

Abstract
Study Objectives:
Kleine-Levin syndrome (KLS) is a rare, mostly sporadic disorder, characterized by intermittent episodes of hypersomnia plus cognitive and behavior disorders. Although its cause is unknown, multiplex families have been described. We contrasted the clinical and biological features of familial versus sporadic KLS.
Methods:
Two samples of patients with KLS from the United States and France (n = 260) were studied using clinical interviews and human leukocyte antigen (HLA) genotyping. A multiplex family contained two or more first- or second-degree affected relatives (familial cases).
Results:
Twenty-one patients from 10 multiplex families (siblings: n = 12, including two pairs of monozygotic twins; parent-child: n = 4; cousins: n = 2; uncle-nephews: n = 3) and 239 patients with sporadic KLS were identified, yielding to 4% multiplex families and 8% familial cases. The simplex and multiplex families did not differ for autoimmune, neurological, and psychiatric disorders. Age, sex ratio, ethnicity, HLA typing, karyotyping, disease course, frequency, and duration of KLS episodes did not differ between groups. Episodes were less frequent in familial versus sporadic KLS (2.3 ± 1.8/y versus 3.8 ± 3.7/y, P = 0.004). Menses triggered more frequently KLS onset in the nine girls with familial KLS (relative risk, RR = 4.12, P = 0.03), but not subsequent episodes. Familial cases had less disinhibited speech (RR = 3.44, P = 0.049), less combined hypophagia/hyperphagia (RR = 4.38, P = 0.006), more abrupt termination of episodes (RR = 1.45, P = 0.04) and less postepisode insomnia (RR = 2.16, P = 0.008). There was similar HLA DQB1 distribution in familial versus sporadic cases and no abnormal karyotypes.
Conclusion:
Familial KLS is mostly present in the same generation, and is clinically similar to but slightly less severe than sporadic KLS.
Citation:
Nguyen QT, Groos E, Leclair-Visonneau L, Monaca-Charley C, Rico T, Farber N, Mignot E, Arnulf I. Familial Kleine-Levin syndrome: a specific entity? SLEEP 2016;39(8):1535–1542.
Keywords: family, hypersomnia, Kleine-Levin syndrome, multiplex, periodic
Significance.
Kleine-Levin syndrome (KLS) is a rare disorder in adolescents, with relapsing-remitting episodes including hypersomnia, mental confusion, derealization, and apathy. The cause is unknown, but exceptional multiplex families have been reported. Here, for the first time, we measured among 260 patients with KLS that 8% had an affected family member. The disease was mostly present in the same generation. Familial KLS was clinically not different (although slightly less severe) from sporadic KLS, with similar HLA DQ distribution and no abnormal karyotypes. This large comparison of phenotype and genotype will support future DNA exome sequencing in these families and the role of geneticcs in KLS pathogenesis.
INTRODUCTION
Kleine-Levin syndrome (KLS) is a rare neurological disorder (approximately two cases per million inhabitants) affecting mostly teenagers and young adults.1 It is characterized by relapsing-remitting episodes of hypersomnia associated with behavioral, mood, and cognitive disturbances, such as confusion, apathy, and derealization. Less frequently, hyperphagia and hypersexuality are also observed during these episodes. The cause of KLS is unknown, in the absence of biological or imaging markers, except for decreased metabolism/per-fusion in the associative cortex and the basal ganglia during episode in functional imaging.2 An association of KLS with human leukocyte antigen (HLA) DQB1*02 suggestive of autoimmune origin has been reported in an European series,3 but not confirmed in two controlled series from the US4 and France.5 Most KLS cases are sporadic, but several multiplex families containing two to six cases have been described, including five families with two affected siblings (a set of twin brothers),6,7 three with brother and sister,8–10 a grandmother, a father and two children,11 a mother and her son,3 a father and his five children,12 and an uncle and his nephew.13 The report of several families for this exceptionally rare disorder suggests genetic vulnerability in some cases. Because large series containing familial cases are lacking, we systematically collected demographic, clinical, and HLA measures in two large samples to compare sporadic and familial cases.
METHODS
Patients
Our study used the KLS database from the Stanford Center for Sleep Sciences and Medicine (Palo Alto, CA, USA) and from the National KLS Reference Center of Pitié-Salpêtrière hospital (Paris, France). All patient had signed an informed consent for clinical and genetic studies and the study was approved by ethic review boards of Stanford University and Pitié Salpetrière Hospital, respectively. Patients used in this study were those consecutively enrolled from 2004 to 2005 in the Stanford Center, and from 2008 to 2015 in the Paris center. All patients met international criteria for primary KLS,14 including: (1) episodes of hypersomnia lasting between 2 days and 4 w, occurring at least once per year, plus at least one of the following symptoms, including megaphagia, hypersexuality, abnormal behavior such as irritability, aggression or odd behavior, and cognitive abnormalities such as feeling of unreality, confusion and hallucination; (2) episodes recurring at least every year; (3) normal alertness, cognitive functioning, and behavior between attacks; and (4) with the disease being not better explained by another sleep disorder, medical or neurological disorder, mental disorder, medication use, or substance use disorder. Note that all patients also met the more recent KLS criteria.15 We did not include patients with atypical associated symptoms, differential and uncertain diagnoses, as well as secondary KLS, defined as the presence of neurological symptoms prior to the first KLS episode and persistent between episodes (Figure 1). A familial case was defined as any patient with KLS having at least one first- or second-degree relative affected with KLS and contrasted to sporadic cases (without any other affected family member, at first or second degree). A simplex family contained only one patient with KLS, and a multiplex family contained at least two patients with KLS, with a first- or second-degree family link.
Figure 1.

Flow chart of the patients.
Investigations
A neurologist experienced in KLS diagnosis interviewed all patients. Subjects completed the Stanford KLS autoquestionnaire (whether in English or in French), which included 280 questions about personal and family history, onset and course of KLS, triggering factors, symptoms during episodes (sleep, cognition, derealization, eating and sexual behavior, psychiatric symptoms, autonomous nervous system symptoms), response to therapies and sleep, eating attitude, depression, and anxiety during asymptomatic periods.4,16 Among the KLS autoquestionnaire questions, we combined the “childish speech,” “repetitive speech,” and “searching one's words” questions into a single composite criterion named “poor speech.” Patients from Paris also completed the Starkstein Apathy scale, in and out of an episode.17 All patients from the two centers had HLA genotyping, which included high- resolution DQB1 and DRB1 typing at Stanford and medium resolution typing of DQB1 in Paris, except for HLA DQB1*06:02, which was looked for in all patients from the two centers. In addition, all French patients had a karyotype.
Statistical Analysis
Measures are reported as mean ± standard deviations or percentages whenever most appropriate. Dichotomous traits were analyzed using a chi-square test. Continuous variables were compared between groups using Student t tests. P values lower than 0.05 were considered as significant.
RESULTS
Frequency of Familial KLS
The initial sample included 279 patients referred for KLS, including 177 patients from Paris and 102 from Stanford (6 French patients common to the two centers were counted in the French center in which they were followed up). After review, 19 patients were excluded for unsure diagnosis (n = 7), atypical KLS (n = 7), differential diagnosis (n = 4; including bipolar disorder, atypical idiopathic hypersomnia, encephalopathy, Nieman-Pick Type C disease) and secondary KLS (n = 1, multiple sclerosis) leading to 260 typical patients. There were 239 patients with sporadic KLS and 21 patients with familial KLS from 10 multiplex families, yielding to 10 of 249 multiplex families (4%) and 21 of 260 familial (8%), of all cases. Family relationship included siblings (n = 12, including two pairs of monozygotic twins), parent-child (n = 4), first-degree cousins (n = 2), and uncle plus two nephews (n = 3). All cases were born from nonconsanguineous parents. Family 4 and Family 9 were previously published.18,19 The 10 family trees are shown in Figure 2. There were seven families with all affected cases in the same generation (siblings, same-generation cousins), two with vertical transmission (parent-child), and one with mixed different and same generations affected members (one uncle and his nephew and niece who were brother and sister). The frequency of familial cases was similar in both the French and USA series.
Figure 2.

Family trees of patients with familial Kleine-Levin syndrome, including patients with Kleine-Levin syndrome diagnosed by the physician (plain black figures), subjects with possible Kleine-Levin syndrome as reported by families (plain gray figures), and subjects without Kleine-Levin syndrome (empty figures). Note that suspected cases reported by families are included on the pedigree but not included in the total count of 21 patients from KLS families.
Demography and Medical History
Ethnicities, sex ratio, mean age at KLS onset, and body mass index were not different between groups (Table 1). There was no difference in the frequency of psychiatric disorders, epilepsy, and neurodegenerative diseases in first-degree relatives, except for a trend toward more attention deficit among relatives of patients with familial versus sporadic KLS.
Table 1.
Demography, ethnicity, familial history, human leukocyte antigen typing and karyotypes in familial versus sporadic Kleine-Levin syndrome.

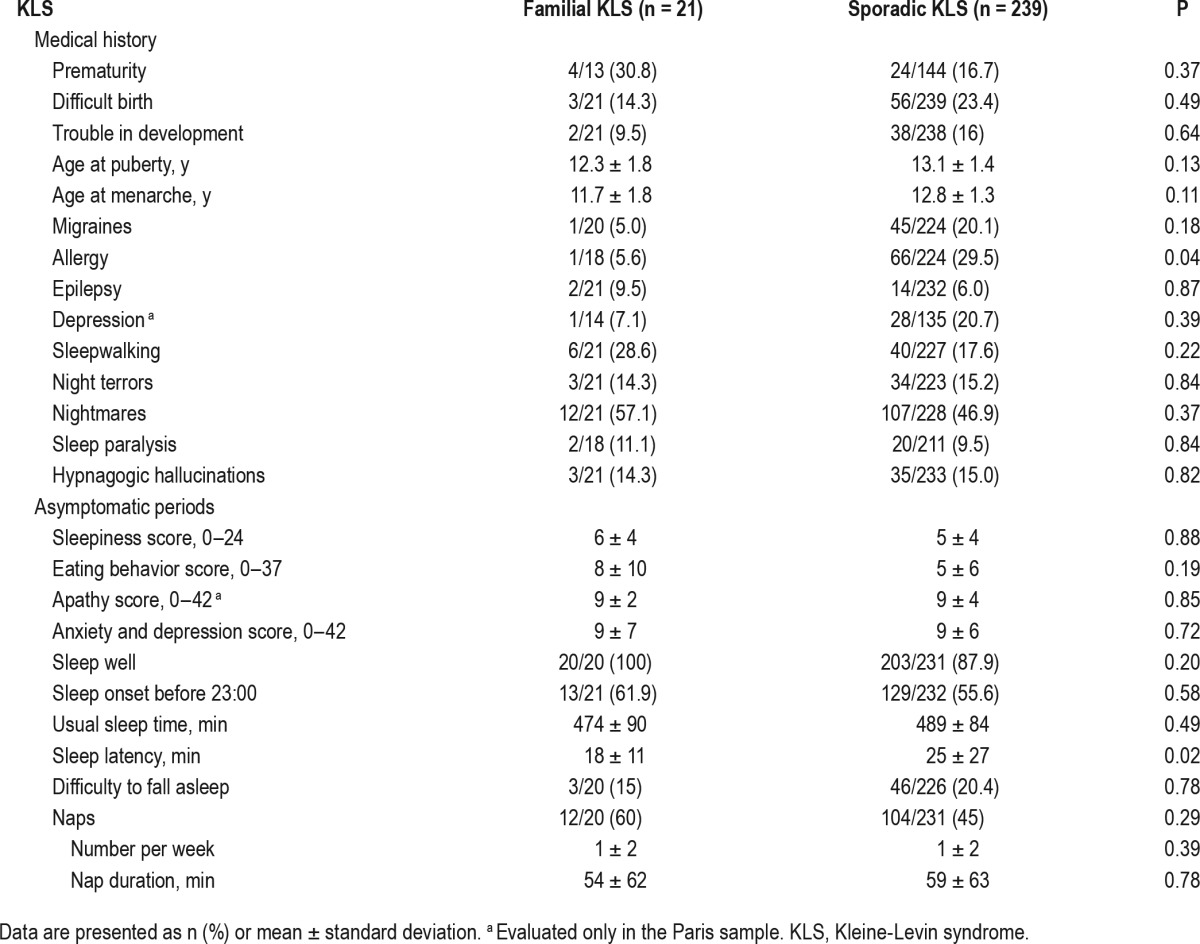
Prior reports of prematurity, difficulties at birth, and developmental difficulties were similar across groups. Age at puberty was similar in both groups. Patients with familial KLS tended to report allergy less frequently, but medical or sleep disorders occurred at the same frequency (Table 2). During asymptomatic periods, patients had normal apathy scores, eating behavior, anxiety and depression scores, and these were not different between familial and sporadic groups. Daytime sleepiness (sleepiness score as well as naps frequency and duration) and the quality and quantity of nighttime sleep were also similar, although a trend for shorter sleep onset latency was reported in familial cases.
Table 2.
Medical history and asymptomatic periods in familial versus sporadic Kleine-Levin syndrome.
Symptoms During KLS Episodes
As indicated in Table 3, 94% of familial and 87% of sporadic cases remembered an event prior to KLS onset (infection head trauma, alcohol intake, menarche) but the trigger involved was not different between groups, except that more frequent menses were reported before the first episode in women with familial versus sporadic KLS (relative risk = 4.13). Menses and other triggering factors were equally associated with KLS relapses in both groups, however. Age at KLS onset and first, longest, shortest, and mean episode durations were similar between groups. The frequency of episodes was lower in patients with familial versus sporadic KLS. Notably, the patients with familial KLS experienced most episodes at different times from their siblings.
Table 3.
Events surrounding onset and disease course in patients with familial versus sporadic Kleine-Levin syndrome.

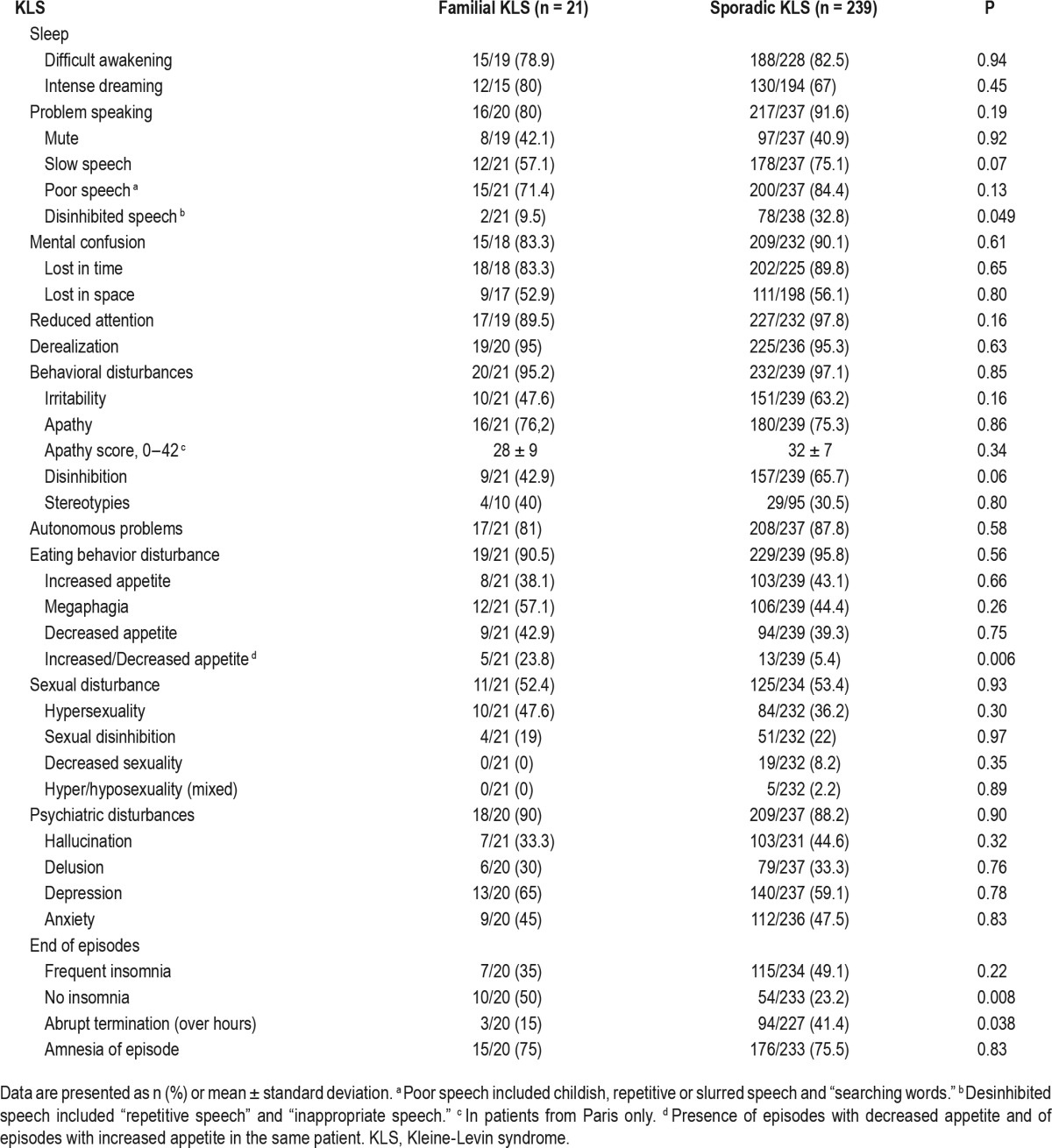
On average, the longest (21 h) and shortest (14–16 h) sleep durations during episodes were similar between groups (data not shown), as well as reports of intense dreaming, difficul-ties to be awakened, and sleep paralysis. As for cognitive symptoms, sporadic patients had more disinhibited language (relative risk = 3.44) and tended to have a slower language delivery than in the familial group, but the frequency of confusion, attention problems, and derealization was equally high in both groups (Table 4). As for behavioral problems, the frequency and severity of apathy as well as the frequency of irritability were similar in both groups, but patients with familial KLS tended to have less sexual disinhibition and had more frequently both increased and/or decreased appetite depending upon episodes (relative risk = 4.38), in contrast with more frequent single kinds of appetite change (eating less or eating more) in patients with sporadic KLS. Psychiatric and autonomous nervous system problems were equally frequent and similar between groups. At the end of episodes, patients with sporadic KLS had more frequently insomnia (relative risk = 1.54), a more rapid termination of episodes (relative risk = 2.76), but a similar degree of retrograde amnesia when compared to familial cases.
Table 4.
Symptoms during episodes of Kleine-Levin syndrome.
Human Leukocytes Class II Antigens, Karyotypes, and Brain Functional Imaging
Frequency and distribution of HLA DQ alleles did not differ between familial and sporadic groups (Table 1). Karyotyping was normal in all patients except for a patient with sporadic KLS who had1.61Mb duplication in Xp22.31 (containing four genes HDHD1, STS, VCX, and PNPLA4). Quantitative polymerase chain reaction confirmed this duplication, which was inherited from his mother (who had no KLS or other neurological and psychiatric disorders). This patient had developmental delay (toileting, language), attention deficit, childhood anxiety, and mild autistic spectrum disorder. The twin sisters (Family 7) and the nontwin sisters (Family 5) both had a similar decreased metabolism in the left temporo-occipital junction in positron emitting tomography with fluorodeoxyglucose during the asymptomatic period. This decreased metabolism in temporo-occipital junction was a common finding during asymptomatic periods in approximately two-thirds of sporadic cases here.
DISCUSSION
In this large joint series of patients with KLS from the US and France, 8% of cases were familial (4% of multiplex families), with two to three cases observed in the same multiplex family. Familial and sporadic cases shared a very similar demographic, clinical, and biological profile. During asymptomatic periods, nothing distinguished familial from sporadic cases. However, the frequency of episodes was lower in patients with familial KLS. Menses preceded more frequently the first KLS episode in girls with familial than sporadic KLS. Several symptoms were less marked in familial than in sporadic cases, including less frequent disinhibited language, as well as more mixed (increased/decreased appetite) eating disorders, and a more progressive termination of episodes, with less frequent postepisode insomnia. This blunted symptomatology may be related to differences in recruitment, as severe index cases are most likely to come to the attention of our center whereas systematic screening of the family cases will identify even milder cases. Nonetheless, symptoms were very similar between index cases and other affected family members including core symptoms of KLS (hypersomnia, derealization, apathy, cognitive problems) and the less frequent associated disinhibited behaviors (megaphagia, hypersexuality) and psychiatric symptoms (delusion, hallucination, depression) symptoms. Similarly, genetic markers (including HLA genotyping and karyotypes) were also similar between groups when tested.
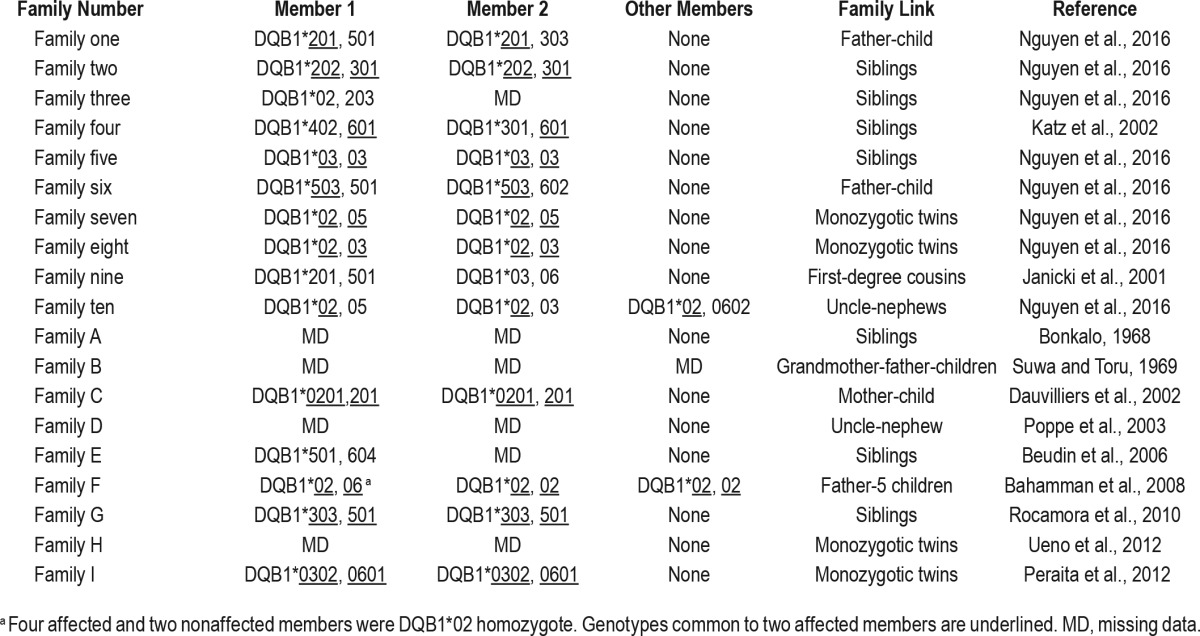
The frequency of familial KLS had been previously evaluated in a series of 120 patients, and estimated at 5% of patients or 1.7% of family members.4 With a prevalence of 2 to 10 patients with KLS per million of inhabitants and five first-degree relatives per proband, we previously estimated an 800- to 4,000-fold increased risk in first-degree relatives. With the new estimate including 21 patients from 10 families among 260 patients, a similar 1,300-fold (800- to 4,000-fold) increased risk in first-degree relatives was calculated. This increase occurred without any evidence of consanguinity or without any differences in ethnicity. Most notably, no relative increase of Jewish origin among French patients with KLS was observed,5 unlike in American patients,4 but as most patients were Caucasians, estimation of familial risk in other ethnicities was not possible. The familial cases were mostly (70%) belonging to the same generation in our series, as for seven of nine previously published cases (78%, Table 5). The low vertical risk must be stressed, as it is useful to reassure KLS patients with regard to their children.
Table 5.
HLA DQB1 genotypes in patients with familial Kleine-Levin syndrome in our study (families with numbers) and in the scientific literature (families with letters).
The more frequent association of KLS onset with menses in familial cases suggests a possible link between female hormones and familial KLS. Sporadic KLS syndrome is noteworthy male predominant (68% to 87%).1 Female cases with episode onset linked to menstruation have been reported by Kleine,20 and later singled out as a distinct syndrome, menstrual-related hypersomnia.21 Because patients have similar symptoms (hypersomnia, megaphagia, mood disorders, hyper-sexuality) as classic KLS,22 the disorder is now considered as a variant form of KLS in most recent classifications of sleep disorders.15 In menstrual-related hypersomnia, relapses are always or almost always associated with menses and/or puerperium, which was not the case in our female patients with familial KLS. It should be noted that other hormone- dependent factors, including menarche and age at puberty, did not differ between familial versus sporadic KLS.
The pathophysiology of KLS is unknown. An autoimmune origin has been suspected, because KLS affects mostly young people, is relapsing-remitting, and is often triggered by unspecific infections at KLS onset and during relapses.23 Narcolepsy type 1, another sleep disorder of autoimmune origin, is strongly associated with HLA DQB1*06:02 and can be triggered by infections (streptococcus and H1N1 influenza) but more rarely familial than KLS.24 In KLS, the DRB1*0301–DQB1*0201 and DRB1*0701–DQB1*0202 haplotypes (when grouped together) were more often encountered in 30 European patients with KLS than in controls,3 but this result was not confirmed in several subsequent larger series from the US,4 France,5 and Taiwan.23 In this study, no specific HLA DQ association was found in familial KLS either. Although familial clustering was observed, the 11 French familial cases had no common karyo-type abnormality, suggesting that familial KLS does not result from any large common chromosomal alteration. Interestingly, however, one patient presented with a micro-duplication of Xp22.31 (inherited from his otherwise healthy mother) but he had sporadic KLS. This duplication has been reported as a possible cause of intellectual disability, behavior abnormalities (autism), hypotonia, and seizures.25 Genes in the duplicated area include HDHD1, VCX, STS (steroid sulfatase, deficient in X-linked ichtyosis), and PNPLA4. High- definition karyo-type and DNA exome sequencing are next needed to explore whether more subtle DNA abnormalities can be found in the multiplex KLS families.
DISCLOSURE STATEMENT
This was not an industry-supported study. The French Kleine-Levin syndrome research program was funded in part by a grant to Dr. Arnulf (PHRC 070138) from the French Ministry of Health. It was sponsored by the Assistance Publique - Hôpitaux de Paris. The American cohort was initiated thanks to a grant from the Kleine-Levin Syndrome Foundation (Boston). Outside this work, Dr. Arnulf received honoraria for board and speaking engagements from UCB Pharma, and reported another study on brain imaging in KLS founded by the KLS Foundation in Boston. Dr. Leclair-Visonneau and Prof. Monaca had travels and congresses paid by UCB Pharma. Dr. Mignot received a grant from the Kleine-Levin syndrome foundation (Boston) to start the KLS clinical and DNA collection. Outside this work, Dr. Mignot received funding and occasional consulting income from Jazz Pharmaceuticals, had a grant from The Lundbeck Foundation, Sunovion and GSK, was consultant for Novo Nordisk, Reset, Merk, Resmed and INC, developed a device with ALPCO, and was speaker's bureau for the Federal Trade Commission. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank the physicians who referred their patients for suspicion of KLS. Authors' contribution: Dr. Nguyen collected the clinical information in a database, performed the statistics and prepared, with Dr. Arnulf, the first manuscript draft. Dr. Groos helped diagnosing patients with KLS and ruled out psychiatric diagnoses. Dr. Leclair-Visonneau and Dr. Monaca diagnosed and referred more than five patients with KLS via the Rare Disease network. Mr. Rico and Mr. Farber recruited patients in USA. Dr. Mignot obtained the grant for collecting the USA KLS cohort and edited the manuscript. Dr. Arnulf obtained the grant for collecting the French patients cohort, diagnosed and collected the Stanford patients, diagnosed, treated, and followed up the French KLS patients (helped by Dr. Groos), and drafted the manuscript. The final manuscript was approved by all coauthors.
REFERENCES
- 1.Arnulf I, Rico T, Mignot E. Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol. 2012;11:918–28. doi: 10.1016/S1474-4422(12)70187-4. [DOI] [PubMed] [Google Scholar]
- 2.Kas A, Lavault S, Habert MO, Arnulf I. Feeling unreal: a functional imaging study in 41 patients with Kleine-Levin syndrome. Brain. 2014;137:2077–87. doi: 10.1093/brain/awu112. [DOI] [PubMed] [Google Scholar]
- 3.Dauvilliers Y, Mayer G, Lecendreux M, et al. Kleine-Levin syndrome: an autoimmune hypothesis based on clinical and genetic analyses. Neurology. 2002;59:1739–45. doi: 10.1212/01.wnl.0000036605.89977.d0. [DOI] [PubMed] [Google Scholar]
- 4.Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63:482–93. doi: 10.1002/ana.21333. [DOI] [PubMed] [Google Scholar]
- 5.Lavault S, Golmard J, Groos E, et al. Kleine-Levin syndrome in 120 patients: differential diagnosis and long episodes. Ann Neurol. 2015;77:529–40. doi: 10.1002/ana.24350. [DOI] [PubMed] [Google Scholar]
- 6.Ueno T, Fukuhara A, Ikegami A, Ohishi F, Kume K. Monozygotic twins concordant for Kleine-Levin syndrome. BMC Neurol. 2012;12:31. doi: 10.1186/1471-2377-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peraita-Adrados R, Vicario JL, Tafti M, Garcia de Leon M, Billiard M. Monozygotic twins affected with kleine-levin syndrome. Sleep. 2012;35:595–6. doi: 10.5665/sleep.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonkalo A. Hypersomnia. A discussion of psychiatric implications based on three cases. Br J Psychiatry. 1968;114:69–75. doi: 10.1192/bjp.114.506.69. [DOI] [PubMed] [Google Scholar]
- 9.Beudin P. Un syndrome de Kleine-Levin : une histoire familiale. Med Som. 2006;3:55–6. [Google Scholar]
- 10.Rocamora R, Gil-Nagel A, Franch O, Vela-Bueno A. Familial recurrent hypersomnia: two siblings with Kleine-Levin syndrome and menstrual-related hypersomnia. J Child Neurol. 2010;25:1408–10. doi: 10.1177/0883073810366599. [DOI] [PubMed] [Google Scholar]
- 11.Suwa K, Toru M. A case of periodic somnolence whose sleep was induced by glucose. Folia Psychiatr Neurol Jpn. 1969;23:253–62. doi: 10.1111/j.1440-1819.1969.tb02878.x. [DOI] [PubMed] [Google Scholar]
- 12.Bahammam A, Gadelrab M, Owais S, Alswat K, Hamam K. Clinical characteristics and HLA typing of a family with Kleine-Levin syndrome. Sleep Med. 2008;9:575–8. doi: 10.1016/j.sleep.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 13.Poppe M, Friebel D, Reuner U, Todt H, Koch R, Heubner G. The Kleine-Levin syndrome - effects of treatment with lithium. Neuropediatrics. 2003;34:113–9. doi: 10.1055/s-2003-41273. [DOI] [PubMed] [Google Scholar]
- 14.American Academy of Sleep Medicine. International Classification of Sleep Disorders. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005. [Google Scholar]
- 15.American Academy of Sleep Medicine. International Classification of Sleep Disorders. 3rd ed. Darien, IL: American Academy of Sleep Medicine; 2014. [Google Scholar]
- 16.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatrica Scandinavica. 1983;67:361–70. doi: 10.1111/j.1600-0447.1983.tb09716.x. [DOI] [PubMed] [Google Scholar]
- 17.Starkstein SE, Mayberg HS, Preziosi TJ, Andrezejewski P, Leiguarda R, Robinson RG. Reliability, validity, and clinical correlates of apathy in Parkinson's disease. J Neuropsychiatry Clin Neurosci. 1992;4:134–9. doi: 10.1176/jnp.4.2.134. [DOI] [PubMed] [Google Scholar]
- 18.Katz JD, Ropper AH. Familial Kleine-Levin syndrome: two siblings with unusually long hypersomnic spells. Arch Neurol. 2002;59:1959–61. doi: 10.1001/archneur.59.12.1959. [DOI] [PubMed] [Google Scholar]
- 19.Janicki S, Franco K, Zarko R. A case report of Kleine-Levin syndrome in an adolescent girl. Psychosomatics. 2001;42:350–2. doi: 10.1176/appi.psy.42.4.350. [DOI] [PubMed] [Google Scholar]
- 20.Kleine W. Periodische Schlafsucht. Monatsschrift fur Psychiatrie und Neurologie. 1925;57:285–320. [Google Scholar]
- 21.Billiard M, Guilleminault C, Dement WC. A menstruation-linked periodic hypersomnia. Kleine-Levin syndrome or new clinical entity? Neurology. 1975;25:436–43. doi: 10.1212/wnl.25.5.436. [DOI] [PubMed] [Google Scholar]
- 22.Billiard M, Jaussent I, Dauvilliers Y, Besset A. Recurrent hypersomnia: a review of 339 cases. Sleep Med Rev. 2011;15:247–57. doi: 10.1016/j.smrv.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Huang Y, Guilleminault C, Lin K, Hwang F, Liu F, Kung Y. Relationship between Kleine-Levin Syndrome and upper respiratory Infection in Taiwan. Sleep. 2012;35:123–9. doi: 10.5665/sleep.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50:S16–22. doi: 10.1212/wnl.50.2_suppl_1.s16. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Shen Y, Köhler U, et al. Interstitial microduplication of Xp22.31: Causative of intellectual disability or benign copy number variant? Eur J Med Genet. 2010;53:93–9. doi: 10.1016/j.ejmg.2010.01.004. [DOI] [PubMed] [Google Scholar]