

Abstract
The in utero environment has the potential to influence epigenetic programming and subsequently the health of offspring. Even though pregnant women living in urban Africa are exposed to multiple chemicals and infectious agents that may impact their developing children, the neonatal epigenome has not been studied in these regions. We assessed whether prenatal exposures to air pollution and maternal human immunodeficiency virus (HIV) are associated with changes to DNA methylation throughout the epigenome using a pilot sample from the Maternal and Child Environmental (MACE) birth cohort, of which 36% of the mothers are HIV positive. Families living in a high air pollution region (south Durban, n=11) and a low air pollution region (north Durban, n=11) with comparable socioeconomic characteristics were selected for analysis. DNA methylation was quantified in cord blood plasma DNA at >430,000 CpG sites using the Infinium HumanMethylation450 BeadChip. Sites associated with living in south Durban or maternal HIV infection (p<0.001) were more likely to be hypomethylated and located in CpG islands. Top differentially methylated sites by region of Durban were enriched in pathways related to xenobiotic metabolism, oxygen and gas transport, and sensory perception of chemical stimuli when performing gene set enrichment testing with LRpath. Differentially methylated sites by maternal HIV status were enriched in cytochrome P450s, pathways involved in detection of chemical stimuli, metabolic processes, and viral regulation and processing. Given the small sample size of the study, future work examining the impact of prenatal exposures to air pollution, maternal infection, and antiviral treatment on the epigenome and downstream health implications is merited in Sub-Saharan African populations.
Keywords: epigenome, air pollution, birth cohort, maternal HIV
INTRODUCTION
In utero exposures lead to epigenetic perturbations that influence early development and in some cases persist throughout the life course, contributing to complex diseases. For example, prenatal and early postnatal exposures to airborne pollutants, cigarette smoke, and allergens are associated with changes to the offspring’s epigenome and subsequent risk for childhood asthma (see reviews (1,2)). In a Belgian birth cohort study, global hypomethylation of placenta DNA and altered mitochondrial DNA methylation were associated with maternal particulate matter (PM2.5) exposure, and the latter mediated the relationship between PM2.5 and decreased total mitochondrial DNA (3,4). In an American mother-child cohort, prenatal polycyclic aromatic hydrocarbon (PAH) exposure was associated with hypermethylation of acyl-CoA synthetase long-chain family member 3 (ACSL3), and this hypermethylation was associated with parental report of asthma by the time children were five years of age (5). As the epigenome is most vulnerable to external exposures during early development, fetuses are vulnerable to epigenetic changes which may influence later-in-life adverse health outcomes (6).
Even though Sub-Saharan African families in urban areas often face multiple stressors (e.g., violence, poverty), infectious agents (e.g., HIV), chemical exposures (e.g. air pollution, e-waste) and malnutrition, there are few epigenetic studies focused on these populations (7). To date, epigenetic epidemiological studies of mothers and children in Sub-Saharan Africa have identified differentially methylated genes in rural Gambian babies and children associated with periconceptual nutrition status (8–11) or maternal aflatoxin exposure during pregnancy (12). In South African, children with fetal alcohol syndrome had altered methylation at imprinted genes compared with healthy children (13). In the Democratic Republic of Congo, maternal stress from ongoing war was associated with differential methylation – including at key genes involved in stress response – among newborns and their mothers (14–16). While these studies are providing needed insight into epigenetically labile regions of the genome and key environmental factors in African populations, they only stem from three major study populations in rural Gambia, South Africa, and the eastern Democratic Republic of Congo. To our knowledge no studies have assessed the impact of in utero air pollution exposure or maternal HIV infection on newborn or children’s epigenomes in Sub-Saharan Africa.
In the present pilot study, the recruitment area includes south Durban in KwaZulu-Natal, South Africa, which has been regarded as one of the most industrialized and polluted regions in Southern Africa (17). A previous study documented a higher prevalence of respiratory outcomes such as asthma and airway hyper-responsiveness among children in this area compared to north Durban, a less industrialized region (18). South Durban is characterized by higher oxides of nitrogen (NOx), sulfur dioxide, and volatile organic compounds among other pollutants compared to north Durban, though both regions have similar ambient PM10 and PM2.5 which hover near WHO guidelines for maximum annual exposure (17). In addition, KwaZulu-Natal has the highest HIV prevalence in South Africa (16.9% in 2012) with the highest risk of exposure among black African females aged between 20 and 34 years (19). All HIV positive pregnant women receiving antenatal care, regardless of their CD4 count, are given a fixed dose combination of antiretroviral therapy from 14 weeks of pregnancy that continues throughout the breastfeeding period (20). Pregnant women in this area thus encounter multiple environmental insults that may impact the growing fetus (e.g., air pollution, maternal HIV infection, and antiviral medication), yet the impact on the epigenome remains to be characterized.
The aim of this pilot study is to assess changes to the neonatal DNA methylome following in utero exposures to airborne pollutants and maternal HIV (infection and treatment). We hypothesize that both factors will alter DNA methylation levels throughout the epigenome quantified with the Infinium HumanMethylation450 BeadChip. We address this hypothesis in a pilot sample from the Maternal and Child Environmental (MACE) birth cohort, comparing 11 newborns from the high ambient pollution south Durban to 11 newborns from the lower pollution north Durban region.
MATERIALS AND METHODS
MACE Cohort
For the pilot stage of the MACE birth cohort study, 100 women were recruited during the third trimester of pregnancy from four public antenatal clinics in close proximity to Air Quality Monitoring Stations in eThekwini Municipality, which is located in KwaZulu-Natal province of South Africa. Half of the participants resided in a heavily industrialized and polluted region (south Durban) and half resided in a lesser polluted area (north Durban) with similar socioeconomic characteristics (21). Women with pregnancy complications (e.g., hypertension, diabetes), multiples, or late first antenatal visits (>34 weeks) were excluded. Further information on recruitment was previously detailed (21,22). Mothers provided informed consent before participating including for genetic analyses, and ethical approval was obtained from the Biomedical and Research Ethics Committee of the University of KwaZulu-Natal.
A subset of mother-child pairs was selected for epigenome-wide analysis (12 each from north and south Durban with the best DNA quality) from among 30 providing cord blood samples. Cord blood samples were limited among the 100 participants due to logistical and compliance limitations at the hospitals where the mothers gave birth.
Demographic and Exposure Data
Demographic (e.g., maternal education, race, age, marital status) and exposure information (e.g., smoking history, exposure to environmental tobacco smoke, occupational chemical exposures) was collected via questionnaire. HIV status of the mothers was assessed at the antenatal clinics. Gestational age, birth weight, and birth outcomes were obtained from patient records. Hospital staff estimated gestational age based on first day of the last menstrual period, and birth weight was measured in the delivery room by trained nurses. Mothers did not receive folate supplementation during pregnancy except for one taking prenatal vitamins. Air pollution exposure is approximated by both region of residency and NOx concentrations. Individual exposures to NOx were estimated based on land use regression modeling (23,24) for all subjects except one (missing necessary data).
DNA Isolation and Treatment
DNA was isolated from umbilical cord blood plasma, as whole blood from the participants was not archived, using the QIAamp DNA Kit (Qiagen), stored at −80°C at the University of KwaZulu-Natal, and shipped overnight on dry ice to the U.S. Double-stranded DNA concentration was determined via the Qubit fluorimetric system. The EZ DNA Methylation kit (Zymo Research Corporation, Irvine, CA) was used for bisulfite conversion of 500 ng of genomic DNA which converts unmethylated cytosines and leaves methylated cytosines unchanged (25). The protocol recommended for downstream Illumina Infinium analysis was followed.
Epigenome-Wide DNA Methylation Analysis
DNA methylation at >485,000 CpG sites throughout the genome was interrogated with the Illumina Infinium HumanMethylation450 BeadChip (‘450K’) (26). Amplification and hybridization of bisulfite converted DNA samples to the beadchips were performed according to manufacturer’s protocol. Twenty-four samples were run across three batches (beadchips). Image data was scanned and recorded via the Illumina iScan with the Methylation NXT scan setting. Raw data from image files was read into R version 3.1 using both ChAMP and methylumi packages (27,28). Raw data was then processed to remove samples and probes with poor quality control, and to adjust for batch effects and biases inherent in the 450K platform. Probes with low detection (p>0.01 or bead count <3 in at least 2 samples) were removed. One subject from the north Durban group that failed to pass quality controls was also excluded. Additional probes were excluded if they: 1) hybridized to the X and Y chromosomes, 2) were known to be cross-reactive (29), or 3) had a SNP within the query site or single base extension position with a minor allele frequency >4% in Africans (or worldwide if African data was not available) leaving a final dataset of 433,782 CpG sites. Dye bias adjustment and background correction were performed using the methylumi package (27). In the ChAMP package (30), beta-mixture quantile normalization was performed to correct for probe type differences (31). The ComBat function was used to correct for batch effects by beadchip (32). Probe locations were annotated with the IlluminaHumanMethylation450kanno.ilmn12.h19 package (33).
Statistical Analysis
All statistics were performed with the R Project for Statistical Computing version 3.1. Univariate statistics for demographic and exposure variables were computed for all subjects (excluding a racial outlier, Supplemental Figure S1) with high quality 450K data (n=22) and stratified by air pollution or maternal HIV exposure group. Fisher exact tests were used to compare demographic and exposure characteristics between groups (north vs. south Durban and maternal HIV positive vs. negative status). All statistical analyses of methylation data utilized ‘M-Values’ which are logit transformed versions of the pre-processed and adjusted beta values from 433,782 CpG sites. Unsupervised clustering by all sites and by the 1000 most variable sites via a multi-dimensional scaling (MDS) algorithm were performed and examined by key categorical variables (e.g., sex, race, region, maternal HIV status, parental smoking). One outlier subject was identified that differed from the rest (the only white subject, Supplemental Figure S1), and all downstream analyses were run without this subject (n=22 in reported analyses).
Multivariable analysis was performed to find differentially methylated sites (DMS) by air pollution exposure or maternal HIV status. The limma package was used to compare methylation at all sites by air pollution exposure group (north Durban, n=11 vs. south Durban, n=11) or maternal HIV status (14 negative vs. 8 positive) while adjusting for gestational age and sex (34). The following covariates were not considered because they were too unbalanced for reliable statistical modeling (e.g., one family in a category): maternal vitamin or supplement use, marital status, mother currently smoking, or mother exposed to smoke at work. The following covariates were tested but not included in the final model as they were associated with methylation at <5 CpG sites at an FDR of 10%: maternal age, previous pregnancies, pre-pregnancy weight, smoke exposure at home, paternal smoking status, maternal smoking in the past, apgar score, delivery type, and use of labor medication(s). Adjusted models with air pollution exposure and HIV status modeled separately or simultaneously were used to identify DMS. Estimates of NOx were also modeled as continuous in subjects with the data (n=21).
Along with identifying DMS by air pollution exposure or maternal HIV status, we examined trends in the data (e.g., hypo- versus hyper-methylation, enrichment in regulatory regions or CpG islands) for CpG sites with p<0.001 or log-fold change>1 or <−1 in exposure and HIV models. Chi-square tests with p-values estimated from 2000 Monte-Carlo simulations were performed to assess enrichment/depletion in annotated regions compared to expected proportion based on all probes.
We performed gene set enrichment testing using the publically-available LRpath program (35). This program uses logistic regression and pre-defined sets of genes (“concepts”) that are related by molecular or biological function or pathway to test the hypothesis that certain concepts have higher significance values (based on raw p-values) for differential methylation compared with other concepts. We utilized log-fold change and p-value data from HIV or air pollution exposure models for 328,040 probes that mapped to known genes (35). Enriched concepts with 10–1000 genes per concept were screened from the databases of KEGG, GO Biological Process, and GO Molecular Function concepts. Overall, this logistic-regression based approach relates the odds of belonging to a given concept with the raw p-value for differential methylation while taking into account the size (e.g., number of genes) of each concept.
RESULTS
Study Population Characteristics
Twenty-two mother-child pairs, 11 from north Durban and 11 from south Durban, were included in the epigenome-wide 450K analysis. Of these, 68% of the newborns were female, 21 families identified as African and 1 as Coloured. 95% of mothers were unmarried, and 86% had never smoked. Table I summarizes these and other characteristics of the study subjects and stratifies results by residency region or maternal HIV status. Demographic variables did not vary significantly by air pollution or maternal HIV exposure groups. NOx levels were higher in south Durban compared with the north (36.1±6.0 vs. 17.8±5.4 ppb, p<0.001 for T test).
Table I.
Study Population Characteristics
| Subjects with 450K Data | Stratified by Air Pollution Exposure Region | Stratified by Maternal HIV Status | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| North (Low) | South (High) | Negative | Positive | |||||||||||||
| N | % | Mean (SD) | N | % | Mean (SD) | N | % | Mean (SD) | N | % | Mean (SD) | N | % | Mean (SD) | ||
| Child | Males | 7 | 32 | 2 | 18 | 5 | 45 | 5 | 36 | 2 | 25 | |||||
| Females | 15 | 68 | 9 | 82 | 6 | 55 | 9 | 64 | 6 | 75 | ||||||
| Race | ||||||||||||||||
| African | 21 | 95 | 11 | 100 | 10 | 91 | 13 | 93 | 8 | 100 | ||||||
| Coloured | 1 | 5 | 0 | 0 | 1 | 9 | 1 | 7 | 0 | 0 | ||||||
| Gestational Age (weeks) | 38.8 (1.4) | 39.2 (1.7) | 38.5 (0.9) | 38.8 (1.5) | 38.9 (1.0) | |||||||||||
| Birth Weight (g) | 3174 (430) | 3140 (410) | 3207 (467) | 3257 (501) | 3058 (193) | |||||||||||
| Maternal | Age (years) | 26.1 (6.6) | 27.0 (6.6) | 25.3 (6.9) | 24.3 (5.6) | 28.6 (7.9) | ||||||||||
| Marital Status | ||||||||||||||||
| Married | 1 | 5 | 0 | 0 | 1 | 9 | 1 | 7 | 0 | 0 | ||||||
| Unmarried | 21 | 95 | 11 | 100 | 10 | 91 | 13 | 93 | 8 | 100 | ||||||
| Previous Pregnancy | ||||||||||||||||
| Yes | 10 | 45 | 4 | 36 | 6 | 55 | 6 | 43 | 4 | 50 | ||||||
| No | 12 | 55 | 7 | 64 | 5 | 45 | 8 | 57 | 4 | 50 | ||||||
| Ever Smoke | ||||||||||||||||
| Yes | 3 | 14 | 2 | 18 | 1 | 9 | 3 | 21 | 0 | 0 | ||||||
| No | 19 | 86 | 9 | 82 | 10 | 91 | 11 | 79 | 8 | 100 | ||||||
| Smoking in the Home | ||||||||||||||||
| Yes | 6 | 27 | 3 | 27 | 3 | 27 | 4 | 29 | 2 | 25 | ||||||
| No | 16 | 73 | 8 | 73 | 8 | 73 | 10 | 71 | 6 | 75 | ||||||
| HIV Status | ||||||||||||||||
| Negative | 14 | 64 | 6 | 55 | 8 | 73 | 14 | 100 | 0 | 0 | ||||||
| Positive | 8 | 36 | 5 | 45 | 3 | 27 | 0 | 0 | 8 | 100 | ||||||
| Air Pollution Exposure | Region | |||||||||||||||
| North Durban (low) | 11 | 50 | 11 | 100 | 0 | 0 | 6 | 43 | 5 | 63 | ||||||
| South Durban (high) | 11 | 50 | 0 | 0 | 11 | 100 | 8 | 57 | 3 | 38 | ||||||
| NOx (ppb) | 26.5 (10.9) | 17.8 (5.4) | 36.1 (6.0) | 26.5 (11.4) | 27.4 (9.7) | |||||||||||
HumanMethylation450 BeadChip (450K)
Models adjusting for gestational age and gender were run to search for DMS among 433,782 CpG sites by 1) air pollution exposure group, 2) NOx levels (n=21 in these models), or 3) maternal HIV status. Results (top DMS) were compared in models assessing any exposure alone or models with HIV and air pollution exposure together. No DMS were identified that were significant at an FDR level of 10%. Given the small sample size, nineteen top DMS by air pollution exposure group and/or NOx levels selected by raw p-value are detailed in Supplemental Table I, including sites in intergenic regions and sites in known genes (e.g., NOVA2, SELK, CNTD2). Twelve of the top DMS by maternal HIV status (p<0.0001 in multiple models) are displayed in Supplemental Table II, including intergenic CpG sites and sites in genes (e.g., POLDIP3, RHOBTB3). No DMS by HIV status remained significant at a FDR of 10%.
No sites were significant following FDR correction. We examined trends in methylation across all sites or groups of sites by air pollution exposure or maternal HIV status (Figures 1 and 2). When comparing methylation of samples from south and north Durban, CpG sites were more likely to be hypomethylated (62.0% of all sites, 77.1% of sites with nominal p<0.001, and 57.4% of sites with log-fold change greater than the absolute value of one [herein referred to as effect size >1]). Likewise, children with HIV positive mothers had more hypomethylated sites (69.2% of all sites, 86.4% of CpG sites with p<0.001, and 65.8% of sites with effect size >1; see Figure 1). Enrichment of sites with p<0.001 or effect size >1 in specific chromosomes, annotation relative to CpG islands, in DNAse I hypersensitive sites, in enhancer regions, in well-characterized differentially methylated regions (DMRs; such as tissue-specific or imprinted gene-specific DMRs), or in specific regulatory features (e.g., promoter or gene associated) was also assessed (data not shown). Among sites with p<0.001 for air pollution exposure group, there was a significant enrichment of sites in CpG islands (46.2% compared to 31.7% of total probes; Figure 2), in DNAse I hypersensitive sites (16.4 vs. 12.6% of all probes), and in DMRs (11.4% vs. 8% of total probes). When looking at top sites by effect sizes, only the significant DMR enrichment remained. Similar analyses by maternal HIV status revealed enrichment of sites with p<0.001 in CpG islands (52.0% vs. 31.7% expected; Figure 2), DNAse I hypersensitive sites (19.0% vs. 12.6% expected), DMRs (16.3% vs. 8% expected), and a paucity of probes in enhancer regions (14.8% vs. 22.4% expected). When comparing probes with effect sizes >1, only the enhancer depletion remained significant (χ2 test, p=0.02).
Figure 1. Enrichment of Hypomethylated Sites among High Air Pollution and Maternal HIV Exposure Groups.
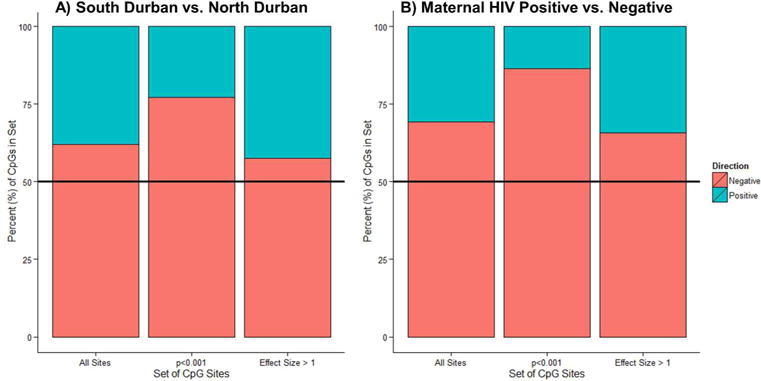
A statistically significant enrichment of hypomethylated sites was observed when comparing cord blood plasma DNA methylation from individuals A) residing in a high air pollution region (south Durban) compared with the low pollution north Durban and B) with maternal HIV exposure compared with uninfected mothers. Relationships were assessed in models adjusting for gestational age, child’s sex, air pollution group, and maternal HIV status (n=22). Percentage of hypomethylated (direction=negative) and hypermethylated (positive) sites are displayed from the following groups of CpG sites: 1) all sites (n=433,782), 2) p<0.001 (n=420 for air pollution, 689 for HIV), or effect size >1 (log fold-change <−1 or >1; n=781 for air pollution analysis, 514 for HIV). A reference line is drawn at 50%.
Figure 2. Enrichment of Differentially Methylated Sites (DMS) in CpG Islands among High Air Pollution and Maternal HIV Exposure Groups.
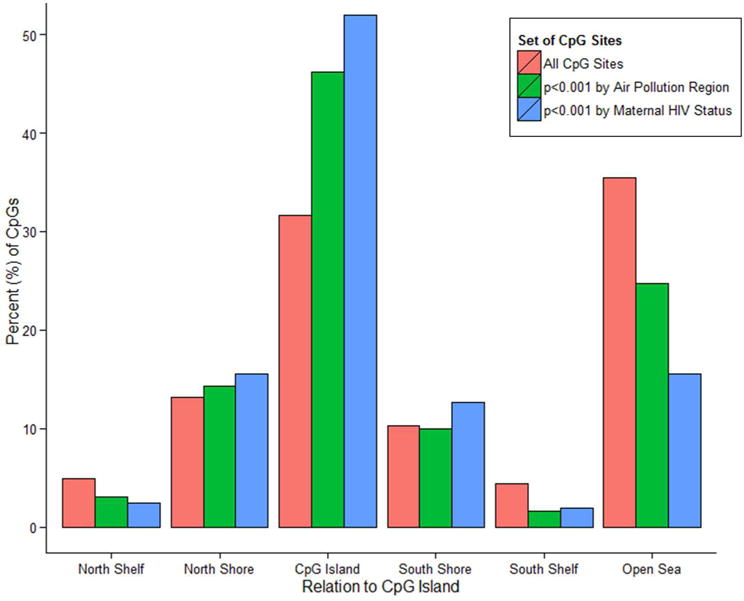
Among the top DMS (p<0.001) by air pollution exposure region (420 CpG sites) or maternal HIV status (689 sites) in models adjusting for both exposures, gestational age, and sex, there was a statistically significant shift in annotation related to CpG islands compared to the expected proportions based on all probes (‘All CpG Sites’). Specifically, DMS were over-represented in CpG islands and depleted in the ‘open sea’ for both in utero exposures.
Pathway Analysis
The program LRpath was used to search databases of gene function and gene pathways (KEGG and GO) for concepts enriched amongst the most DMS (35). Thirty-four concepts or pathways were enriched by air pollution exposure group with (FDR <0.1 see Supplemental Table III for full details; 22 pathways had FDR <0.05, Table II). Enriched functions and pathways include those related to xenobiotic metabolism (e.g., cytochrome P450), oxygen and gas transport, and olfactory and sensory perception of chemical stimuli. Pathways enriched amongst DMS by maternal HIV status include 67 with FDR <10% (Supplemental Table IV) and 26 <5% (Table III). Differential methylation in pathways involved in sensory perception and detection of chemical stimuli, cytochrome P450, and various metabolic processes was enriched. Eleven gene groups related to viral regulation and processing and the innate immune response to viral infections were enriched for higher methylation among individuals whose mothers had HIV with FDR 10%.
Table II. Top Gene Ontology or Function Pathways Enriched in Air Pollution Exposure Analysis.
Concepts significant at a 5% FDR level using LRpath gene set enrichment testing are displayed.
| Concept Name | Concept Database | # Genes in Concept | FDR q-value | Direction† |
|---|---|---|---|---|
| Ascorbate and aldarate metabolism | KEGG | 19 | 0.003 | ↑ |
| oxygen transport | GOBP | 11 | 0.005 | ↓ |
| Pentose and glucuronate interconversions | KEGG | 24 | 0.007 | ↓ |
| Drug metabolism - cytochrome P450 | KEGG | 62 | 0.013 | ↓ |
| Drug metabolism - other enzymes | KEGG | 39 | 0.017 | ↓ |
| oxygen binding | GOMF | 32 | 0.019 | ↓ |
| Retinol metabolism | KEGG | 54 | 0.024 | ↓ |
| Other types of O-glycan biosynthesis | KEGG | 37 | 0.024 | ↓ |
| gas transport | GOBP | 15 | 0.025 | ↓ |
| detection of chemical stimulus involved in sensory perception | GOBP | 357 | 0.025 | ↑ |
| RNA splicing, via endonucleolytic cleavage and ligation | GOBP | 10 | 0.029 | ↓ |
| detection of chemical stimulus | GOBP | 388 | 0.029 | ↑ |
| cellular glucuronidation | GOBP | 13 | 0.029 | ↓ |
| uronic acid metabolic process | GOBP | 14 | 0.029 | ↓ |
| glucuronate metabolic process | GOBP | 14 | 0.029 | ↓ |
| Metabolism of xenobiotics by cytochrome P450 | KEGG | 63 | 0.030 | ↓ |
| sensory perception of chemical stimulus | GOBP | 403 | 0.031 | ↑ |
| glucuronosyltransferase activity | GOMF | 22 | 0.031 | ↓ |
| olfactory receptor activity | GOMF | 320 | 0.031 | ↑ |
| detection of stimulus involved in sensory perception | GOBP | 396 | 0.032 | ↑ |
| negative regulation of intrinsic apoptotic signaling pathway in response to DNA damage | GOBP | 23 | 0.033 | ↑ |
| detection of chemical stimulus involved in sensory perception of smell | GOBP | 320 | 0.047 | ↑ |
Up direction signifies that most genes in the concept were hypermethylated in the south Durban group compared with the north Durban group. Vice versa for the down direction.
Table III. Top Gene Ontology or Function Pathways Enriched in Maternal HIV Status Analysis.
Concepts significant at a 5% FDR level using LRpath gene set enrichment testing are displayed.
| Concept Name | Concept Database | # Genes in Concept | FDR q-value | Direction† |
|---|---|---|---|---|
| olfactory receptor activity | GOMF | 320 | 3.83E-36 | ↓ |
| detection of chemical stimulus involved in sensory perception | GOBP | 357 | 7.59E-36 | ↓ |
| detection of chemical stimulus involved in sensory perception of smell | GOBP | 320 | 1.91E-35 | ↓ |
| sensory perception of chemical stimulus | GOBP | 403 | 1.91E-35 | ↓ |
| detection of stimulus involved in sensory perception | GOBP | 396 | 3.19E-35 | ↓ |
| sensory perception of smell | GOBP | 344 | 8.15E-35 | ↓ |
| detection of chemical stimulus | GOBP | 388 | 1.62E-34 | ↓ |
| Olfactory transduction | KEGG | 337 | 1.33E-30 | ↓ |
| detection of stimulus | GOBP | 553 | 1.06E-29 | ↓ |
| sensory perception | GOBP | 758 | 5.31E-24 | ↓ |
| G-protein coupled receptor activity | GOMF | 705 | 9.80E-22 | ↓ |
| Ascorbate and aldarate metabolism | KEGG | 19 | 3.32E-06 | ↓ |
| Retinol metabolism | KEGG | 54 | 7.46E-06 | ↓ |
| Pentose and glucuronate interconversions | KEGG | 24 | 7.46E-06 | ↓ |
| Metabolism of xenobiotics by cytochrome P450 | KEGG | 63 | 1.81E-05 | ↓ |
| glucuronosyltransferase activity | GOMF | 22 | 1.83E-05 | ↓ |
| Starch and sucrose metabolism | KEGG | 38 | 3.49E-05 | ↓ |
| Drug metabolism - cytochrome P450 | KEGG | 62 | 3.49E-05 | ↓ |
| Steroid hormone biosynthesis | KEGG | 47 | 3.49E-05 | ↓ |
| Porphyrin and chlorophyll metabolism | KEGG | 33 | 7.42E-05 | ↓ |
| Other types of O-glycan biosynthesis | KEGG | 37 | 1.86E-04 | ↓ |
| Drug metabolism - other enzymes | KEGG | 39 | 0.001 | ↓ |
| cellular glucuronidation | GOBP | 13 | 0.002 | ↓ |
| uronic acid metabolic process | GOBP | 14 | 0.002 | ↓ |
| glucuronate metabolic process | GOBP | 14 | 0.002 | ↓ |
| Taste transduction | KEGG | 48 | 0.045 | ↓ |
Up direction signifies that most genes in the concept were hypermethylated in the maternal HIV positive group compared with the negative group. Vice versa for the down direction.
DISCUSSION
In a pilot study using the South African birth cohort, MACE, we observed DNA methylation changes associated with prenatal exposures to airborne pollutants and maternal HIV infection using an epigenome-wide platform, the Infinium HumanMethylation450 BeadChip. To our knowledge, this is one of the first studies in an urban Sub-Saharan African cohort to assess the neonatal epigenome, a much needed area of research given the range of environmental factors encountered by African populations including infectious diseases, chemical exposures, and malnutrition. The increasing prevalence of non-communicable diseases in African populations, which may partially be explained by environmentally-induced epigenetic perturbations early in life as posited by the Developmental Origins of Health and Disease (DOHaD) hypothesis, further necessitates research in this area (7,36).
No significant DMS were observed with an FDR level of 10%. As this was a pilot study with a small sample size, it is possible but not guaranteed that increasing the sample size would reveal significant DMS at a 10% FDR level. Even so within this pilot dataset, intriguing trends in the data by raw p-value, direction and annotation relative to CpG islands among top DMS, and enriched pathways among the sites using LRpath were observed for both group comparisons (air pollution and maternal HIV status). When comparing direction of methylation difference between the high air pollution exposure group or the maternal HIV positive group with their respective reference groups, hypomethylation was more commonly observed (Figure 1). Hypomethylation is typically linked to increased gene expression, especially within CpG islands and promoter regions (37). Interestingly, the top DMS by air pollution or maternal HIV exposure group were more likely to be found within CpG islands and other regulatory regions (DNAse I hypersensitive sites (38) and well-characterized DMRs). Hypomethylation is also problematic in intergenic regions and leads to genomic instability (39–41). Air pollution and antiviral treatments may modify the DNA methylome via oxidative stress which reduces the efficiency of the one-carbon metabolism pathway, thereby reducing the amount of available methyl donors necessary to establish and maintain DNA methylation (42,43). Given that wide-spread modification of the neonatal DNA methylome in regulatory regions could impact gene expression throughout development and potentially beyond and genome-wide hypomethylation can lead to genomic instability, the combined impact of maternal air pollution and HIV exposures on the epigenome and downstream health effects merits further study in a larger cohort.
In terms of specific CpG sites associated with air pollution exposure, none survived correction for multiple testing. Top DMS by raw p-value (Supplemental Table I) were found in genes with potential relevance to early development and response to exposure including the antioxidant selenoprotein SELK, GGT5 which is involved in leukotriene synthesis, and the osteogenesis-regulator PBX1. Pathway analysis via LRpath revealed significantly enriched gene concept groups after correction for multiple hypothesis testing that may confer continued protection against environmental agents (e.g., air pollution) encountered in utero. For example, pathways involved in xenobiotic metabolism and detection of chemical stimuli were enriched amongst samples from the high air pollution region. Pregnant women and fetuses have altered biotransformation capabilities, which are an increased risk for the developing fetus in terms of epigenetic reprogramming in response to environmental insults (6,44). Our study adds to the growing body of literature that suggests transplacental exposure to components of ambient pollution modifies the epigenome and may contribute to risk for adverse outcomes in childhood such as asthma (3–5,45). Previous studies have provided evidence for the impact of ambient pollution components (e.g., PAHs, PM2.5, PM10) on DNA methylation at global markers (repetitive elements) in adult blood leukocytes (46,47) and at the FOXP3 gene in children’s regulatory T cells, which was related to immunity and atopic status in the same children (48). Similar to trends observed in the present study, primarily hypomethylated DMS were discovered among both healthy and asthmatic children in a heavily polluted area of the Czech Republic compared with a less polluted region using an epigenome-wide platform examining >27,000 CpG sites (49).
In terms of specific sites associated with maternal HIV status, the top represented genes by nominal p-value included transcription and translation regulators (TULP2, ZNF398, POLDIP3) and a modifier of mitochrondrial DNA topology (TOP1MT), though none were significant with a FDR of 10%. Twenty-seven pathways were enriched according to LRpath analysis, specifically for hypomethylation with a FDR level of 5% (Table III), and enriched concepts primarily involved detection of chemical stimuli, olfactory signaling, xenobiotic metabolism, and other metabolic functions. Increased expression in these pathways may enable offspring to cope with exposures to maternal antiviral therapies. Additional concepts enriched with an FDR of 10% include processes involved in viral processing and regulation, and this may be a result of treatment exposure or a response to viral exposure. The few previous studies examining the impact of HIV and maternal antiviral therapies on the neonatal epigenome suggest that antiviral treatment and not infection itself is the driver of change (50,51). Heterochromatin differences were observed in lymphocytes from children with prenatal nucleoside reverse transcriptase inhibitor (NRTI) exposure compared with children without NRTI exposure regardless of maternal HIV status (51). Likewise, maternal anti-viral therapy was associated with DNA hypomethylation at repetitive elements, a global marker, in newborn peripheral blood mononuclear cells (PBMCs) compared with newborns from untreated HIV-positive mothers (50). Of the eight HIV positive mothers in the MACE cohort, at the time of birth seven took Nevaripine (a non-NRTI), one additionally took azidothymidine (a NRTI), and one was untreated. The impact of common antiviral medication used by African populations with high HIV prevalence on children’s health through perinatal epigenetic perturbation needs further exploration.
Evidence for the functional and phenotypic impact of exposure-induced epigenetic changes is growing. In a longitudinal study, an association between prenatal PAH exposure and ACSL3 hypermethylation in umbilical cord blood leukocytes was linked to asthma development in children by five years of age (5). In a cross-sectional study, blood leukocyte DNA methylation at TNF mediated the association between phthalate metabolite levels and asthma in children (52). Thus, the need to better understand the impact of multiple co-exposures and stressors (e.g., to nutritional deficiencies, maternal antiviral therapies, air pollution, additional environmental chemicals, socioeconomic status-related stress) on the neonatal epigenome and the downstream implications for development of disease later in life (i.e., DOHaD) is imperative. Furthermore, development of epigenetic biomarkers by establishing DMS reflective of certain exposures or outcomes would be useful for research, diagnostic, and potentially therapeutic purposes in regions with limited resources for full-scale epigenome analyses.
While we are unique in assessing epigenetic changes by both prenatal air pollution and maternal HIV exposure in a South African pilot cohort using an epigenome-wide platform, this study had several limitations. Along with small sample size, due to archived resources available for the study, DNA was isolated from cord blood plasma. Thus, our ability to compare findings to studies using blood leukocytes or to extrapolate findings to other tissues of the body is extremely limited. Top DMS by nominal p-value could not be validated for functional relevance (e.g., gene expression analysis) as the samples were not preserved and stored for RNA isolation. Our estimation of air pollution exposure (by region or by NOx levels) did not include specific levels of certain air pollutants that have been shown to impact the epigenome (e.g., PAHs, PM2.5). Additionally, we were not able to tease apart the epigenetic differences due to maternal HIV infection versus antiviral therapy.
CONCLUSION
In conclusion, epigenetic modification by prenatal exposures to common stressors in urban African populations such as air pollution and maternal HIV (infection or treatment) may lead to epigenetic modifications in neonates. Due to limitations of this study, we recommend further examination of these key in utero exposures, their impact on the epigenome, and the continued impact on downstream growth and disease development in future Sub-Saharan African cohorts.
Supplementary Material
ENVIRONMENTAL IMPACT STATEMENT.
South Durban is a heavily industrialized area in South Africa that is known to have high ambient air pollution and greater prevalence of respiratory complications (e.g., childhood asthma) among residents compared with the non-industrial north Durban. Prenatal air pollution exposure has the potential to impact epigenetic programming and influence disease risk in the offspring throughout the lifespan. Thus, we compared DNA methylation at >430,000 sites across the epigenome in newborns from south Durban with those from north Durban. In addition to air pollution exposure, we also examined associations of DNA methylation with maternal HIV infection, as co-exposures to both the virus and antiviral treatment are relevant health concerns for neonates from many African cities including Durban.
Acknowledgments
The authors wish to thank the participants of the MACE cohort and the nursing staff at the participating clinics and hospitals. The authors also acknowledge the University of Michigan DNA Sequencing Core for running the 450K BeadChip. This research was supported by the following funding sources: the Medical Research Council of South Africa (MRC) and the Michigan Lifestage Environmental Exposures and Disease (M-LEEaD) Core Center (Grant No. P30 ES017885). JMG was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (Award No. 2UL1TR000433). The contents of this publication are solely the responsibility of the grantee and do not necessarily represent the official views of the NIH.
Footnotes
No conflict of interest is declared.
References
- 1.de Planell-Saguer M, Lovinsky-Desir S, Miller RL. Epigenetic regulation: the interface between prenatal and early-life exposure and asthma susceptibility. Environ Mol Mutagen. 2014 Apr;55(3):231–243. doi: 10.1002/em.21836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martino D, Prescott S. Epigenetics and prenatal influences on asthma and allergic airways disease. Chest. 2011 Mar;139(3):640–647. doi: 10.1378/chest.10-1800. [DOI] [PubMed] [Google Scholar]
- 3.Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics. 2015 May 21;10(6):536–544. doi: 10.1080/15592294.2015.1048412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssen BG, Godderis L, Pieters N, Poels K, Kici Ski M, Cuypers A, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. 2013 Jun 7;10(1):22. doi: 10.1186/1743-8977-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One. 2009;4(2):e4488. doi: 10.1371/journal.pone.0004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts JS, Dolinoy DC, Tarini BA. Emerging issues in public health genomics. Annu Rev Genomics Hum Genet. 2014;15:461–480. doi: 10.1146/annurev-genom-090413-025514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobbs A, Ramsay M. Epigenetics and the burden of noncommunicable disease: a paucity of research in Africa. Epigenomics. 2015 Jun;7(4):627–639. doi: 10.2217/epi.15.17. [DOI] [PubMed] [Google Scholar]
- 8.Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014 Apr 29;5:3746. doi: 10.1038/ncomms4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silver MJ, Kessler NJ, Hennig BJ, Dominguez-Salas P, Laritsky E, Baker MS, et al. Independent genomewide screens identify the tumor suppressor VTRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Biol. 2015 Jun 11;16:118. doi: 10.1186/s13059-015-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010 Dec 23;6(12):e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khulan B, Cooper WN, Skinner BM, Bauer J, Owens S, Prentice AM, et al. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: a study of a unique resource in the Gambia. Hum Mol Genet. 2012 May 1;21(9):2086–2101. doi: 10.1093/hmg/dds026. [DOI] [PubMed] [Google Scholar]
- 12.Hernandez-Vargas H, Castelino J, Silver MJ, Dominguez-Salas P, Cros MP, Durand G, et al. Exposure to aflatoxin B1 in utero is associated with DNA methylation in white blood cells of infants in The Gambia. Int J Epidemiol. 2015 Aug;44(4):1238–1248. doi: 10.1093/ije/dyv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masemola ML, van der Merwe L, Lombard Z, Viljoen D, Ramsay M. Reduced DNA methylation at the PEG3 DMR and KvDMR1 loci in children exposed to alcohol in utero: a South African Fetal Alcohol Syndrome cohort study. Front Genet. 2015 Mar 10;6:85. doi: 10.3389/fgene.2015.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodney NC, Mulligan CJ. A biocultural study of the effects of maternal stress on mother and newborn health in the Democratic Republic of Congo. Am J Phys Anthropol. 2014 Oct;155(2):200–209. doi: 10.1002/ajpa.22568. [DOI] [PubMed] [Google Scholar]
- 15.Mulligan CJ, D’Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012 Aug;7(8):853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kertes DA, Kamin HS, Hughes DA, Rodney NC, Bhatt S, Mulligan CJ. Prenatal Maternal Stress Predicts Methylation of Genes Regulating the Hypothalamic-Pituitary-Adrenocortical System in Mothers and Newborns in the Democratic Republic of Congo. Child Dev. 2016 Jan-Feb;87(1):61–72. doi: 10.1111/cdev.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naidoo R, Gqaleni N, Batterman S, Robins TG. South Durban Health Study. Final Project Report. 2007 [Google Scholar]
- 18.Naidoo RN, Robins TG, Batterman S, Mentz G, Jack C. Ambient pollution and respiratory outcomes among schoolchildren in Durban, South Africa. SAJCH. 2013 Jul 31;7(4):127–134. doi: 10.7196/sajch.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simbayi LC, Shisana O, Rehle T, Onoya D, Jooste S, Zungu N, et al. South African National HIV Prevalence, Incidence and Behaviour Survey, 2012. Human Sciences Research Council. 2014 doi: 10.2989/16085906.2016.1153491. [DOI] [PubMed] [Google Scholar]
- 20.South African HIV Clinicians Society. Fixed-dose combination for adults accessing antiretroviral therapy. S Afr J HIV Med. 2013;14(1 Suppl):41–43. [Google Scholar]
- 21.Reddy P, Naidoo RN, Chuturgoon A, Asharam K, Naidoo D, Phulukdaree A, et al. Correlation between glutathione S-transferase Mu 1 (GSTM1) and glutathione S-transferase pi gene (GSTP1) polymorphisms and markers of inflammatory stress in pregnant females. J Toxicol Environ Health Sci. 2013;5(3):52–59. [Google Scholar]
- 22.Reddy P, Naidoo RN, Chuturgoon A, Asharam K, Phulukdaree A, Gounden S. Effect of the TNF α-308 polymorphism on birth outcomes among South African women. J Adv Biomed Sci. 2014;1(1):21–26. [Google Scholar]
- 23.Briggs DJ, de Hoogh C, Gulliver J, Wills J, Elliott P, Kingham S, et al. A regression-based method for mapping traffic-related air pollution: application and testing in four contrasting urban environments. Sci Total Environ. 2000 May 15;253(1–3):151–167. doi: 10.1016/s0048-9697(00)00429-0. [DOI] [PubMed] [Google Scholar]
- 24.Ryan PH, LeMasters GK. A review of land-use regression models for characterizing intraurban air pollution exposure. Inhal Toxicol. 2007;19(Suppl 1):127–133. doi: 10.1080/08958370701495998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001 Jul 1;29(13):E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dedeurwaerder S, Defrance M, Bizet M, Calonne E, Bontempi G, Fuks F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform. 2013 Aug 29;15(6):929–941. doi: 10.1093/bib/bbt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis S, Du P, Bilke S, Triche T, Bootwalla M. Methylumi: Handle Illumina Methylation Data 2012. R Package version 2.2.0. 2012 [Google Scholar]
- 28.Morris TJ, Beck S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 2015 Jan 15;72:3–8. doi: 10.1016/j.ymeth.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013 Feb;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, et al. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014 Feb 1;30(3):428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013 Jan 15;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007 Jan;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 33.Hansen KD. IlluminaHumanMethylation450kanno.ilmn12.h19: Annotation for Illumina’s 450k methylation arrays. R package version 0.2.1 [Google Scholar]
- 34.Smyth GK. Limma: Linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry RA, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor, R. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 35.Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics. 2009 Jan 15;25(2):211–217. doi: 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ. Developmental origins of non-communicable disease: implications for research and public health. Environ Health. 2012 Jun 27;11:42. doi: 10.1186/1476-069X-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012 May 29;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 38.Shu W, Chen H, Bo X, Wang S. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011 Sep 1;39(17):7428–7443. doi: 10.1093/nar/gkr443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene. 2008 Jan 10;27(3):404–408. doi: 10.1038/sj.onc.1210631. [DOI] [PubMed] [Google Scholar]
- 40.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007 Jan;1775(1):138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Ching T, Ha J, Song MA, Tiirikainen M, Molnar J, Berry MJ, et al. Genome-scale hypomethylation in the cord blood DNAs associated with early onset preeclampsia. Clin Epigenetics. 2015 Mar 13;7(1):21. doi: 10.1186/s13148-015-0052-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kloypan C, Srisa-Art M, Mutirangura A, Boonla C. LINE-1 hypomethylation induced by reactive oxygen species is mediated via depletion of S-adenosylmethionine. Cell Biochem Funct. 2015 Aug;33(6):375–384. doi: 10.1002/cbf.3124. [DOI] [PubMed] [Google Scholar]
- 43.Koczor CA, Jiao Z, Fields E, Russ R, Ludaway T, Lewis W. AZT-induced mitochondrial toxicity: an epigenetic paradigm for dysregulation of gene expression through mitochondrial oxidative stress. Physiol Genomics. 2015 Oct;47(10):447–454. doi: 10.1152/physiolgenomics.00045.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nahar MS, Kim JH, Sartor MA, Dolinoy DC. Bisphenol A-associated alterations in the expression and epigenetic regulation of genes encoding xenobiotic metabolizing enzymes in human fetal liver. Environ Mol Mutagen. 2014 Apr;55(3):184–195. doi: 10.1002/em.21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbstman JB, Tang D, Zhu D, Qu L, Sjodin A, Li Z, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect. 2012 May;120(5):733–738. doi: 10.1289/ehp.1104056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo L, Byun HM, Zhong J, Motta V, Barupal J, Zheng Y, et al. Effects of short-term exposure to inhalable particulate matter on DNA methylation of tandem repeats. Environ Mol Mutagen. 2014 May;55(4):322–335. doi: 10.1002/em.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Byun HM, Motta V, Panni T, Bertazzi PA, Apostoli P, Hou L, et al. Evolutionary age of repetitive element subfamilies and sensitivity of DNA methylation to airborne pollutants. Part Fibre Toxicol. 2013 Jul 15;10:28. doi: 10.1186/1743-8977-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hew KM, Walker AI, Kohli A, Garcia M, Syed A, McDonald-Hyman C, et al. Childhood exposure to ambient polycyclic aromatic hydrocarbons is linked to epigenetic modifications and impaired systemic immunity in T cells. Clin Exp Allergy. 2015 Jan;45(1):238–248. doi: 10.1111/cea.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossnerova A, Tulupova E, Tabashidze N, Schmuczerova J, Dostal M, Rossner P, Jr, et al. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat Res. 2013 Jan-Feb;:741–742. 18–26. doi: 10.1016/j.mrfmmm.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Marsit CJ, Brummel SS, Kacanek D, Seage GR, 3rd, Spector SA, Armstrong DA, et al. Infant Peripheral Blood Repetitive Element Hypomethylation Associated with Antiretroviral Therapy in utero. Epigenetics. 2015 Jun 11;10(8):708–716. doi: 10.1080/15592294.2015.1060389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Senda S, Blanche S, Costagliola D, Cibert C, Nigon F, Firtion G, et al. Altered heterochromatin organization after perinatal exposure to zidovudine. Antivir Ther. 2007;12(2):179–187. [PubMed] [Google Scholar]
- 52.Wang IJ, Karmaus WJ, Chen SL, Holloway JW, Ewart S. Effects of phthalate exposure on asthma may be mediated through alterations in DNA methylation. Clin Epigenetics. 2015 Mar 15;7(1):27. doi: 10.1186/s13148-015-0060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.