

In this issue of Biological Psychiatry, Warnault et al. (1) investigated the role of a point mutation in the brain-derived neurotrophic factor (BDNF) gene from valine 68 (Val68) to methionine (Met68) (mouse homolog of human Met66BDNF allele) in the risk of developing alcoholism using a knock-in approach in C57BL/6 mice. The mice expressing Met68BDNF showed excessive alcohol consumption compared with wild type Val68BDNF mice. Treatment of Met68BDNF mice with the tropomyosin receptor kinase B (trkB) agonist LM22A-4 or overexpression of Val68BDNF in the ventromedial prefrontal cortex resulted in the attenuation of alcohol consumption. Thus, results discussed in the manuscript clearly suggest that humans carrying the Met66BDNF allele appear to be at high risk for developing alcoholism and also pinpoint a potential therapeutic agent for treating alcohol use disorders.
Alcoholism is a brain disease that is associated with repetitive cycles of uncontrollable drinking (2, 3). Two major states of alcoholism have been implicated in the initiation and maintenance of alcohol use disorders, termed positive and negative affective states (2–5). The positive affective state is related to the euphoric and rewarding properties of ethanol, whereas the negative affective state refers to alleviation of the negative emotional state that develops during withdrawal. The latter has been conceptualized in the field as the “dark side of addiction” (2–5). Several cellular mechanisms involving corticotropin-releasing factor, neuropeptide Y, gamma-aminobutyric, glutamate, and dopaminergic systems within key brain structures have been identified to underlie the neurobiological basis of both positive and negative reinforcement properties of alcohol addiction (2–6). One other cellular molecule appears to be BDNF, and related signaling mechanisms acting within specific brain circuitry regulate synaptic plasticity associated with alcohol drinking behaviors (3–6). When I started my research on BDNF about 10 years ago, knowledge of the molecule’s role in brain diseases was still in its infancy compared with present time. We now know that BDNF is a crucial brain signaling protein that is integral to many signaling pathways and whose deficiency in key neural circuits is associated with development of psychiatric disorders, including alcoholism (3–7). It is still unclear how and in which cell types BDNF functions in the brain, but available evidence suggests that BDNF via tropomyosin receptor kinase B (trkB) regulates synaptic plasticity via activation of several signaling systems, most prominently mitogen-activated protein (MAP) kinases leading to activation of the gene transcription factor cAMP-responsive element binding (CREB) protein (5,6). BDNF signaling has been implicated in neurogenesis, neuronal differentiation, and maturation (4–7). The BDNF gene is complex and has several exons, and via alternative splicing, all exons splice to a common exon to encode similar proteins with discrete cellular functions (7). BDNF exons are also regulated epigenetically via histone modifications and DNA methylation, and BDNF expression is also posttranscriptionally regulated by small non-coding RNAs known as microRNAs (4, 6, 7). BDNF and regulatory mechanisms are altered by acute and chronic ethanol exposure in various brain structures, such as the prefrontal cortex, striatum, amygdala, and hippocampus, and are involved in alcohol use disorders (3–6).
In this issue of Biological Psychiatry, Warnault et al. (1) report novel findings demonstrating a direct role of Valine 68 to Methionine BDNF gene polymorphism (Met68BDNF) in excessive alcohol drinking behaviors in mice. This polymorphism generally causes decreased activity-dependent release of BDNF leading to its decreased function (1, 8). Met68BDNF in mice is homologous to the human Met66BDNF that has been implicated in the predisposition of several psychiatric disorders, including addiction (1, 8). Warnault et al. (1) also found that over-expression of the wild-type form of BDNF (Val68BDNF) in the ventromedial prefrontal cortex as well as treatment of Met68BDNF mice with a trkB receptor agonist was able to attenuate alcohol drinking behaviors. Furthermore, Met68BDNF mice showed a compulsive pattern of drinking, as evidenced by the inability of quinine addition to attenuate alcohol drinking. Together, these data provide evidence implicating the involvement of the BDNF gene polymorphism (miceMet68 and humanMet66) in the development of alcoholism (1). Interestingly, Met68BDNF mice do not exhibit anxiety or depression-like behaviors indicating these animals may be drinking to achieve euphoric effects (positive affective state) of alcohol and not for the alleviation of negative affective state. However, it’s interesting to note that both humans and mice carrying the Met66BDNF polymorphism show impaired fear extinction (8). Further studies are needed to dissect out the role of other brain circuits such as the amygdala, dorsal striatum, and hippocampus, in regulating alcohol-drinking behaviors in Met68BDNF mice.
Several studies in the field (1, 3–6,9) indicate that deficits of BDNF levels in medial prefrontal cortex (mPFC), striatum (dorsolateral striatum), hippocampus, and amygdala (central nucleus of amygdala [CeA] and medial nucleus of amygdala [MeA]) are clearly involved in regulating alcohol consumption (Figure 1). For example, BDNF in these brain structures serves as a homeostatic factor in regulating alcohol-drinking behaviors (3–6). It has been shown that lower BDNF expression in the CeA and MeA, due to higher expression of histone deacetylase2 (HDAC2) -mediated deficits in histone acetylation, have been shown to regulate anxiety and alcohol-drinking behaviors in alcohol preferring (P) as compared with alcohol non-preferring (NP) rats (4). It has also been shown that decreased BDNF, and associated decreased MAP kinase and CREB phosphorylation and expression of activity-regulated cytoskeleton associated protein (Arc) in the CeA are involved in alcohol preference and dependence (4, 5). These mechanisms are associated with synaptic remodeling events, such as reductions in dendritic spines in the CeA and MeA that are normalized by the inhibition of HDAC2 specifically in the CeA or by treatment with HDAC inhibitors (4). Furthermore, studies also found that a history of alcohol dependence produces decreased expression of BDNF in the mPFC, amygdaloid (CeA, MeA), and dorsal striatal brain structures in preclinical models (3–6, 9). It has been reported that decreased expression of BDNF in mPFC is regulated by increased expression of microRNAs (miR-206 and miR30a-5p) during alcohol exposure, and these mechanisms may be involved in regulating dependence-induced alcohol drinking behaviors (6,9). Interestingly, infusion of BDNF or increasing BDNF via inhibition of above miR30a-5p in the mPFC was able to attenuate alcohol-drinking behaviors (3, 6). Alcohol dependence-induced reductions in BDNF expression in the CeA and MeA are associated with anxiety-like behaviors in rats (4, 5). Correcting the BDNF deficits by the infusion of exogenous BDNF directly into CeA or by systemic pan-HDAC inhibitor treatment (trichostatin A) was able to attenuate anxiety-like behaviors during alcohol withdrawal after chronic exposure (4, 5). The epigenetic regulation of BDNF also plays a crucial role in early life adversity, such as adolescent alcohol exposure, which produces a persistent decrease in the expression of BDNF and Arc and dendritic spines in the CeA and MeA due to HDAC2-mediated deficits in histone acetylation in adulthood. These changes are associated with adolescent alcohol exposure-induced anxiety and alcohol drinking behaviors in adulthood that are attenuated by HDAC inhibitor treatment (10). As discussed by Warnault et al. (1), low blood levels of BDNF have been associated with the severity of alcoholism in clinical populations. All these studies support the replication of convergent molecular findings on BDNF that highlight the role of BDNF in the regulation of alcohol consumption (Figure 1).
Figure 1.
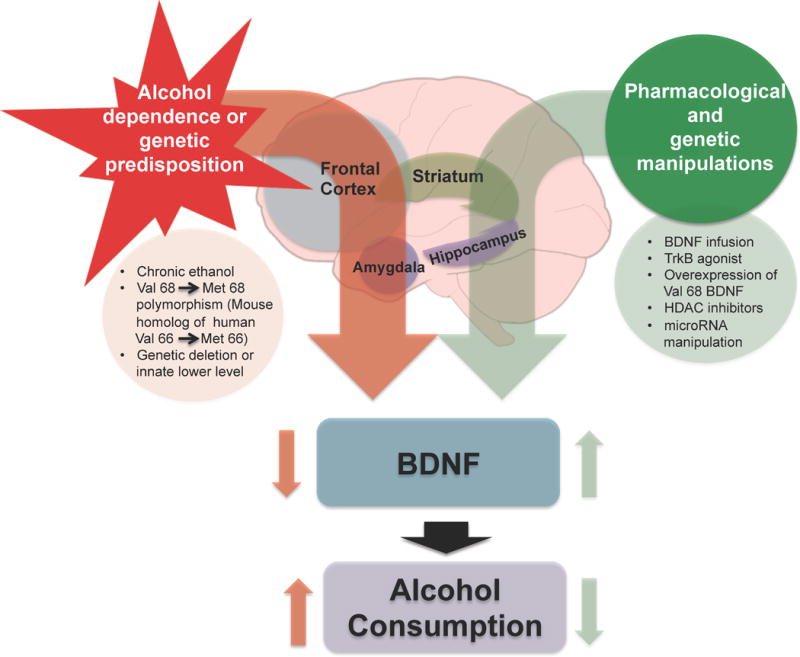
This model shows the involvement of decreased brain-derived neurotrophic factor (BDNF) in various brain regions (frontal cortex, amygdala, hippocampus, and dorsal striatum) in promoting alcohol intake. Decreased BDNF could be due to genetic deletion of the BDNF gene, methionine 68BDNF (Met68BDNF) polymorphism, or chronic ethanol exposure-induced BDNF reduction. Innately lower expression of BDNF in the central (CeA) and medial nucleus of amygdala (MeA) has been associated with excessive alcohol drinking behaviors. On the other hand, manipulations that lead to increases in the expression of BDNF in given brain circuitry (frontal cortex, amygdala, hippocampus, and dorsal striatum) are associated with decreases in alcohol intake in various preclinical models. For example, directly infusing BDNF in the prefrontal cortex or CeA is able to attenuate drinking and anxiety-like behaviors developed during ethanol withdrawal. Also, over-expression of wild type Valine68BDNF (Val68BDNF) in the ventral prefrontal cortex of Met68BDNF (mouse homolog of human Met66BDNF) mice was able to decrease excessive alcohol intake. Interestingly, infusion of HDAC2 siRNA in the CeA of alcohol-preferring rats was able to correct the deficits in BDNF levels and attenuated excessive alcohol drinking behaviors. Together, these studies support the notion that BDNF gates alcohol drinking behaviors (1, 3–6, 9, 10).
In summary, the new data presented in the current issue of Biological Psychiatry by Warnault et al. (1), clearly support the hypothesis that a deficiency in BDNF due to the Met68BDNF polymorphism has an essential role in promoting alcohol consumption and further advances the field by bringing forth mechanistic evidence that a BDNF gene polymorphism plays a role in excessive and compulsive alcohol drinking. This study also suggests that human studies are necessary in order to identify ‘at risk’ populations, so that better treatment strategies can be developed that, via regulation of BDNF signaling can treat or prevent alcoholism (Figure 1).
Acknowledgments
The research work in the laboratory of SCP was supported by the National Institute on Alcohol Abuse and Alcoholism Grant Nos. AA-019971 (Neurobiology of Adolescent Drinking in Adulthood Project), AA-010005, AA-021662, AA-013341, and P50 AA-022538 and the Department of Veterans Affairs Merit Review Grant No.I01BX000143 and Senior Research Career Scientist award.
Footnotes
Disclosures
SCP reports that a US patent application entitled “Histone acetyl transferase activators and histone deacetylase inhibitors in the treatment of alcoholism” (serial number 60/848237, filed on September 29th, 2006) is currently pending.
References
- 1.Warnault V, Darcq E, Morisot N, Phamluong K, Wilbrecht L, Massa SM, et al. The BDNF valine 68 to methionine polymorphism increases compulsive alcohol drinking in mice that is reversed by tropomyosin receptor kinase B activation. Biol Psychiatry. 2016;79:463–473. doi: 10.1016/j.biopsych.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui C, Noronha A, Warren KR, Koob GF, Sinha R, Thakkar M, et al. Brain pathways to recovery from alcohol dependence. Alcohol. 2015;49:435–452. doi: 10.1016/j.alcohol.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyzar EJ, Pandey SC. Molecular mechanisms of synaptic remodeling in alcoholism. Neurosci Lett. 2015;601:11–19. doi: 10.1016/j.neulet.2015.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci. 2010;67:73–88. doi: 10.1007/s00018-009-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Logrip ML, Barak S, Warnault V, Ron D. Corticostriatal BDNF and alcohol addiction. Brain Res. 2015;1628:60–67. doi: 10.1016/j.brainres.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 8.Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tapocik JD, Barbier E, Flanigan M, Solomon M, Pincus A, Pilling A, et al. microRNA-206 in rat medial prefrontal cortex regulates BDNF expression and alcohol drinking. J Neurosci. 2014;34:4581–4588. doi: 10.1523/JNEUROSCI.0445-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey SC, Sakharkar AJ, Tang L, Zhang H. Potential role of adolescent alcohol exposure-induced amygdaloid histone modifications in anxiety and alcohol intake during adulthood. Neurobiol Dis. 2015;82:607–619. doi: 10.1016/j.nbd.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]