between Spc110p molecules (Figure 7). Spc110p assembles at the SPB dynamically in G1/S phases of mitosis and then becomes stabilized during G2, suggesting that regulation of Spc110p recruitment and/or assembly is coupled to the cell cycle (Yoder et al., 2003).
The potential mismatch between the 6-symmetric Spc42p lattice and the 6.5-fold symmetry of the γTuRC raises important questions as to how Spc110p assembly at the SPB is coupled to γTuRC assembly. Although γTuRCs could contain either six γTuSCs, leading to the presentation of 12 γ-tubulins and a gap, or seven γTuSCs with a half γTuSC overlap (Kollman et al., 2010), cryo-EM tomography of intact SPBs indicates that in vivo, γTuRCs contain seven γTuSCs (Kollman et al., 2015). In one possible model, the sixfold symmetry would remain coherent all the way from the central plaque to the γTuRC, with a hexamer of Spc110p dimers the functional unit interacting with γTuSCs to form γTuRCs. An additional factor would then be required to stabilize the seventh γTuSC to complete the γTuRC. Alternatively, the sixfold symmetry of the Spc42p lattice could break down as it propagates through the flexible coiled-coil domain of Spc110p, possibly due to predicted breaks in the coiled-coil register, allowing Spc110p oligomer sizes greater than a hexamer of dimers. This would allow each γTuSC to interact with an Spc110p dimer.
In a higher-resolution cryo-EM map of γTuSC helical filaments, we observed density that appears to connect part of the Spc110p coiled-coil with the γ-tubulin in the ring below (Kollman et al., 2015). This suggests that the seventh γTuSC in a γTuRC is cooperatively stabilized beyond the principal Spc97/98p–Spc110p interactions, favoring formation of heptameric γTuRCs over smaller assemblies.
In vivo, we showed via an engineered Spc110p-targeting system that Spc110p assemblies larger than dimers favor stable association of γTuSC with Spc110p. Given that dimeric Spc110p is able to induce γTuSC assembly but with substantially weaker Kd (Figure 3, D and E), it is not surprising that we observe low-level γTuSC recruitment to dimeric Spc110p in vivo.
The role of Spc110p phosphorylation
Overall our results indicate that Spc110p higher-order oligomerization is the primary driver of γTuRC assembly. Although Spc110p phosphorylation does affect γTuRC assembly, our results point to a generally mild effect. (Figures 1C and 2, D and E). However, Spc110p is subject to a highly complex mixture of phosphorylation events, with many known phosphorylation sites attributed to at least two kinases: cyclin-Cdk1 and Mps1 (Huisman et al., 2007; Keck et al., 2011; Lin et al., 2011). We are unable to account for the effects of all of these modifications in our study, and the relevant combination and sequence of phosphorylation events required for γTuRC assembly and MT nucleation has not been systematically explored. However, from our in vivo experiments, in which both dimeric and tetrameric Spc110p are subject to the same posttranslational modifications, it is clear that Spc110p phosphorylation in the absence of higher-order oligomerization is insufficient to support γTuRC assembly under physiological conditions.
Previous observations provide some constraints on which kinases may regulate Spc110p-dependent γTuRC assembly and at which points in the cell cycle they must act. Mps1p phosphorylates Spc110p after S-phase onset, as cdc4 mutant cells, which arrest at the G1/S transition, lack the Mps1p-dependent phosphorylated isoform of Spc110p (Friedman et al., 1996). However, nuclear MTs are present at cdc4 mutant SPBs (Byers and Goetsch, 1974). The same is true of cdc53 mutant cells, which arrest before S phase through the same mechanism as cdc4 (Mathias et al., 1996). This indicates that Mps1p phosphorylation, represented in the 5D phosphomimetic mutation (Figure 1C), is unlikely to be required for γTuRC assembly and instead acts after the formation of γTuRCs to regulate other aspects of γTuRC function. This leaves open the role of cyclin-Cdk1 phosphorylation of Spc110p, although Spc110p shows strong preferential phosphorylation by S-phase Clb5-Cdk1 (Loog and Morgan, 2005), suggesting that it primarily affects γTuRC function after onset of S phase, potentially in tandem with Mps1p.
In our in vivo Spc110p-targeting assay, we observed γTuSC association with Spc110p only in cells arrested in M phase by treatment with the microtubule-depolymerizing agent nocodazole (Figure 5). Activation of the spindle assembly checkpoint under nocodazole arrest might establish a state favoring γTuRC formation, although the precise mechanism responsible requires further investigation.
Implications of Spc110p:γTuSC interactions mediated by the conserved centrosomin motif 1
Using truncation mutants, we determined that the N-terminal 111 residues of Spc110p are dispensable for γTuSC binding in vitro and that the minimal binding domain contains at least residues 112–147 (Figure 6). In vivo, a truncation mutant lacking the first 34 residues was viable (Supplemental Figure S6B). This indicates that the Spc110/Pcp1 motif (Lin et al., 2014) at residues 15–27 is not required for γTuRC assembly and function in vitro or in vivo. However, the more extensive 111-residue deletion mutant was not viable (Supplemental Figure S6, C and D). The binding affinity of this mutant for γTuSC was approximately threefold weaker than that of full-length Spc110p (Figure 6, B and C), suggesting that the mutant interaction was too weak for normal function in vivo. Alternatively, this domain of Spc110p might be required for interaction with additional, essential regulatory factors. Consistent with this, much of the N-terminus lacks clear density in cryo-EM reconstructions of the γTuSC-Spc110p filament (Kollman et al., 2015), suggesting that this domain might not stably associate with γTuSC and instead might serve to recruit additional factors. Given that 14 of 31 known phosphorylation sites on Spc110p are located within the 111-residue N-terminal domain (Keck et al., 2011) and none in residues 112–147, it might be the case that Spc110p contains a core γTuSC-binding domain coupled to a regulatory domain that is heavily phosphorylated.
Spc110p residues 112–147 are conserved with γTuRC-binding proteins, including human CDK5RAP2 (Choi et al., 2010), S. pombe Mto1 and Pcp1 (Flory et al., 2002; Samejima et al., 2008), Aspergillus nidulans apsB (Zekert et al., 2010), and Drosophila melanogaster centrosomin (Terada et al., 2003). This raises the possibility that the interactions between γTuRCs and their binding proteins, as well as localization-dependent assembly, are conserved in metazoans. In S. pombe, the Mto1/2 complex has been suggested to play a very similar role to Spc110p in assembling the γTuRC (Lynch et al., 2014). In D. melanogaster cells in which the γTuRC-specific components Dgrip75, Dgrip128, Dgrip163, and GCP71WD were depleted by RNA interference, localization of γTuSC to centrosomes but not the spindle is maintained (Verollet et al., 2006). In this state, which resembles budding yeast in that only γTuSC components are present, soluble γTuRCs are not observed, but MTs are still nucleated from centrosomes. We hypothesize that under these conditions, γTuRC assembly might be facilitated by centrosomin or other centrosome-localized γTuRC-binding proteins, similar to the case with Spc110p in budding yeast. The evolution of the γTuRC-specific components might thus have been driven by a need for stable γTuRC self-assembly allowing MT nucleation at sites distinct from centrosome-localized γTuRC assembly factors, relying on attachment factors such as augmin to mediate nucleation within the spindle (Goshima et al., 2008) or AKAP450 and GM-210 at the Golgi (Rios et al., 2004; Rivero et al., 2009).
MATERIALS AND METHODS
Protein expression constructs
Baculoviral constructs for γTuSC expression were prepared as described (Choy et al., 2009; Vinh et al., 2002). All E. coli expression vectors for Spc110p were prepared by standard PCR and restriction enzyme–based cloning methods, except for GCN4 coiled-coil fusion constructs, which were prepared by overlap extension PCR. The Spc110 fragment was prepared by PCR with primers designed to introduce a region of sequence overlap with GCN4-p1 (dimer) or -LI (tetramer). The GCN4-LI and -p1 fragments were constructed from a series of overlapping synthetic oligonucleotides as described (Hoover and Lubkowski, 2002). Spc110p1-220-5D-dimer and -tetramer constructs were synthesized (Life Technologies, Carlsbad, CA) and cloned into either pGEX-6P-2 for expression as a GST fusion or pET28 with an N-terminal 6His tag and TEV protease cleavage site.
Protein expression and purification
All procedures were carried out at 4°C. Buffer HBn is 40 mM HEPES, pH 7.5, 1 mM MgCl2, and 1 mM ethylene glycol tetraacetic acid (EGTA) with n mM KCl. For expression in E. coli, cultures were grown in terrific broth at 37°C to OD600 ≈ 0.4 and then cooled to 18°C before induction of expression at OD600 = 0.6–0.8 with 100 μM isopropyl β-d-thiogalactoside for 16–18 h. Cells were then harvested by centrifugation and further processed for protein purification or flash frozen in liquid nitrogen and stored at −80°C. All cells were lysed with an Emulsiflex C3 cooled with a 4°C water bath (Avestin, Ottawa, Canada).
γTuSC.
Sf9 cells were coinfected with baculovirus encoding GST-Spc1101-220, γ‑tubulinS48C/S153C (Kollman et al., 2015), Spc97-YFP, and Spc98-CFP and grown for 48 h. We opted for the γ-tubulinS48C/S153C mutation because it allows the use of oxidizing agent as a positive control for γTuRC assembly, whereas it behaves like wild-type γTuSC in the presence of reducing agent (5 mM dithiothreitol [DTT] in our experiments). Cells were harvested by centrifugation and washed with phosphate-buffered saline containing 1 mM phenylmethylsulfonyl fluoride before flash freezing in liquid nitrogen. Cells were lysed in HB100 with 5 mM DTT, 0.5% Tween-20, 1× cOmplete protease inhibitor, EDTA-free (Roche Diagnostics, Indianapolis, IN), and 0.1% phosphatase inhibitor cocktails 2 and 3 (Sigma-Aldrich, St. Louis, MO) before glutathione affinity purification and anion exchange chromatography as described (Vinh et al., 2002). γTuSC was buffer exchanged into HB100 with 5 mM DTT and 10% glycerol using a PD10 desalting column (GE Healthcare Life Sciences, Piscataway, NJ) before flash freezing in liquid nitrogen and storage at −80°C.
GST-Spc1101-220.
Baculovirus-expressed GST-Spc110p1-220 was purified along with γTuSC as described. After the anion exchange step, fractions containing GST-Spc110p1-220 were further purified by size exclusion chromatography on a Superdex 200 16/60pg (GE Healthcare Life Sciences). E. coli–derived GST-Spc1101-220 was expressed in BL21(DE3) Codon Plus-RIL. Cells were lysed in HB300 lysis buffer (HB300 with 1 mM DTT, 0.5% Tween-20, and 1× cOmplete protease inhibitor cocktail). Lysate was clarified by ultracentrifugation at 125,000 × g, and the supernatant was bound to Glutathione Sepharose-4B (GE Healthcare Life Sciences) for 3 h with gentle agitation. The resin was packed into a column, washed with 10 CV HB300 lysis buffer and then 10 CV HB300. Protein was eluted by suspending the resin in 1 CV elution buffer (25 mM Tris, pH 7.8, 100 mM KCl, 1 mM DTT, 25 mM reduced glutathione) with gentle agitation for 10 min. The eluate was removed, and then the elution procedure was repeated three additional times. The pooled eluate was further purified by anion exchange chromatography followed by size exclusion chromatography as shown in Table 1.
TABLE 1:
Spc110p purification procedures.
| Construct | Dialysis buffer | Ion exchange | Size exclusion |
|---|---|---|---|
| Baculovirus-expressed GST-Spc110p1-220 | Not applicable | MonoQ 10/300 GL | Superdex 200 16/60pg |
| E. coli–expressed GST-Spc1101-220 | 25 mM Tris, pH 7.8, 250 mM KCl, 1 mM DTT | MonoQ 10/300 GL | Superdex 200 16/60pg |
| Untagged Spc110p1-220 | 25 mM potassium phosphate, pH 6, 250 mM KCl, 1 mM DTT | MonoS 10/300 GL | Superdex 75 16/60pg |
| Spc110p1-220 dimer | HB250, 1 mM DTT | HiTrap SP | Superdex 200 16/60pg |
| Spc110p1-220 tetramer | HB250, 1 mM DTT | HiTrap SP | Superdex 200 16/60pg |
| Spc110p35-220 tetramer | HB250, 1 mM DTT | HiTrap SP | Superdex 200 16/60pg |
| Spc110p112-220 tetramer | HB250, 1 mM DTT | HiTrap SP | Superdex 75 16/60pg |
| Spc110p183-220 tetramer | 50 mM potassium phosphate, pH 8.0, 250 mM KCl, 1 mM DTT | MonoQ 10/300 GL | Superdex 75 16/60pg |
Spc110p1-220-dimer, -tetramer, and -truncation mutants.
Proteins were expressed in E. coli BL21(DE3) CodonPlus-RIL. Cells were lysed in nickel-nitriloacetic acid (NiNTA) lysis buffer (50 mM potassium phosphate, pH 8.0, 500 mM KCl, 10 mM imidazole, 1 mM DTT, cOmplete protease inhibitor cocktail, and 0.5% Tween-20). Lysates were clarified by ultracentrifugation at 125,000 × g, and the supernatants were applied to NiNTA Superflow resin with gentle agitation for 1 h. The resin was packed into a column, washed with 10 CV lysis buffer followed by 10 CV wash buffer (50 mM potassium phosphate, pH 8.0, 250 mM KCl, 25 mM imidazole, 1 mM DTT, and cOmplete protease inhibitor cocktail), and then eluted with 4 × 1 CV elution buffer (wash buffer with 250 mM imidazole). Eluate was pooled, and TEV protease was added to remove 6His tags and dialyzed overnight into the buffer indicated in Table 1. After dialysis, protein was applied to the indicated ion exchange column (Table 1) and eluted with a linear gradient of 0.25–1 M KCl. Fractions containing Spc110p were pooled, concentrated to ∼2 ml, and applied to the indicated size exclusion column and eluted with HB250 with 10% glycerol and 1 mM DTT (Table 1). Fractions containing Spc110p were pooled, concentrated, flash frozen in liquid nitrogen, and stored at −80°C.
Spc110p1-220.
Untagged Spc110p1-220 was expressed as a 6His–maltose-binding protein (MBP)–3C protease cleavage site fusion from the vector H-MBP-3C (Alexandrov et al., 2001) in E. coli BL21(DE3) CodonPlus-RIL. The purification proceeded as for Spc110p1-220 dimer, except that the His-MBP tag was cleaved with 3C protease. Additional chromatography steps are shown in Table 1.
Spc110p147-220 tetramer.
Because we could not obtain Spc110p147-220 tetramer in soluble form, we purified it from inclusion bodies under denaturing conditions. Protein was expressed in E. coli BL21(DE3) CodonPlus-RIL. After lysis using an Emulsiflex C3 in NiNTA lysis buffer, inclusion bodies were pelleted by ultracentrifugation at 125,000 × g and then dissolved in pH 8.0 solubilization buffer (8 M urea, 100 mM potassium phosphate, 10 mM Tris). Solubilized inclusion bodies were incubated with NiNTA superflow resin with gentle agitation for 1 h, washed with 10 CV pH 6.3 solubilization buffer, and eluted with 4 CV pH 5.9 solubilization buffer and then 4 CV pH 4.5 solubilization buffer. Protein was refolded by dropwise dilution into a 10× volume of refolding buffer (HB250, 10% glycerol, 400 mM l-arginine, 1 mM DTT). Refolded protein was concentrated by capture on NiNTA Superflow resin, which was then washed with 10 CV wash buffer and eluted with 4 × 1 CV elution buffer. Eluate was pooled and TEV protease added, and it was dialyzed overnight into HB250, 10% glycerol, and 1 mM DTT. After dialysis, protein was concentrated to ∼2 ml, applied to a Superdex 75 16/60 pg size exclusion column, and eluted with HB250, 10% glycerol, and 1 mM DTT. Fractions containing Spc110p147-220 tetramer were pooled, concentrated, flash frozen in liquid nitrogen, and stored at −80°C.
Quantifying efficiency of γTuSC purification
Our purification method selects for γTuSC that is competent to interact with Spc110p. To quantify the fraction of γTuSC within baculovirus-infected insect cells that is capable of interacting with Spc110p, we performed a series of γTuSC purifications with varying amounts of glutathione-Sepharose resin. The high-speed supernatant and flowthrough fractions were analyzed by Western blotting for each condition. γTuSCCFP/YFP and GST-Spc110p1‑220 were detected by α-GFP (1:4000; A-11122; Life Technologies) and α-GST (1:8000; G7781, Sigma-Aldrich), respectively, imaged by fluorophore-conjugated goat α-rabbit secondary antibody (1:2000; 926-68021; Licor Biosciences, Lincoln, NE). Blots were scanned using a Licor Odyssey scanner. Band intensities were quantified using Fiji (Schindelin et al., 2012).
FRET assay
The TB150 buffer (50 mM Tris, pH 7.0, 150 mM KCl) used previously (Lin et al., 2014) led to high levels of Spc110p-independent FRET, so we used a HEPES buffer (40 mM HEPES, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 5 mM DTT), in which γTuSC was better behaved (Supplemental Figure S3C). Proteins were exchanged into assay buffer (HBn with 10% glycerol, 5 mM DTT, and 0.1% phosphatase inhibitor cocktails 2 and 3) using Zeba desalting spin columns (Pierce, Rockford, IL). For assays with Spc110 dimer and tetramer, γTuSC was in assay buffer with 100 mM KCl, and Spc110 was in assay buffer with 250 mM KCl. Spc110 and γTuSC were combined to give a final KCl concentration of 150 mM. For other assays, proteins were prepared in assay buffer with 150 mM KCl. Reactions were assembled in black, clear-bottom, 384-well plates (3655; Corning, Corning, NY) in assay buffer with 0.1 mg/mL bovine serum albumin using a Mantis liquid dispenser (Formulatrix, Waltham, MA) and mixed by pipetting. Reactions were sealed and incubated for 15 min at 25°C. Fluorescence spectra were recorded with a Spectramax M5 plate reader (Molecular Devices, Sunnyvale, CA) with excitation at 420 nm and emission recorded from 460 to 600 nm in 5-nm steps through a 455-nm long-pass filter. Photomultiplier tube sensitivity was set to automatic, and 100 readings were averaged per well.
FRET data analysis
Background spectra from samples containing no fluorophore were subtracted from experimental spectra, and then spectra were decomposed into CFP and YFP components by least squares fitting as a linear combination of CFP and YFP basis spectra (Zimmermann, 2005) using scripts written in R (Supplemental Figure S1, A–C; R Core Team, 2013). Correction for direct excitation of the YFP acceptor (i.e., YFP signal not attributable to FRET) was determined by measuring γTuSCYFP emission spectra with excitation at 420 nm in the absence of CFP (Supplemental Figure S1E). Spectra were recorded at varying γTuSCYFP concentrations and then fitted as a linear combination of YFP and buffer blank basis spectra (Supplemental Figure S1, D and E). The YFP fluorescence intensity due to direct excitation was plotted as a function of γTuSCYFP concentration and fitted by linear least squares. This yielded the correction term YFPcorr = 0.40[γTuSC] − 0.75 (Supplemental Figure S1F). FRET was calculated as
 |
Binding-curve fitting was performed by nonlinear least squares in R using either a simple binding model,
 |
or, when γTuSC concentration was very close to the calculated Kd, a tight-binding model (Pollard, 2010),
 |
where the free parameters FRETmin and FRETmax are the minimal and maximal FRET, respectively, and Kd is the dissociation constant.
All FRET data are the average of three technical replicates, with error bars indicating SD.
Computational simulation of γTuRC assembly pathway
With the goal of obtaining Kd values, we modeled the γTuRC assembly process up to a γTuSC tetramer (Figure 4B) in the presence of Spc110p1-220 tetramer as a system of ODEs with a fixed kon = 105 M−1 s−1 and variable koff. Because we sought to fit equilibrium FRET data with the ODE model, the absolute magnitudes of kon and koff are not physically meaningful. Only their ratio, Kd = koff /kon, is taken into account and is determined by numerical optimization of koff. Subscripts to γTuSC and Spc110p represent their oligomeric state:
 |
 |
 |
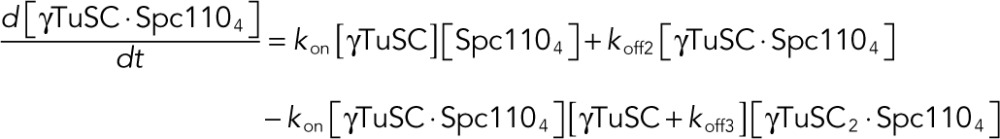 |
 |
 |
The system of ODEs was solved numerically using the deSolve package in R (Soetaert et al., 2010) to obtain equilibrium concentrations of each species. Simulated FRET values were calculated as follows:
 |
where eq denotes an equilibrium concentration, i denotes initial concentration, β relates concentrations of FRET-producing species to FRET units, and α is the baseline FRET from γTuSC in the absence of Spc110p.
To fit the model to experimental FRET data, we minimized an objective function giving the sum of squared residuals between simulated and experimental FRET data using the L-BFGS-B algorithm implemented in the R function optim. A lower bound of zero was used for all parameters. Free parameters include koff1, koff2, koff3, koff5, α, and β. koff4 was defined in terms of kon, koff1, koff2, and koff3 based on the thermodynamic cycle shown in Figure 4B as follows:
 |
Using the nominal γTuSC concentrations in the optimization procedure did not yield satisfactory fits. We reasoned that the model is extremely sensitive to the initial γTuSC concentration because of the appearance of terms with second-power dependence on both γTuSC and γTuSC2⋅Spc1104 concentration in the system of ODEs. Thus the initial lowest γTuSC concentration was included as an additional free parameter in the model. The higher γTuSC concentrations were then scaled according to the mean total fluorescence (i.e., summed across all wavelengths of a spectrum) at each concentration (Supplemental Figure S4A). The optimization changed the initial lowest γTuSC concentration from a nominal 10 to 16 nM. After deriving an initial set of parameter estimates by manual parameter adjustment followed by one round of computational optimization, we randomized the initial estimates 100 times within bounds threefold less or greater than the initial estimate (Supplemental Figure S4, B–I). The parameters from the best fit achieved from this procedure are given in Figure 4B. Additional rounds of parameter randomization did not improve the fit.
SEC-MALS and analytical size exclusion
MALS analysis was performed with WTC-050S5 (Wyatt Technology, Santa Barbara, CA) or KW-804 (Shodex, New York, NY) size exclusion columns on an Ettan liquid chromatography system (GE Healthcare Life Science) with inline DAWN HELEOS MALS and Optilab rEX differential refractive index detectors (Wyatt Technology). Data were analyzed using ASTRA VI software (Wyatt Technology). Size exclusion chromatography was performed with HB150 or HB250 with 5 mM DTT. Analytical size exclusion was performed on the Ettan liquid chromatography system with Superdex 200 PC 3.2/30 column (GE Healthcare Life Science) with a flow rate of 40 μl/min.
Blue native PAGE
Samples in HB150 with 5 mM DTT and 10% glycerol were separated on NativePAGE Novex 3-12% bis-Tris gels in NativePAGE running buffer along with NativeMark size standards (Life Technologies). The cathode buffer contained 0.02% Coomassie brilliant blue G-250. Electrophoresis was performed at 4°C at 150 V for 1 h and then 250 V until the dye front reached the bottom of the gel. Gels were then fixed by microwaving in 40% methanol and 10% acetic acid and incubating for 15 min and then destained by microwaving in 8% acetic acid and incubating until bands appeared on a clear background. Gels were then washed in water.
Negative-stain electron microscopy
γTuSC or γTuSC-Spc110p complexes at 75 nM in assay buffer were applied to glow-discharged, carbon-coated, 400-mesh copper grids as 2-μl drops and incubated for 30 s. Excess sample was blotted away, and the grid was washed quickly with two drops of water and then stained with 0.75% uranyl formate for 30 s. After removing excess stain, grids were air dried before imaging with a Tecnai 12 (FEI, Hillsboro, OR) operating at 120 kV. Images were acquired with ∼1.5-μm defocus on a 4k × 4k charge-coupled device camera (Gatan, Pleasanton, CA) with a pixel size of 2.21 Å. Particles were picked in 128 ×128–pixel (γTuSC) or 180 × 180–pixel (γTuSC-Spc110p complexes) boxes using e2boxer.py, part of EMAN2 (Tang et al., 2007). Reference-free two-dimensional class averages were generated using e2refine2d.py. Particles belonging to indistinct class averages were discarded using e2evalparticles.py. The final class averages were generated after several cycles of class averaging followed by discarding of bad particles.
Microtubule nucleation assay
S. cerevisiae tubulin was overexpressed and purified as described (Johnson et al., 2011). γTuSC and Spc110p1‑220 dimer or tetramer, prepared in the same manner as for the FRET assay, were combined at 10× final concentration in a 1:4 M ratio (calculated on a dimer basis) and incubated at room temperature for 15 min. γTuSC:Spc110p1-220-dimer or -tetramer complexes and S. cerevisiae tubulin were diluted at the appropriate concentrations into microtubule assembly buffer (80 mM K–1,4-piperazinediethanesulfonic acid [PIPES], pH 6.9, 125 mM KCl, 20% glycerol, 1 mM EGTA, 1 mM MgCl2, 1 mM GTP) on ice. Reactions were incubated at 30°C for 20 min, fixed for 3 min in 10 volumes of 1% glutaraldehyde in BRB80 (80 mM K-PIPES, pH 6.9, 1 mM EGTA, 1 mM MgCl2), and then diluted 10 times into BRB80 (final volume 1.5 ml). A 1-ml amount of the resulting fixed reactions was layered onto 20% glycerol/BRB80 cushions and centrifuged for 45 min, 24,000 × g, onto 18-mm round coverslips. Microtubules were visualized on the coverslips by immunofluorescence with fluorescein isothiocyanate–mouse anti–α-tubulin (F2168; Sigma-Aldrich), and 5–10 fields of microtubules were counted for each experiment.
In vivo γTuSC recruitment assay
The in vivo recruitment assay (Figure 5) used wide-field fluorescence microscopy to monitor the binding of γTuSC, tagged with Spc97p-mCherry, to Spc110p1-220 that was C-terminally tagged with GFP and lacI. Spc110p was visualized as GFP puncta localized to a lacO array positioned on chromosome XII. Colocalization of GFP and mCherry fluorescence was quantified using Imaris software (Bitplane, South Windsor, CT). Colocalization was measured as the total number of GFP puncta in the nucleus that were within 0.5 μm of mCherry puncta. Any GFP puncta within 0.5 μm of the SPB were excluded.
The Spc110p1-220-dimer and -tetramer regions were derived from the same plasmids used for expression in E. coli. The GFP-lacI sequence was derived from pGVH60 (Bystricky et al., 2005). This yeast-integrating plasmid also provided the backbone for integration of the SPC110 chimera at the ADE2 locus. Expression in S. cerevisiae used a β-estradiol–inducible expression system (McIsaac et al., 2013). The Z4EV promoter element was derived from pMN10. The gene encoding the Z4EV artificial transcription factor (from DBY12395) was PCR amplified and integrated at the CAN1 locus into strain KGY315 (Greenland et al., 2010). An array of 256 copies of the lacO sequence was integrated on the right arm of chromosome XII within the intergenic region between TRX1 and PDC1, using pGM22. pGM22 contains the KpnI–SacI fragment from pLI831(Muller, 1996) cloned into pSB11 (a gift from Sue Biggins, Fred Hutchinson Cancer Research Center). Spc97p was tagged with mCherry at the C-terminus using pBS34 and standard protocols (depts.washington.edu/yeastrc/pages/pBS34.html).
Strains used in this study were GMY128 (ade2-1oc/ade2-1::Z4EVpr-SPC1101-220-GCN4-p1-GA-GFP-LacI-ADE2; ADE3/ADE3; can1-100/can1-100::NatMX-ACT1pr-Z4EV; his3-11,15/his3-11,15; leu2-3112/leu2-3112; trp1-1/trp1-1; ura3-1/ura3-1; ChrXII-R/ChrXII-R::lacO-TRP1; SPC97-mCherry::HphMX/SPC97-mCherry::hphMX), GMY129 (same as GMY128 except ade2-1oc/ade2-1::Z4EVpr-SPC1101-220-GCN4-LI-GA-GFP-LacI-ADE2), and KYY90 (same as GMY128 except ade2-1oc/ade2-1::Z4EVpr-SPC1101–2205D-GCN4-LI-GA-GFP-LacI-ADE2).
Cells grown at 30°C to mid log phase in yeast extract/peptone/dextrose (YPD) were incubated for 30 min with 100 nM β-estradiol, and then nocodazole (15 μg/ml) was added and incubation continued for 1 h. Cells were washed, resuspended in YPD with 15 μg/ml nocodazole without estradiol, and incubated for another 1.5 h. Cells were washed to remove YPD, resuspended in synthetic defined (SD) medium, and mounted on a 1% SeaKem LE agarose in SD pad. Fluorescence microscopy images were taken using a DeltaVision system (Applied Precision, Issaquah, WA) with an IX70 inverted microscope (Olympus, Tokyo, Japan), a Uplan Apo 100× oil objective (1.35 numerical aperture), and a CoolSnap HQ digital camera (Photometrics, Tucson, AZ) as previously described (Muller et al., 2005).
Red/white plasmid shuffle assay
To evaluate whether N-terminal truncations of Spc110 were functional, we used a red/white plasmid shuffle system as described previously (Tien et al., 2013). Strain HSY2-12C (MATa ade2-1oc ade3Δ can1-100 his3-11,15 leu2-3112 lys2Δ::HIS3 spc110Δ::TRP1 trp1-1 ura3-1) harboring 2-μ plasmid pHS26 (ADE3 LYS2 SPC110; Sundberg et al., 1996) was transformed with mutant derivatives of plasmid pHS29 (CEN6 ARSH4 URA3 SPC110; Sundberg et al., 1996). These plasmids, pKY20-21, carried N-terminal deletions of Spc110p that removed up to amino acid residue V34 and K111, respectively. They were constructed using the QuikChange Multi Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA) with pHS29 as a DNA template and primers that spanned the region to be deleted. pKY176 bearing Spc110Δ111 with an added N-terminal nuclear localization sequence was constructed by QuikChange using an oligonucleotide encoding the NLS from pBS41 (GenBank accession KF177452). The plasmids were transformed into HSY2-12C and selected for growth on SD –Ura low-Ade plates. Deletions that rendered SPC110 nonfunctional were dependent on pHS26 for viability and grew as solid red colonies. If the pKY plasmids encoded a functional SPC110, then cells would lose pHS26 and the colonies would sector white.
Supplementary Material
Acknowledgments
We gratefully acknowledge many helpful discussions with members of the Agard and Davis labs, as well as with our collaborators on the Yeast Centrosome–Structure, Assembly, and Function Program Project grant in the labs of Chip Asbury, Ivan Rayment, Andrej Sali, Mark Winey, and Sue Jaspersen. We thank Sanford J. Silverman, Robert S. McIsaac, Gaby Schaefer, Susan Gasser, and Sue Biggins for plasmids and strains used in this study. We thank the lab of Jonathan Weissman for the use of the Licor Odyssey scanner and Michael Braunfeld and Cameron Kennedy for assistance with electron microscopy and computational resources. We acknowledge financial support from the Howard Hughes Medical Institute (D.A.A.), the National Institute of General Medical Sciences (R01 GM031627 to D.A.A.; P41 GM103533 to T.D.; and P01 GM105537 to D.A.A., T.D.), a National Science Foundation Graduate Research Fellowship Grant (1144247 to A.S.L.), and a University of California, San Francisco, Discovery Fellowship (A.S.L.).
Abbreviations used:
- CM1
centrosomin motif 1
- γTuRC
γ-tubulin ring complex
- γTuSC
γ-tubulin small complex
- MT
microtubule
- MTOC
microtubule-organizing center
- SPB
spindle pole body.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0072) on May 25, 2016.
REFERENCES
- Adams IR, Kilmartin JV. Localization of core spindle pole body (SPB) components during SPB duplication in saccharomyces cerevisiae. J Cell Biol. 1999;145:809–823. doi: 10.1083/jcb.145.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A, Dutta K, Pascal SM. MBP fusion protein with a viral protease cleavage site: one-step cleavage/purification of insoluble proteins. BioTechniques. 2001;30:1194. doi: 10.2144/01306bm01. [DOI] [PubMed] [Google Scholar]
- Bullitt E, Rout MP, Kilmartin JV, Akey CW. The yeast spindle pole body is assembled around a central crystal of Spc42p. Cell. 1997;89:1077–1086. doi: 10.1016/s0092-8674(00)80295-0. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harb Symp Quant Biol. 1974;38:123–131. doi: 10.1101/sqb.1974.038.01.016. [DOI] [PubMed] [Google Scholar]
- Bystricky K, Laroche T, van Houwe G, Blaszczyk M, Gasser SM. Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J Cell Biol. 2005;168:375–387. doi: 10.1083/jcb.200409091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Liu P, Sze SK, Dai C, Qi RZ. CDK5RAP2 stimulates microtubule nucleation by the γ-tubulin ring complex. J Cell Biol. 2010;191:1089–1095. doi: 10.1083/jcb.201007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RM, Kollman JM, Zelter A, Davis TN, Agard DA. Localization and orientation of the gamma-tubulin small complex components using protein tags as labels for single particle EM. J Struct Biol. 2009;168:571–574. doi: 10.1016/j.jsb.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorenzini M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- Elliott S, Knop M, Schlenstedt G, Schiebel E. Spc29p is a component of the Spc110p subcomplex and is essential for spindle pole body duplication. Proc Natl Acad Sci USA. 1999;96:6205–6210. doi: 10.1073/pnas.96.11.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlemann S, Neuner A, Gombos L, Gibeaux R, Antony C, Schiebel E. An extended gamma-tubulin ring functions as a stable platform in microtubule nucleation. J Cell Biol. 2012;197:59–74. doi: 10.1083/jcb.201111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory MR, Morphew M, Joseph JD, Means AR, Davis TN. Pcp1p, an Spc110p-related calmodulin target at the centrosome of the fission yeast schizosaccharomyces pombe. Cell Growth Differ. 2002;13:47–58. [PubMed] [Google Scholar]
- Friedman DB, Kern JW, Huneycutt BJ, Vinh DB, Crawford DK, Steiner E, Scheiltz D, Yates J, 3rd, Resing KA, Ahn NG, et al. Yeast Mps1p phosphorylates the spindle pole component Spc110p in the N-terminal domain. J Biol Chem. 2001;276:17958–17967. doi: 10.1074/jbc.M010461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Sundberg HA, Huang EY, Davis TN. The 110-kD spindle pole body component of saccharomyces cerevisiae is a phosphoprotein that is modified in a cell cycle-dependent manner. J Cell Biol. 1996;132:903–914. doi: 10.1083/jcb.132.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Goshima G, Mayer M, Zhang N, Stuurman N, Vale RD. Augmin: a protein complex required for centrosome-independent microtubule generation within the spindle. J Cell Biol. 2008;181:421–429. doi: 10.1083/jcb.200711053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenland KB, Ding H, Costanzo M, Boone C, Davis TN. Identification of saccharomyces cerevisiae spindle pole body remodeling factors. PLoS One. 2010;5:e15426. doi: 10.1371/journal.pone.0015426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet V, Knibiehler M, Gregory-Pauron L, Remy M, Chemin C, Raynaud-Messina B, Bon C, Kollman JM, Agard DA, Merdes A, et al. Crystal structure of γ-tubulin complex protein GCP4 provides insight into microtubule nucleation. Nat Struct Mol Biol. 2011;18:915–919. doi: 10.1038/nsmb.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: An automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman SM, Smeets MFMA, Segal M. Phosphorylation of Spc110p by Cdc28p-Clb5p kinase contributes to correct spindle morphogenesis in S. cerevisiae. J Cell Sci. 2007;120:435–446. doi: 10.1242/jcs.03342. [DOI] [PubMed] [Google Scholar]
- Johnson V, Ayaz P, Huddleston P, Rice LM. Design, overexpression, and purification of polymerization-blocked yeast alphabeta-tubulin mutants. Biochemistry. 2011;50:8636–8644. doi: 10.1021/bi2005174. [DOI] [PubMed] [Google Scholar]
- Keck JM, Jones MH, Wong CCL, Binkley J, Chen D, Jaspersen SL, Holinger EP, Xu T, Niepel M, Rout MP, et al. A cell cycle phosphoproteome of the yeast centrosome. Science. 2011;332:1557–1561. doi: 10.1126/science.1205193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Schiebel E. Spc98p and Spc97p of the yeast gamma-tubulin complex mediate binding to the spindle pole body via their interaction with Spc110p. EMBO J. 1997;16:6985–6995. doi: 10.1093/emboj/16.23.6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Schiebel E. Receptors determine the cellular localization of a gamma-tubulin complex and thereby the site of microtubule formation. EMBO J. 1998;17:3952–3967. doi: 10.1093/emboj/17.14.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Greenberg CH, Li S, Moritz M, Zelter A, Fong KK, Fernandez J, Sali A, Kilmartin J, Davis TN, et al. Ring closure activates yeast γTuRC for species-specific microtubule nucleation. Nat Struct Mol Biol. 2015;22:132–137. doi: 10.1038/nsmb.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Merdes A, Mourey L, Agard DA. Microtubule nucleation by gamma-tubulin complexes. Nat Rev Mol Cell Biol. 2011;12:709–721. doi: 10.1038/nrm3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Polka JK, Zelter A, Davis TN, Agard DA. Microtubule nucleating γ-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature. 2010;466:879–882. doi: 10.1038/nature09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Zelter A, Muller EG, Fox B, Rice LM, Davis TN, Agard DA. The structure of the gamma-tubulin small complex: implications of its architecture and flexibility for microtubule nucleation. Mol Biol Cell. 2008;19:207–215. doi: 10.1091/mbc.E07-09-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TC, Gombos L, Neuner A, Sebastian D, Olsen JV, Hrle A, Benda C, Schiebel E. Phosphorylation of the yeast gamma-tubulin Tub4 regulates microtubule function. PLoS One. 2011;6:e19700. doi: 10.1371/journal.pone.0019700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TC, Neuner A, Schlosser YT, Scharf AN, Weber L, Schiebel E. Cell-cycle dependent phosphorylation of yeast pericentrin regulates gamma-TuSC-mediated microtubule nucleation. Elife. 2014;3:e02208. doi: 10.7554/eLife.02208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- Lynch EM, Groocock LM, Borek WE, Sawin KE. Activation of the gamma-tubulin complex by the Mto1/2 complex. Curr Biol. 2014;24:896–903. doi: 10.1016/j.cub.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias N, Johnson SL, Winey M, Adams AE, Goetsch L, Pringle JR, Byers B, Goebl MG. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol Cell Biol. 1996;16:6634–6643. doi: 10.1128/mcb.16.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, Oakes BL, Wang X, Dummit KA, Botstein D, Noyes MB. Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic Acids Res. 2013;41:e57. doi: 10.1093/nar/gks1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudjou M, Bordes N, Paintrand M, Bornens M. Γ-tubulin in mammalian cells: The centrosomal and the cytosolic forms. J Cell Sci. 1996;109:875–887. doi: 10.1242/jcs.109.4.875. [DOI] [PubMed] [Google Scholar]
- Muller EG. A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol Biol Cell. 1996;7:1805–1813. doi: 10.1091/mbc.7.11.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller EG, Snydsman BE, Novik I, Hailey DW, Gestaut DR, Niemann CA, O’Toole ET, Giddings TH, Jr, Sundin BA, Davis TN. The organization of the core proteins of the yeast spindle pole body. Mol Biol Cell. 2005;16:3341–3352. doi: 10.1091/mbc.E05-03-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Vinh DB, Crawford DK, Davis TN. A genetic analysis of interactions with Spc110p reveals distinct functions of Spc97p and Spc98p, components of the yeast gamma-tubulin complex. Mol Biol Cell. 1998;9:2201–2216. doi: 10.1091/mbc.9.8.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. A guide to simple and informative binding assays. Mol Biol Cell. 2010;21:4061–4067. doi: 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team R: a language and environment for statistical computing. 2013. R Foundation for Statistical Computing. Available at http://www.R-project.org/ (accessed 30 September 2013)
- Rios RM, Sanchis A, Tassin AM, Fedriani C, Bornens M. GMAP-210 recruits gamma-tubulin complexes to cis-golgi membranes and is required for golgi ribbon formation. Cell. 2004;118:323–335. doi: 10.1016/j.cell.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation at the cis-side of the golgi apparatus requires AKAP450 and GM130. EMBO J. 2009;28:1016–1028. doi: 10.1038/emboj.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samejima I, Miller VJ, Groocock LM, Sawin KE. Two distinct regions of Mto1 are required for normal microtubule nucleation and efficient association with the gamma-tubulin complex in vivo. J Cell Sci. 2008;121:3971–3980. doi: 10.1242/jcs.038414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Lourenco PC, Snaith HA. Microtubule nucleation at non-spindle pole body microtubule-organizing centers requires fission yeast centrosomin-related protein mod20p. Curr Biol. 2004;14:763–775. doi: 10.1016/j.cub.2004.03.042. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetaert K, Petzoldt T, Setzer WR. Solving differential equations in R: Package deSolve. J Stat Software. 2010;33:1–25. [Google Scholar]
- Sundberg HA, Goetsch L, Byers B, Davis TN. Role of calmodulin and Spc110p interaction in the proper assembly of spindle pole body compenents. J Cell Biol. 1996;133:111–124. doi: 10.1083/jcb.133.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Terada Y, Uetake Y, Kuriyama R. Interaction of aurora-A and centrosomin at the microtubule-nucleating site in drosophila and mammalian cells. J Cell Biol. 2003;162:757–763. doi: 10.1083/jcb.200305048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien JF, Fong KK, Umbreit NT, Payen C, Zelter A, Asbury CL, Dunham MJ, Davis TN. Coupling unbiased mutagenesis to high-throughput DNA sequencing uncovers functional domains in the Ndc80 kinetochore protein of saccharomyces cerevisiae. Genetics. 2013;195:159–170. doi: 10.1534/genetics.113.152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verollet C, Colombie N, Daubon T, Bourbon HM, Wright M, Raynaud-Messina B. Drosophila melanogaster gamma-TuRC is dispensable for targeting gamma-tubulin to the centrosome and microtubule nucleation. J Cell Biol. 2006;172:517–528. doi: 10.1083/jcb.200511071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh DB, Kern JW, Hancock WO, Howard J, Davis TN. Reconstitution and characterization of budding yeast gamma-tubulin complex. Mol Biol Cell. 2002;13:1144–1157. doi: 10.1091/mbc.02-01-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder TJ, Pearson CG, Bloom K, Davis TN. The Saccharomyces cerevisiae spindle pole body is a dynamic structure. Mol Biol Cell. 2003;14:3494–3505. doi: 10.1091/mbc.E02-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekert N, Veith D, Fischer R. Interaction of the aspergillus nidulans microtubule-organizing center (MTOC) component ApsB with gamma-tubulin and evidence for a role of a subclass of peroxisomes in the formation of septal MTOCs. Eukaryotic Cell. 2010;9:795–805. doi: 10.1128/EC.00058-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T. Spectral imaging and linear unmixing in light microscopy. Adv Biochem Eng Biotechnol. 2005;95:245–265. doi: 10.1007/b102216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.