

Abstract
Study Objectives:
Cross-sectional studies report a correlation between slow wave sleep (SWS) duration and insulin sensitivity (SI) in children and adults. Suppression of SWS causes insulin resistance in adults but effects in children are unknown. This study was designed to determine the effect of SWS fragmentation on SI in children.
Methods:
Fourteen pubertal children (11.3–14.1 y, body mass index 29th to 97th percentile) were randomized to sleep studies and mixed meal (MM) tolerance tests with and without SWS disruption. Beta-cell responsiveness (Φ) and SI were determined using oral minimal modeling.
Results:
During the disruption night, auditory stimuli (68.1 ± 10.7/night; mean ± standard error) decreased SWS by 40.0 ± 8.0%. SWS fragmentation did not affect fasting glucose (non-disrupted 76.9 ± 2.3 versus disrupted 80.6 ± 2.1 mg/dL), insulin (9.2 ± 1.6 versus 10.4 ± 2.0 μIU/mL), or C-peptide (1.9 ± 0.2 versus 1.9 ± 0.1 ng/mL) levels and did not impair SI (12.9 ± 2.3 versus 10.1 ± 1.6 10−4 dL/kg/min per μIU/mL) or Φ (73.4 ± 7.8 versus 74.4 ± 8.4 10−9 min−1) to a MM challenge. Only the subjects in the most insulin-sensitive tertile demonstrated a consistent decrease in SI after SWS disruption.
Conclusion:
Pubertal children across a range of body mass indices may be resistant to the adverse metabolic effects of acute SWS disruption. Only those subjects with high SI (i.e., having the greatest “metabolic reserve”) demonstrated a consistent decrease in SI. These results suggest that adolescents may have a unique ability to adapt to metabolic stressors, such as acute SWS disruption, to maintain euglycemia. Additional studies are necessary to confirm that this resiliency is maintained in settings of chronic SWS disruption.
Citation:
Shaw ND, McHill AW, Schiavon M, Kangarloo T, Mankowski PW, Cobelli C, Klerman EB, Hall JE. Effect of slow wave sleep disruption on metabolic parameters in adolescents. SLEEP 2016;39(8):1591–1599.
Keywords: glucose, insulin, puberty, slow wave sleep
Significance.
Cross-sectional studies in children and adults and a small number of interventional studies in adults suggest that short slow wave sleep (SWS) duration and SWS fragmentation are risk factors for metabolic syndrome. The current studies, which represent the first SWS disruption studies with metabolic phenotyping to be conducted in children, demonstrate that a single night of SWS disruption does not diminish insulin sensitivity in pubertal children, challenging the existing dogma based on studies in adults. These results imply that adolescents may have a unique metabolic resiliency to acute sleep disruption; follow-up studies will be important in settings of more prolonged SWS restriction or disruption.
INTRODUCTION
Epidemiologic studies have demonstrated that both chronic short sleep and poor sleep quality are associated with an increased risk of obesity and type 2 diabetes mellitus (T2DM) in adults. Consistent with these cross-sectional observations, metabolic abnormalities have also been observed in adults after either acute sleep restriction or sleep fragmentation in a controlled laboratory setting.1 Importantly, recent work has suggested that some of these effects are related to specific disruption of slow wave sleep (SWS). SWS is the deepest stage of sleep and is defined by the presence of low-frequency, high-amplitude waveforms known as slow waves or delta waves on electroencephalogram (EEG) tracings. Categorical sleep stage scoring has traditionally been used to identify SWS. However, use of spectral analysis to quantify slow wave activity (SWA) provides a more precise measure of sleep depth and quality. A connection between SWS and metabolism is supported by some, but not all,2–5 cross-sectional studies demonstrating an inverse correlation between the amount of SWS or SWA and body mass index (BMI),6 waist circumference,7 and hemoglobin A1C,8 and a positive correlation between SWS and lean body mass.9 Moreover, selective suppression of SWS with preservation of total sleep time decreases insulin sensitivity (SI) in adults.10,11 The strong association between disturbed sleep and metabolic abnormalities in adults has led to the hypothesis that poor sleep hygiene is a modifiable risk factor for obesity and insulin resistance.1 As sleep restriction (i.e., multiple nights with insufficient sleep) has reached epidemic proportions among teenagers,12 it is important to determine the effects of reduced and/or disordered sleep on glucose homeostasis in pubertal children.
Two physiologic changes occur during puberty that may exacerbate the effect of reduced or fragmented sleep in adolescents. The first is that there is a more than 30% decline in SI during puberty, even among lean adolescents.13 The second is that, beginning at 11 to 12 y of age and continuing through late adolescence, there is a steep decline in the amount of SWS,14,15 the sleep stage now implicated in maintaining SI in adults.11 Despite these dynamic changes in SI and sleep during normal adolescence, cross-sectional studies of sleep, body weight, and metabolism in teenagers have not consistently demonstrated an association between habitual sleep duration and body weight16 or SI.17–21 In studies in which sleep parameters were objectively determined via in-hospital polysomnography (PSG), however, investigators observed correlations between SWS and insulin secretion,22 SWS and SI,23 and SWA and SI.24 In the only interventional sleep study thus far in an adolescent population,25 investigators observed a decrease in SI after 3 days of sleep restriction to only 4 h a night. Of interest, the total amount of time spent in SWS was preserved during this intervention, indicating the importance of further studies of the relationship between SWS and SI in this population.
To critically address the hypothesis that SWS is an important determinant of SI during puberty, we conducted SWS disruption studies using controlled auditory stimuli followed by a mixed meal tolerance test (MMTT) in a group of healthy adolescents.
METHODS
Subjects
Fourteen healthy children were studied. All subjects were pubertal as determined by Tanner staging and orchidometry, and all girls were premenarchal. Subjects had no signs or symptoms of diabetes and were not on any medications that influence glucose or insulin levels. One subject was on medications that may affect sleep and metabolism (methylphenidate and sertraline) but was not excluded because sleep parameters in the non-disrupted study fell within the normal range, the medication doses did not change between study visits, and exclusion of her data points did not change the results of any analyses. Subjects with known sleep disorders or suspected to have obstructive sleep apnea based on results of a validated sleep questionnaire26 were excluded. Some polysomnographic and clinical data, including the characteristics of the study subjects, have been described previously.27 The study was approved by the Partners Human Research Committee. Signed informed assent and consent were obtained from each subject and parent.
Experimental Protocol
Subjects underwent two PSG sleep studies spaced 2 mo apart, with and without SWS disruption in random order, in the Clinical Research Center of Massachusetts General Hospital, as previously described.27 PSG was performed according to standard methodology using an EEG (total of six frontal, central and occipital leads), electro-oculogram (EOG), electromyo-gram (EMG), electrocardiogram (ECG), and pulse oximetry recordings (ALICE LE PSG system, Sleepware software, Phillips Respironics, Murrysville, PA). PSG recording began 10 min before lights out based on subject and parent reports of habitual bedtime (21:00–22:30) and continued until spontaneous awakening the following morning (05:00–07:30). An intravenous (IV) catheter was inserted upon admission and connected to a long line for remote blood sampling throughout the night.
During the SWS disruption night, auditory stimuli (3 sec 1,500 Hz tones at 40 dB increasing in 10-dB increments to a maximum of 100 dB followed by 18 sec of noise simulating a knock on the door at 75 dB) were delivered via a speaker placed at the head of the bed (iHome iP3 stereo speaker system, SDI Technologies, Inc, Rahway, NJ) for 8 h, as previously described.27 Stimuli were delivered whenever at least two delta waves (≤ 4 Hz) appeared in a 15-sec PSG recording interval and were followed by tactile stimuli if the subject did not arouse. Subjects were allowed to sleep undisturbed after the 8-h intervention until spontaneous awakening the next morning. Two subjects repeated the disruption night because of insufficient disruption (250–280 min SWS) during the first visit related to technical difficulties.
All subjects ate dinner before lights out. Food and drink were not permitted from 21:00 until the MMTT was completed the following morning. The MMTT was conducted within 1 h of final awakening to assess glucose tolerance. The MMTT is well tolerated by children, presents a more physiologic stimulus than either oral or IV glucose tolerance tests, and has been validated against hyperglycemic clamp studies.28,29 At time 0, subjects consumed a weight-based dose (6 cc/kg; maximum 360 cc) of Boost High Protein Nutritional Energy Drink [68% carbohydrate, 17% protein, and 15% fat] (Mead-Johnson) in under 5 min. Plasma glucose, insulin, and C-peptide levels were measured at time 0, 10, 20, 30, 60, 90, 120, 150, and 180 min. One subject did not tolerate the MMTT (nausea) during the disruption night so only fasting hormone levels were used in disrupted and non-disrupted sleep comparisons. Study nurses ensured that subjects remained awake for the duration of the MMTT.
We analyzed overnight heart rate variability (HRV), measured seated blood pressure and heart rate at the end of the study visit, and serum cortisol at 06:00–08:00, as previous studies have suggested that an increase in sympathetic tone or cortisol secretion during sleep disruption may be responsible for the ensuing insulin resistance.
Plasma glucose and cortisol were measured using the Architect Integrated ci8200 Chemistry and Immunochemistry Analyzer (Abbott Diagnostics, Abbott Park, IL), respectively, and plasma insulin and C-peptide were measured with the Roche E170 immunoanalyzer.
Data Analysis
Sleep Scoring
The sleep recordings were visually scored by a single registered PSG technician according to American Academy of Sleep Medicine criteria30 in 30-sec epochs as stages of nonrapid eye movement (NREM; N1, N2, or N3), rapid eye movement (REM), or wake. The arousal index (AI) was defined as the number of arousals per hour of sleep. Sleep latency was defined as the duration of time between lights out and the first epoch of sleep, and sleep efficiency was defined as the percent of time in bed spent asleep. Wake after sleep onset was defined as minutes of wake after the first sleep epoch until natural awakening.
EEG Analysis
Spectral analysis was used to analyze total SWA in the 0.5–4 Hz range during stages N2 and N3 using SpectralTrainFig (https://github.com/DennisDean/SpectralTrainFig) within Matlab (MathWorks, Inc., version R2013b, Natick, MA). Artifacts were removed after being identified either within SpectralTrainFig or as any delta activity at least five times the average delta activity in N2 and N3 for the entire sleep opportunity. NREM episodes were defined as the SWA between epochs of REM sleep using a modified Feinberg and Floyd method.31 If the subject's sleep opportunity contained a “skipped” first REM episode, which is common in adolescents, an epoch of REM sleep was inserted (iREM) at either the first epoch of wake or at the nadir of SWA between visually identified NREM episodes (modified “Jenni and Carskadon criteria”).32 The SWA in each NREM episode was expressed as a percentage of total night SWA to facilitate between-subject comparisons.
HRV Analysis
HRV was calculated using a continuous single-lead ECG recording from each 8-h sleep study. Kubios HRV software v2.233 was used to remove artifacts and to compute low-frequency band power (0.04–0.15 Hz), a marker of sympathetic activity, and high-frequency band power (0.15–0.4 Hz), a marker of vagal activity.
Assessment of Beta-Cell Function and Insulin Sensitivity
The homeostatic model assessment of insulin resistance (HOMA-IR), a surrogate estimate of basal SI, was calculated using the formula: fasting plasma insulin (μIU/mol) × fasting plasma glucose (mmol/L)/22.5.34 Beta cell responsiveness (Φ) and SI were assessed from serial insulin, C-peptide, and glucose measurements during the MMTT using the oral C-peptide and glucose minimal models, respectively.35 These models have been validated using hyperglycemic clamp studies28,29 and radio-labeled glucose analogs in adults36 and adolescents.37 The C-peptide minimal model determines Φ to basal glucose levels (Φb) and to a nutrient load (Φt) and accounts for subject age, height, weight, and BMI. Total beta cell responsivity to a meal (Φt) is partitioned into two components: a dynamic response (Φd, response to the acute rise in blood glucose during a meal), and a static response (Φs, response to the glucose level achieved above basal after a delay, T). SI, estimated from the glucose minimal model, reflects both insulin-mediated glucose disposal (tissue uptake) and inhibition of hepatic gluconeogenesis. The total disposition index (DI), which measures Φ in the context of the prevailing SI, is calculated as the product of Φt and SI.
Statistical Methods
Differences in sleep architecture, SWA, HRV, and metabolic parameters between the disrupted and non-disrupted nights were compared using analysis of covariance with a treatment (disruption versus no disruption) effect. The areas under the curve (AUC) for the glucose, insulin, and C-peptide responses to the MMTT were calculated using the trapezoidal rule. The relationships between sleep parameters and SI were assessed using Pearson correlation. Data that were not normally distributed were log-transformed for analysis. Data are expressed as mean ± standard error [range] unless otherwise indicated, and P < 0.05 is considered significant.
RESULTS
Baseline Characteristics
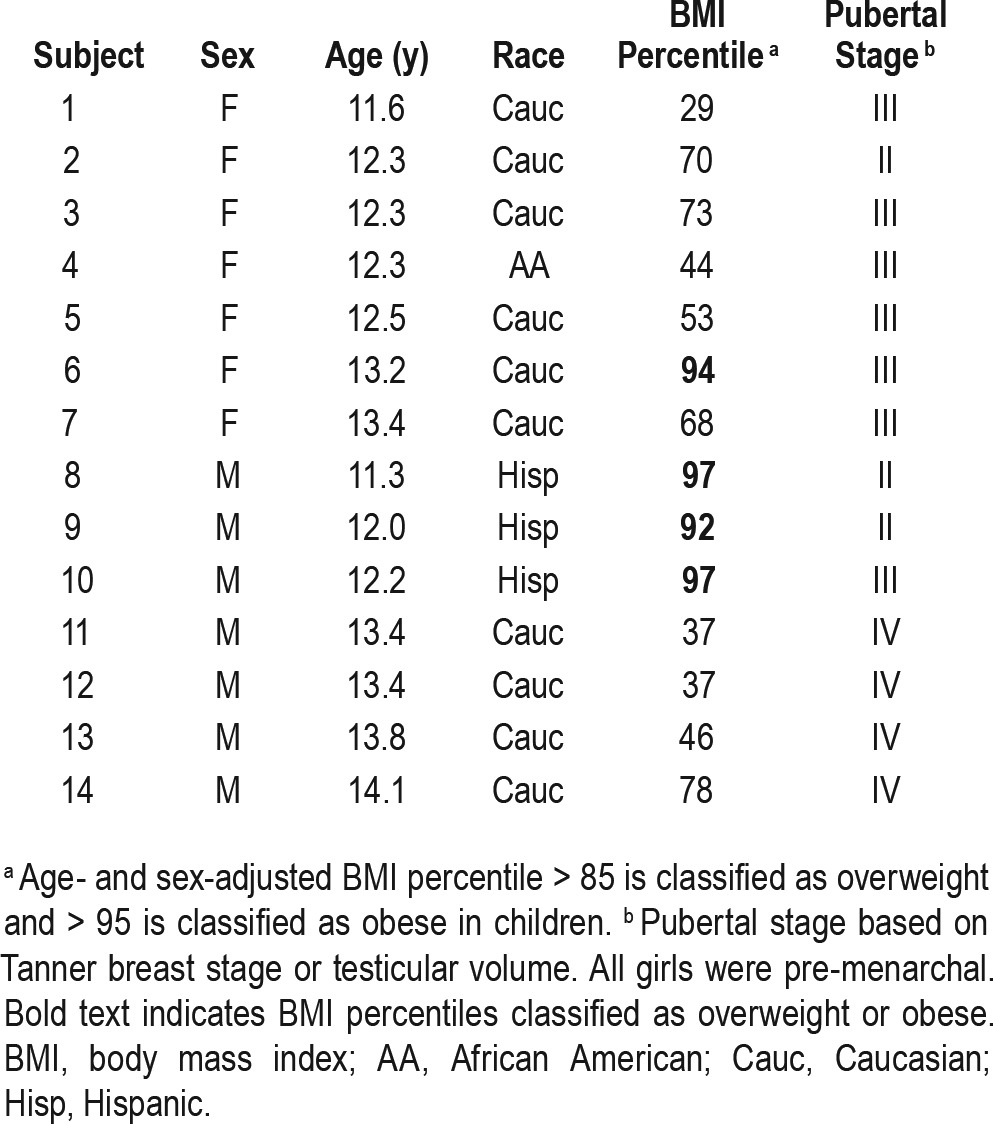
Seven boys, ages 11.3–14.1 y, with testicular volumes of 4–15 mL (Tanner II-IV) and 7 premenarchal girls, ages 11.6–13.4 y, with Tanner II-III breasts participated (Table 1). Subjects were predominantly non-Hispanic Caucasians with a wide range of BMIs (29th to 97th percentile for age and sex). There were no changes in BMI percentiles or Tanner staging between study visits.
Table 1.
Subject characteristics.
Standard Polysomnographic Metrics
During the non-disrupted sleep night, subjects slept for 8.2 ± 0.1 h and demonstrated normal sleep architecture with 32.7 ± 2.5% of time spent in SWS (N3) (Table 2 and Figure 1A). Sleep efficiency, sleep latency, and AIs were consistent with normative data in adolescents38 and there were no apneic episodes.
Table 2.
Comparison of sleep parameters during the non-disrupted and disrupted sleep nights.

Figure 1.

Sleep stages (wake, REM, inserted REM (iREM), N1, N2, and N3) (top) and slow wave activity (bottom) in a representative subject during 1 night of non-disrupted (A) and 1 night of disrupted (B) sleep. Visits were spaced 2 mo apart. Note the consolidated periods of deep sleep (N3) across the night during normal sleep (A) in contrast to the N3 fragmentation during the disrupted sleep night (B). The start and end of each of the first two NREM episodes are denoted in boxes above the slow wave activity in the bottom panels. NREM, nonrapid eye movement; REM, rapid eye movement.
During the disrupted night, 68.1 ± 10.7 [20–120] auditory or tactile stimuli were delivered. These 8 h of disrupted SWS were followed by a brief duration of recovery sleep (23.9 ± 6.5 min) [0–72] before natural awakening. There was a 40.0 ± 8.0% [−11.5 to 84.0] decrease in %SWS (P < 0.001) relative to the non-disrupted sleep night, and the SWS that remained was fragmented into short bouts (1.3 ± 0.2 min [0.7–3.2] versus 7.1 ± 0.8 min [3.8–13.2] in the non-disrupted night; P < 0.001) due to frequent arousals (Table 2). SWS was replaced by lighter sleep (N1 and N2) with no change in %REM or %wake after sleep onset (Table 2, Figure 1B) such that the overall percentage of time spent in NREM was preserved (disrupted: 71.6 ± 2.2% [54.1–78.6] versus non-disrupted: 73.3 ± 2.4% [56.3–83.9], P = 0.4).
Slow Wave Activity
During the non-disrupted sleep night, total delta power was 2.7 × 106 μV2, with SWA predominating in the first two NREM episodes (70%) [55–95] (Figure 1A), consistent with previous studies in this demographic.14,24 Sleep disruption decreased total delta power by 38.8 ± 5.9% [−6.5 to 65.9] (Figure 1B).
Beta Cell Function and Insulin Action
Subjects began the MMTT at 06:00–07:45 with no difference in timing between study visits. All subjects had normal (< 100 mg/dL) fasting blood glucoses the morning after the night of non-disrupted sleep (76.8 ± 2.4 mg/dL [57–91]) and disrupted sleep (80.6 ± 2.1 mg/dL [68–93]) with no difference between the two visits (Figure 2). Fasting insulin levels were also normal (< 15 μIU/mL) with the exception of one obese boy in whom fasting hyperinsulinemia (> 20 μIU/mL) was observed after both visits (non-disrupted 22.3 μIU/mL, disrupted 25 μIU/mL) and a second obese boy with fasting hyperinsulinemia (21.6 μIU/mL) restricted to the non-disrupted sleep visit. There were no differences in fasting glucose, insulin (non-disrupted: 9.2 ± 1.6 [2.0–22.3], disrupted: 10.4 ± 2.0 μIU/mL [4.2–35]), or C-peptide (non-disrupted: 1.9 ± 0.2 [1.0–3.3], disrupted: 1.9 ± 0.1 ng/mL [1.2–3.1]) levels or in the corresponding postprandial AUCs between study visits (glucose AUC: 17,167.5 ± 524.6 [13,275.0–19,920.0] versus 16,802.7 ± 952.3 [3,675.5–36,126.0] [non-disrupted versus disrupted], insulin AUC 9,805.5 ± 2,995.9 [3,985.5– 46,702.5] versus 9,447.7 ± 2,361.9 [3,675.5–36,126.0], C-peptide AUC 961.1 ± 86.7 [650.0–1,912.5] versus 943.9 ± 91.7 [667.0–1,938.5]) (Figure 2).
Figure 2.
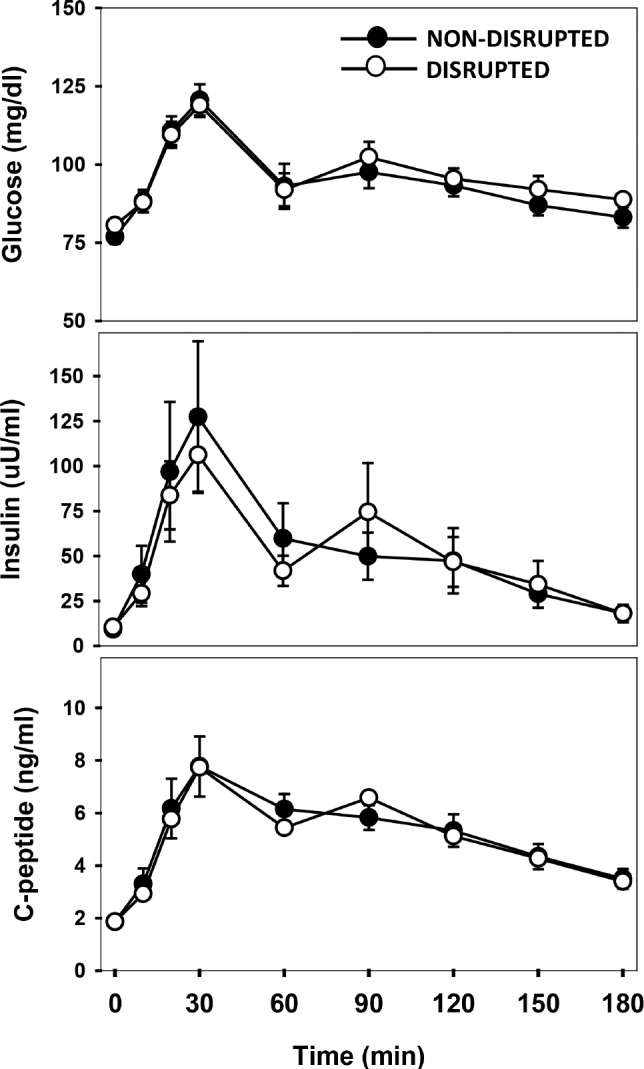
Slow wave sleep fragmentation in pubertal children did not alter fasting (t = 0 min) glucose, insulin, or C-peptide levels or the responses to a mixed meal tolerance test (t = 10, 20, 30, 60, 90, 120, 150, and 180 min) performed within 1 h of waking (○) compared with results after a night of non-disrupted sleep (●). Data are presented as mean ± standard error.
Beta cell responsivity (Φ) to baseline glucose levels and to the MM challenge was maintained after SWS disruption (Figure 3A). SI, as determined by glucose minimal modeling and HOMA-IR (non-disrupted: 1.8 ± 0.3 [0.4–4.3] versus disrupted 2.1 ± 0.5 [0.8–7.8]), was also preserved following one night of sleep disruption resulting in stable total disposition indices (DI) (Figure 3B). Results from both study visits were comparable to those previously reported in healthy adolescents with similar anthropometrics.37
Figure 3.

(A) Dynamic, static, basal, and total components of beta cell responsivity, and (B) insulin sensitivity and the disposition index after non-disrupted (left) and disrupted (right) sleep nights calculated from serum glucose, insulin, and C-peptide responses to mixed meal tolerance tests using minimal modeling. Individual-level data for both sleep conditions are connected by a line; off-set circles indicate mean ± standard error of the mean.
Correlations between Sleep Parameters and Insulin Sensitivity
A greater BMI percentile was associated with lower SI (β = - 0.2, P = 0.006). There was no correlation between minutes spent in SWS and SI (β = 0.04, P = 0.1) or between percent change in SWS (min) and percent change in SI (β = 0.03, P = 0.6). A correlation between the percent change in delta power and the percent change in SI was explained by a single outlier. There was an interaction between the percent difference in SI between the two studies and SI during the non-disrupted night, such that only the most insulin sensitive subjects were vulnerable to the adverse metabolic effects of sleep disruption: the subjects in the highest SI tertile (SI range 16.5–31.3 × 10−4 dL/kg/min per μIU/mL) demonstrated a 21–73% decrease in SI after sleep disruption, whereas the remaining subjects did not demonstrate a consistent response (Figure 3).
Biomarkers of Sympathetic Activation and Stress
ECG low-frequency power (sympathetic activity) increased by 29.6 ± 11.3% [−21.0 to 147.5] and high-frequency power (vagal activity) decreased by 15.9 ± 7.5% [−34.6 to 82.2] during the night of sleep disruption relative to non-disrupted sleep (Table 2). There were no differences in mean blood pressure (non-disrupted 67 ± 6 mm Hg [60–81] versus disrupted 79 ± 2 [64–90]), resting heart rate (86 ± 4 beats/min [67–122] versus 86 ± 4 [56–119]), or cortisol levels (7.8 ± 0.8 mcg/dL [2.6–11.5] versus 7.0 ± 0.7 [3.2–11.8]) in the morning after the two study nights.
DISCUSSION
The idea of sleep as a modifiable risk factor for obesity and insulin resistance is an attractive one that is supported by epidemiological literature and a small number of interventional sleep disruption studies. Most of these studies have been limited to adults, yet it is adolescents who would be predicted to be at the highest risk for insulin resistance and T2DM because of the “perfect storm” of chronic short sleep, a physiologic decline in SWS,14,15 and a physiologic peak in insulin resistance during puberty.13 The current studies were designed to fill this scientific gap by investigating the effect of SWS disruption on SI in pubertal subjects. Despite our prediction that adolescents would be more sensitive to SWS disruption than adults, we found that adolescents across a range of BMIs appear to be metabolically resilient to acute restriction and fragmentation of SWS.
Although previous interventional studies have demonstrated that even 2 to 3 nights of SWS or total sleep restriction results in metabolic derangements in adults, with effects on glucose homeostasis, orexigenic and anorexigenic hormones, food preferences, and portion sizes,39 the research protocols employed dramatically altered sleep structure to an extent unlikely to occur in everyday life. The single interventional study conducted in pediatric subjects also employed a severe sleep restriction protocol of 4 h of sleep for 3 days with no opportunity for daytime naps25 in contrast to the current protocol in which sleep disruption was more modest and likely closer to naturalistic conditions. In addition, a meal of mixed macro-nutrients (the MMTT) was chosen over an oral or IV glucose tolerance test in the current studies to more closely emulate habitual nutrient intake while still providing a robust assessment of SI.
In the current studies, the adolescent subjects had lower SI and slightly higher beta cell responsivity after 1 night of undisturbed sleep compared with results of MMTTs in normal weight adults,40 consistent with the known increase in insulin resistance in adolescents. Greater baseline insulin resistance, however, did not translate into greater metabolic vulnerability to SWS disruption; basal SI, postprandial SI, and post-prandial beta cell responsivity were maintained despite a 40% decrease in SWS. Most adolescents avert diabetes despite a profound decrease in SI during puberty.13 The decrease in SI is countered by a compensatory increase in insulin secretion41; however, the beta cell response is quite modest and cannot fully explain the preservation of glucose homeostasis during puberty. Thus, adolescent physiology must invoke additional adaptive responses to defend against the physiologic increase in SI, perhaps at the level of the insulin receptor and downstream signaling pathways in the liver, muscle, and/or adipose tissue that have yet to be identified. The current studies suggest that a similar adaptive mechanism may help adolescents defend against the metabolic derangements associated with SWS disruption in adults.
The pathophysiologic links between SWS suppression and metabolic abnormalities in adults are unknown. One hypothesis is that these changes are mediated by increased sympathetic activity that induces insulin resistance by limiting peripheral blood flow, and hence insulin and glucose delivery, to skeletal muscle and adipose tissue.42 SWS is associated with a nadir in systemic sympathetic activity (SA)43 and analyses of nocturnal HRV, a very sensitive marker of SA, suggest that severe SWS disruption in adults increases SA during the night.44 Some11,44 but not all45 studies in adults suggest that sympathetic hyperactivity may persist in the daytime after SWS disruption, although a study using muscle microneurography, which provides a direct readout of SA, found no evidence for increased SA in the daytime after SWS disruption.45 In the current study in adolescents, there was evidence of sympathetic hyperactivity during the night of sleep disruption, as seen in adults, but metabolic parameters were not disrupted. Other potential mediators of insulin resistance include increased cortisol, adipokines, and inflammatory cytokines; however, no SWS disruption studies11,44,45 including our own have thus far demonstrated clinically significant changes in any of these variables.
It is possible that our subjects remained insulin-sensitive not because of their unique pubertal physiology but because SWS was not suppressed to the same degree achieved in previous studies in adults. The current sleep disruption protocol was modeled after that of Tasali and colleagues,11 who demonstrated that auditory-based sleep disruption decreases SWS by 90% in lean adult subjects. The same protocol applied to adolescent subjects in the current studies produced a more modest (40%) decrease in SWS, consistent with the greater homeostatic sleep drive that exists in early adolescence compared with adulthood.46 Moreover, because subjects resumed SWS immediately after being aroused, a greater decrease in SWS may not be achievable in adolescents without inducing full awakenings. In light of Tasali et al.'s finding of a 25% decrease in SI after 3 nights of severe SWS disruption, it is possible that there is a dose-response relationship between SWS and glucose metabolism whereby SI is preserved if the amount of SWS remains above a certain threshold, a concept first introduced by Herzog et al.10 The increased homeostatic sleep drive among adolescents, however, also suggests that severe SWS disruption, to the extent achieved experimentally in adults, is unlikely to occur among teenagers in everyday life. We cannot exclude the possibility that metabolic abnormalities would manifest in adolescents after more prolonged sleep disruption, as may occur in sleep apnea or as a result of environmental noise.
It is of interest that only the most insulin-sensitive adolescent subjects demonstrated a consistent decrease in SI following SWS disruption. Previous studies that reported a decrease in SI after SWS suppression also differed from the current studies in that they included healthy, lean adults and specifically excluded the overweight or obese. In contrast, the adolescent subjects studied herein represented a wide range of BMIs (approximately one-third overweight or obese). Therefore, one explanation for the discrepant SI results is that SWS disruption only impairs SI in individuals who are highly insulin sensitive and has negligible effects in insulin-resistant individuals. This further suggests that SWS disruption may create insulin resistance by interfering with some of the same physiologic pathways that have already been inhibited by obesity such as endoplasmic reticulum stress, mitochondrial dysfunction, and ectopic lipogenesis mediated by inflammatory cytokines, free fatty acids, and nitric oxide.47 Studies in transgenic mice have also demonstrated that obesity impairs the brain's ability to sense increased glucose or insulin levels and to respond appropriately by decreasing hepatic glucose output.48 This finding is of particular interest because these glucose-sensing neurons (located in the arcuate nucleus) project to the ventrolateral preoptic nucleus,49 which plays a critical role in promoting and maintaining NREM sleep.50 Thus, the link between SWS disruption and insulin resistance is likely to be multifactorial with effects on the liver, muscle, adipose tissue, and even the brain.
The current study has several limitations that may help inform the design of future studies in adolescents. The intensive nature of the research protocol limited the size of our sample and thus we were not able to tease out the effects of sex, pubertal stage, or BMI. Furthermore, data derived from a predominantly middle-class, Caucasian population may not be generalizable to minority groups who are at greater risk of T2DM and who may have different sleep patterns and sleep environments. Although we were careful to exclude subjects with sleep-disordered breathing and/or associated symptoms (e.g., snoring, excessive daytime sleepiness, or inattention and hyperactivity), we did not control for potential differences in sleep duration and quality in the 2 mo between study visits, which may have also been influenced by the time of year (e.g., school versus summer). Most importantly, this and other SWS disruption studies in adults cannot address whether chronic SWS disruption has the same detrimental metabolic effects as short-term disruption or if compensatory physiological changes develop over time. Of note, Broussard and colleagues recently demonstrated that 2 nights of recovery sleep was sufficient to reverse the metabolic abnormalities that developed after 4 nights of severe sleep restriction in healthy men.51 These results provide some reassurance that intermittent and short reprieves from chronic sleep restriction, and perhaps from sleep disruption, may be sufficient to avert T2DM.
In conclusion, we have now demonstrated that SI is preserved in adolescents following selective, acute SWS fragmentation. These data provide preliminary evidence that adolescents may be uniquely poised to respond to metabolic stressors, such as SWS disruption, to maintain euglycemia and further suggests that only those adolescents who are highly insulin sensitive, and therefore at low risk for T2DM, are susceptible to the metabolic insult of SWS disruption. Both interventional and observational studies of chronic sleep disruption in a larger group of adolescents are necessary to confirm and generalize these important findings.
DISCLOSURE STATEMENT
This was not an industry supported study. Dr. Shaw received support from the NIH (K23HD073304-02 and the National Institute of Diabetes and Digestive Diseases [NIDDK]-sponsored Boston Area Diabetes Endocrinology Research Center Pilot Research Grant Award), the Pediatric Endocrine Society, and Harvard Catalyst (The Harvard Clinical and Translational Science Center [Award #1UL1TR001102-01] and financial contributions from Harvard University and its affiliated academic health care centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Center for Research Resources or the National Institutes of Health). Drs. McHill, Klerman, and Cobelli are funded by the NIH (T32HL007901; K24HL105664, R01HL114088, R01GM105018, P01AG009975, R21HD086392, NSBRI HFP02802, HFP04201; and R01 DK-78646 and R01 DK-82396, respectively). This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences [NIEHS] (1SI2ES025429-01, Lasker Clinical Research Scholar Award to Dr. Shaw). Dr. Hall has received research support from Marathon Pharmaceuticals. Dr. Vienne has consulted for Celgene International Sárl. The other authors have indicated no financial conflicts of interest. Clinical studies were conducted in the Clinical Research Unit at Massachusetts General Hospital, Boston, MA. Analyses were performed at NIEHS, Brigham and Women's Hospital, and the University of Padova. This study does not meet the FDAAA 801 definition of an “applicable clinical trial” and as such was not registered at clinicaltrials.gov.
ACKNOWLEDGMENTS
The authors thank the Clinical Research Center staff and the sleep technicians for their support in conducting these studies and Dr. Amir Lahav for expertise in creating auditory stimuli.
REFERENCES
- 1.Arble DM, Bass J, Behn CD, et al. Impact of sleep and circadian disruption on energy balance and diabetes: a summary of workshop discussions. Sleep. 2015;38:1849–60. doi: 10.5665/sleep.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint J, Kothare SV, Zihlif M, et al. Association between inadequate sleep and insulin resistance in obese children. J Pediatr. 2007;150:364–9. doi: 10.1016/j.jpeds.2006.08.063. [DOI] [PubMed] [Google Scholar]
- 3.Chamorro R, Ferri R, Algarin C, Garrido M, Lozoff B, Peirano P. Sleep cyclic alternating pattern in otherwise healthy overweight school-age children. Sleep. 2014;37:557–60. doi: 10.5665/sleep.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Resta O, Foschino Barbaro MP, Bonfitto P, et al. Low sleep quality and daytime sleepiness in obese patients without obstructive sleep apnoea syndrome. J Intern Med. 2003;253:536–43. doi: 10.1046/j.1365-2796.2003.01133.x. [DOI] [PubMed] [Google Scholar]
- 5.Vgontzas AN, Tan TL, Bixler EO, Martin LF, Shubert D, Kales A. Sleep apnea and sleep disruption in obese patients. Arch Intern Med. 1994;154:1705–11. [PubMed] [Google Scholar]
- 6.Rao MN, Blackwell T, Redline S, Stefanick ML, Ancoli-Israel S, Stone KL. Association between sleep architecture and measures of body composition. Sleep. 2009;32:483–90. doi: 10.1093/sleep/32.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theorell-Haglow J, Berne C, Janson C, Sahlin C, Lindberg E. Associations between short sleep duration and central obesity in women. Sleep. 2010;33:593–8. [PMC free article] [PubMed] [Google Scholar]
- 8.Feupe SF, Frias PF, Mednick SC, McDevitt EA, Heintzman ND. Nocturnal continuous glucose and sleep stage data in adults with type 1 diabetes in real-world conditions. J Diabetes Sci Technol. 2013;7:1337–45. doi: 10.1177/193229681300700525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro CM, Catterall J, Warren P, et al. Lean body mass and non-rapid eye movement sleep. Br Med J. 1987;294:22. doi: 10.1136/bmj.294.6563.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herzog N, Jauch-Chara K, Hyzy F, et al. Selective slow wave sleep but not rapid eye movement sleep suppression impairs morning glucose tolerance in healthy men. Psychoneuroendocrinology. 2013;38:2075–82. doi: 10.1016/j.psyneuen.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 11.Tasali E, Leproult R, Ehrmann DA, Van CE. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105:1044–9. doi: 10.1073/pnas.0706446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Sleep Foundation. Washington, D.C.: 2006. Sleep in America Poll. https://sleepfoundation.org/sites/default/files/2006_summary_of_findings.pdf. [Google Scholar]
- 13.Amiel SA, Sherwin RS, Simonson DC, Lauritano AA, Tamborlane WV. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med. 1986;315:215–9. doi: 10.1056/NEJM198607243150402. [DOI] [PubMed] [Google Scholar]
- 14.Campbell IG, Feinberg I. Longitudinal trajectories of non-rapid eye movement delta and theta EEG as indicators of adolescent brain maturation. Proc Natl Acad Sci U S A. 2009;106:5177–80. doi: 10.1073/pnas.0812947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarokh L, Carskadon MA. Developmental changes in the human sleep EEG during early adolescence. Sleep. 2010;33:801–9. doi: 10.1093/sleep/33.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Beydoun MA, Wang Y. Is sleep duration associated with childhood obesity? A systematic review and meta-analysis. Obesity(Silver Spring) 2008;16:265–74. doi: 10.1038/oby.2007.63. [DOI] [PubMed] [Google Scholar]
- 17.Berentzen NE, Smit HA, Bekkers MB, et al. Time in bed, sleep quality and associations with cardiometabolic markers in children: the Prevention and Incidence of Asthma and Mite Allergy birth cohort study. J Sleep Res. 2014;23:3–12. doi: 10.1111/jsr.12087. [DOI] [PubMed] [Google Scholar]
- 18.Javaheri S, Storfer-Isser A, Rosen CL, Redline S. Association of short and long sleep durations with insulin sensitivity in adolescents. J Pediatr. 2011;158:617–23. doi: 10.1016/j.jpeds.2010.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel MC, Shaikh WA, Singh SK. Association of sleep duration with blood glucose level of Gujarati Indian adolescents. Indian J Physiol Pharmacol. 2012;56:229–33. [PubMed] [Google Scholar]
- 20.Sung V, Beebe DW, Vandyke R, et al. Does sleep duration predict metabolic risk in obese adolescents attending tertiary services? A cross-sectional study. Sleep. 2011;34:891–8. doi: 10.5665/SLEEP.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews KA, Dahl RE, Owens JF, Lee L, Hall M. Sleep duration and insulin resistance in healthy black and white adolescents. Sleep. 2012;35:1353–8. doi: 10.5665/sleep.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koren D, Levitt Katz LE, Brar PC, Gallagher PR, Berkowitz RI, Brooks LJ. Sleep architecture and glucose and insulin homeostasis in obese adolescents. Diabetes Care. 2011;34:2442–7. doi: 10.2337/dc11-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Li AM, Au CT, et al. Association between sleep architecture and glucose tolerance in children and adolescents. J Diabetes. 2015;7:10–5. doi: 10.1111/1753-0407.12138. [DOI] [PubMed] [Google Scholar]
- 24.Armitage R, Lee J, Bertram H, Hoffmann R. A preliminary study of slow-wave EEG activity and insulin sensitivity in adolescents. Sleep Med. 2013;14:257–60. doi: 10.1016/j.sleep.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klingenberg L, Chaput JP, Holmback U, et al. Acute sleep restriction reduces insulin sensitivity in adolescent boys. Sleep. 2013;36:1085–90. doi: 10.5665/sleep.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chervin RD, Hedger K, Dillon JE, Pituch KJ. Pediatric sleep questionnaire (PSQ): validity and reliability of scales for sleep-disordered breathing, snoring, sleepiness, and behavioral problems. Sleep Med. 2000;1:21–32. doi: 10.1016/s1389-9457(99)00009-x. [DOI] [PubMed] [Google Scholar]
- 27.Shaw ND, Butler JP, Nemati S, et al. Accumulated deep sleep is a powerful predictor of LH pulse onset in pubertal children. J Clin Endocrinol Metab. 2015;100:1062–70. doi: 10.1210/jc.2014-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacha F, Gungor N, Arslanian SA. Measures of beta-cell function during the oral glucose tolerance test, liquid mixed-meal test, and hyperglycemic clamp test. J Pediatr. 2008;152:618–21. doi: 10.1016/j.jpeds.2007.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalla MC, Campioni M, Polonsky KS, et al. Two-hour seven-sample oral glucose tolerance test and meal protocol: minimal model assessment of beta-cell responsivity and insulin sensitivity in nondiabetic individuals. Diabetes. 2005;54:3265–73. doi: 10.2337/diabetes.54.11.3265. [DOI] [PubMed] [Google Scholar]
- 30.Iber C, Ancoli-Israel S, Chesson A, Quan SF. 1st ed. Westchester, IL: American Academy of Sleep Medicine; 2007. The AASM manual for the scoring of sleep and associated events: rules, terminology, and technical specifications. [Google Scholar]
- 31.Mankowski PW, Phillips AJ, Klerman EB. New methods for defining NREM/REM sleep cycles in human sleep episodes. Sleep. 2015;38:A137. (Abstract Suppl) [Google Scholar]
- 32.Jenni OG, Carskadon MA. Spectral analysis of the sleep electroencephalogram during adolescence. Sleep. 2004;27:774–83. [PubMed] [Google Scholar]
- 33.Tarvainen MP, Niskanen JP, Lipponen JA, Ranta-Aho PO, Karjalainen PA. Kubios HRV--heart rate variability analysis software. Comput Methods Programs Biomed. 2014;113:210–20. doi: 10.1016/j.cmpb.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 34.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 35.Cobelli C, Dalla MC, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes. 2014;63:1203–13. doi: 10.2337/db13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalla MC, Caumo A, Basu R, Rizza R, Toffolo G, Cobelli C. Minimal model estimation of glucose absorption and insulin sensitivity from oral test: validation with a tracer method. Am J Physiol Endocrinol Metab. 2004;287:E637–43. doi: 10.1152/ajpendo.00319.2003. [DOI] [PubMed] [Google Scholar]
- 37.Sunehag AL, Man CD, Toffolo G, Haymond MW, Bier DM, Cobelli C. beta-Cell function and insulin sensitivity in adolescents from an OGTT. Obesity. 2009;17:233–9. doi: 10.1038/oby.2008.496. [DOI] [PubMed] [Google Scholar]
- 38.Katz ES, D'Ambrosio CM. Pediatric obstructive sleep apnea syndrome. Clin Chest Med. 2010;31:221–34. doi: 10.1016/j.ccm.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Leproult R, Van CE. Role of sleep and sleep loss in hormonal release and metabolism. Endocr Dev. 2010;17:11–21. doi: 10.1159/000262524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basu R, Dalla Man C, Campioni M, et al. Effects of age and sex on postprandial glucose metabolism: differences in glucose turnover, insulin secretion, insulin action, and hepatic insulin extraction. Diabetes. 2006;55:2001–14. doi: 10.2337/db05-1692. [DOI] [PubMed] [Google Scholar]
- 41.Caprio S, Plewe G, Diamond MP, et al. Increased insulin secretion in puberty: a compensatory response to reductions in insulin sensitivity. J Pediatr. 1989;114:963–7. doi: 10.1016/s0022-3476(89)80438-x. [DOI] [PubMed] [Google Scholar]
- 42.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab. 2009;296:E11–21. doi: 10.1152/ajpendo.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med. 1993;328:303–7. doi: 10.1056/NEJM199302043280502. [DOI] [PubMed] [Google Scholar]
- 44.Stamatakis KA, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest. 2010;137:95–101. doi: 10.1378/chest.09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sayk F, Teckentrup C, Becker C, et al. Effects of selective slow-wave sleep deprivation on nocturnal blood pressure dipping and daytime blood pressure regulation. Am J Physiol Regul Integr Comp Physiol. 2010;298:R191–7. doi: 10.1152/ajpregu.00368.2009. [DOI] [PubMed] [Google Scholar]
- 46.Jenni OG, Achermann P, Carskadon MA. Homeostatic sleep regulation in adolescents. Sleep. 2005;28:1446–54. doi: 10.1093/sleep/28.11.1446. [DOI] [PubMed] [Google Scholar]
- 47.Martyn JA, Kaneki M, Yasuhara S. Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology. 2008;109:137–48. doi: 10.1097/ALN.0b013e3181799d45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parton LE, Ye CP, Coppari R, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–32. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 49.Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–90. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–9. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- 51.Broussard JL, Wroblewski K, Kilkus JM, Tasali E. Two nights of recovery sleep reverses the effects of short-term sleep restriction on diabetes risk. Diabetes Care. 2016;39:e40–1. doi: 10.2337/dc15-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]