

SUMMARY
Oncogene-induced DNA methylation-mediated transcriptional silencing of tumor suppressors frequently occurs in cancer, but the mechanism and functional role of this silencing in oncogenesis is not fully understood. Here, we show that oncogenic epidermal growth factor receptor (EGFR) induces silencing of multiple unrelated tumor suppressors in lung adenocarcinomas and glioblastomas by inhibiting the DNA demethylase TET oncogene family member 1 (TET1) via the C/EBPα transcription factor. After oncogenic EGFR inhibition, TET1 binds to tumor suppressor promoters and induces their re-expression through active DNA demethylation. Ectopic expression of TET1 potently inhibits lung and glioblastoma tumor growth, and TET1 knockdown confers resistance to EGFR inhibitors in lung cancer cells. Lung cancer samples exhibited reduced TET1 expression or TET1 cytoplasmic localization in the majority of cases. Collectively, these results identify a conserved pathway of oncogenic EGFR-induced DNA methylation-mediated transcriptional silencing of tumor suppressors, which may have therapeutic benefit for oncogenic EGFR-mediated lung cancers and glioblastomas.
Graphical Abstract
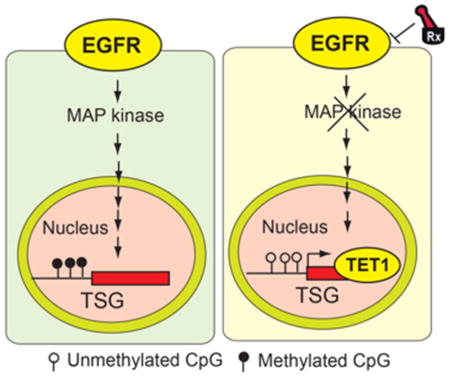
eTOC Text
Forloni et al. find that oncogenic EGFR induces silencing of tumor suppressor genes by repressing the DNA demethylase TET1 in lung cancer and glioblastoma. This mechanism is necessary for tumor growth and determining the response to EGFR inhibitors.
INTRODUCTION
Normal cells undergo multiple genetic and epigenetic alterations (DNA methylation and/or histone modification-based modifications) to become cancerous (Baylin and Jones, 2011; Vogelstein et al., 2013), and DNA methylation-mediated transcriptional gene silencing (hereafter referred to as epigenetic silencing) of tumor suppressor genes (TSGs) has been reported in numerous cancers (Baylin and Jones, 2011; Baylin and Ohm, 2006). Previous studies showed that oncogenes instruct epigenetic silencing of specific TSGs and pro-apoptotic genes (Gazin et al., 2007; Palakurthy et al., 2009; Wajapeyee et al., 2013). Oncogenic KRAS was shown to induce epigenetic silencing of the Fas pro-apoptotic gene via ordered recruitment of transcriptional repressors in mouse NIH3T3 cells (Gazin et al., 2007; Wajapeyee et al., 2013). Another study reported that oncogenic KRAS engages a completely different group of proteins to induce epigenetic silencing of TSGs in colon cancer cells, which confers the CpG island methylator phenotype (Serra et al., 2014). Oncogene-induced epigenetic silencing is probably influenced by several factors, including the oncogene type, organism and species, and cancer type.
Epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein and one of the four members of the erbB family of tyrosine kinase receptors (Lurje and Lenz, 2009). Deregulated EGFR signaling due to oncogenic mutations in the EGFR gene or EGFR gene amplification is associated with the genesis of numerous human cancers, including lung, brain, breast, prostate, pancreatic, and ovarian cancers (Foley et al., 2010; Herbst et al., 2008; Huang et al., 2009; Sheng and Liu, 2011; Traish and Morgentaler, 2009; Troiani et al., 2012). EGFR is mutated in a subset of lung adenocarcinomas, and EGFR inhibitors are now used to treat lung cancer patients with tumors harboring EGFR mutations (Politi et al., 2015).
Here, we demonstrate that oncogenic EGFR epigenetically silences multiple unrelated TSGs in lung cancer and glioblastoma multiforme (GBM) cells via transcriptional downregulation of the active DNA demethylase TET1. We also show that TET1 exerts a tumor-suppressive effect on lung and GBM cells, and TET1 re-expression following oncogenic EGFR inhibition is required to elicit a response to EGFR tyrosine kinase inhibitors (TKIs) in lung cancer.
RESULTS
Oncogenic EGFR Induces Epigenetic Silencing of Diverse TSGs in Lung Cancer Cells
Oncogenic EGFR is mutated in approximately 15% of lung adenocarcinomas and several other cancer types (Foley et al., 2010; Herbst et al., 2008; Huang et al., 2009). The role of oncogenic EGFR in inducing epigenetic silencing of TSGs and its mechanism of action are not known. Therefore, we investigated whether oncogenic EGFR can induce epigenetic silencing of TSGs in lung cancer cells, analyzed the molecular mechanism, and evaluated the implications of EGFR-induced epigenetic silencing of TSGs in the biology and treatment of cancer.
We tested EGFR-induced epigenetic silencing of TSGs in EGFR-mutant lung adenocarcinoma in two isogenic lung adenocarcinoma cell lines, HCC827/Del and HCC827/Del-TM. These cells were generated by expressing either EGFR-Del747-752 (Del) or EGFR-Del747-752-T790M (Del-TM) mutant construct, respectively, in the HCC827 cell line, and have been characterized in previous studies (Costa et al., 2007; Kobayashi et al., 2006). HCC827/Del and HCC827/Del-TM cells were treated with the DNA methyltransferase (DNMT) inhibitor decitabine and the histone deacetylase inhibitor vorinostat, and changes in gene expression were analyzed by microarray to identify genes that were epigenetically silenced. Treatment of HCC827/Del cells with decitabine and vorinostat altered the expression of a large number of genes. However, only 57 genes were commonly upregulated in both HCC827/Del and HCC827/Del-TM cells following treatment with decitabine and vorinostat (Figure 1A and Table S1), suggesting that these genes were epigenetically silenced specifically by mutant EGFR. Fifteen of the 57 re-expressed genes were reported previously to have tumor suppressor activities in either lung cancer or other cancer types (Table S1).
Figure 1. Oncogenic EGFR is necessary for epigenetic silencing of TSGs in lung cancer cells (See also Figure S1).

(A) (Left) Venn diagram shows the number of genes that are commonly or specifically upregulated in HCC827/Del (DEL) or HCC827/Del-TM (DEL-TM) cells after decitabine and vorinostat treatment. (Right) Heatmap of indicated genes in HCC-827/Del (DEL) and HCC827/Del-TM (DEL-TM) cells after decitabine and vorinostat treatment. (B–E) HCC827/Del cells were treated with gefitinib (0.1 μM), afatinib (0.1 μM), or DMSO for 48 h. (B) mRNA expression for the indicated TSGs was measured by RT-qPCR. Gene expression was normalized with respect to GAPDH mRNA expression. Relative expression for the indicated genes in drug-treated cells compared to DMSO-treated cells is plotted. (C) DNA methylation of CpG islands in the promoters of the indicated genes was measured using Me-DIP analysis. The percent of CpG island DNA methylation for the promoters of the indicated genes in drug-treated cells is compared with that of DMSO-treated cells. (D) RNA polymerase II enrichment on TSG promoters was measured via ChIP assay. Relative enrichment in drug-treated cells is compared with that of DMSO-treated cells. (E) Histone H3 lysine 9 acetylation (H3K9-Ac) marks on the indicated gene promoters were measured. Relative amounts of H3K9-Ac marks in drug-treated cells are shown relative to those observed in DMSO-treated cells. (F) The indicated EGFR-mutant lung cancer cell lines were treated with afatinib (0.1 μM) or DMSO for 48 h. mRNA expression levels for the indicated TSGs were measured via RT-qPCR. Gene expression was normalized with respect to GAPDH mRNA expression. Relative expression levels for the indicated genes in drug-treated cells are compared with those of DMSO-treated cells. *p < 0.05.
We hypothesized that the re-expressed TSGs in EGFR-mutant lung cancer cell lines treated with decitabine and vorinostat were epigenetically silenced by oncogenic EGFR. To test this hypothesis, we treated HCC827/Del and HCC827/Del-TM cells with the EGFR inhibitor gefitinib, or the second-generation EGFR inhibitor afatinib, and analyzed the expression of five unrelated TSGs (ANGPTL4, ARNT2, ATF3, NDRG1, and NDRG4) that were re-expressed after treatment with decitabine and vorinostat in both HCC827/Del and HCC827/Del-TM cells (Figure S1A and Table S1). Gefitinib treatment induced the re-expression of all five TSGs specifically in HCC827/Del cells, whereas afatinib treatment inhibited oncogenic EGFR signaling in both HCC827/Del and HCC827/Del-TM cells and induced the re-expression of all five TSGs in both cell lines (Figures 1B and S1B). Consistent with the re-expression of TSGs, oncogenic EGFR inhibition reduced promoter DNA hypermethylation (Figures 1C and S1C) and enrichment of RNA pol II on TSG promoters (Figures 1D and S1D), whereas oncogenic EGFR inhibition increased the acetylation mark of histone H3 lysine 9 (Figures 1E and S1E). We generalized these results by analyzing the expression of these five TSGs in a panel of EGFR-mutant lung cancer cell lines. Treatment of HCC827/Del and HCC827/Del-TM cells with afatinib induced the re-expression of all five TSGs in multiple unrelated EGFR-mutant lung cancer cell lines (Figure 1F), which provided additional support for our hypothesis. Collectively, these results demonstrate that oncogenic EGFR can epigenetically silence several unrelated TSGs in multiple EGFR-mutant lung cancer cell lines.
EGFR is Sufficient to Induce Epigenetic Silencing of TSGs in Lung Cancer Cells
We investigated whether oncogenic EGFR was sufficient to establish epigenetic silencing of TSGs in lung cancer cells by cloning the promoter of the tumor suppressor NDRG4 upstream of either a firefly luciferase gene or a blasticidin-resistance gene. These constructs were stably and individually transfected into the EGFR-mutant cell line HCC827/Del and control HeLa cells. The results indicated that the NDRG4 promoter-luciferase reporter and the NDRG4 promoter-blasticidin-resistance gene constructs were silenced in HCC827/Del cells but not in HeLa cells (Figures 2A and 2B).
Figure 2. EGFR is sufficient to induce epigenetic silencing of TSGs and inhibits expression of TSGs via the MAPK pathway (See also Figure S1).

(A) NDRG4 promoter-luciferase reporter construct was stably transfected into HCC827/Del or HeLa cells, and luciferase activity was measured at the indicated time points after treatment with gefitinib (0.1 μM) for 48 h. (B) NDRG4 promoter-blasticidin—resistance gene construct was stably transfected into HCC827/Del or HeLa cells, and colonies were formed in the presence or absence of gefitinib (0.1 μM) under blasticidin selection. Representative wells are shown. (C) Bisulfite sequencing of the NDRG4 promoter driving luciferase expression in HCC827/Del cells under the indicated conditions. Open circles represent unmethylated CpGs; filled circles represent methylated CpGs. (D and E) HCC827/Del cells were treated with Wortmannin (a PI3K inhibitor) (10 μM), U0126 (a MEK inhibitor) (10 μM), or ruxolitinib (a JAK inhibitor) (10 μM) for 48 h. (D) Expression levels of the indicated proteins were analyzed via immunoblot analyses. (E) Expression levels of the indicated TSGs were measured via RT-qPCR. Relative expression levels for the indicated TSGs in drug-treated cells are compared with those of DMSO-treated cells. Gene expression was normalized with respect to GAPDH mRNA expression. (F) CpG SDIP assay. The percent of promoter CpG island DNA methylation in drug-treated cells was compared with that observed in DMSO-treated cells. (G) RNA Pol II recruitment was analyzed for the indicated gene promoters. Fold-enrichment of RNA Pol II in drug-treated cells relative to that in DMSO-treated cells is shown. *p < 0.05.
To confirm that oncogenic EGFR establishes the epigenetic silencing of NDRG4 in HCC827/Del cells, we treated the cells with a low dose of gefitinib that was sufficient to inhibit oncogenic EGFR (Figure S1F). Gefitinib treatment stimulated luciferase activity and increased the number of blasticidin-resistant colonies in HCC827/Del cells but not in HeLa cells (Figures 2A and 2B). Similar results were obtained when these cells were treated with decitabine and vorinostat (Figure S1G), and in HCC827/Del-TM cells treated with afatinib (Figures S1H and S1I). Consistent with the luciferase assay results, bisulfite sequencing of the NDRG4 promoter cloned upstream of luciferase revealed that gefitinib treatment reduced CpG island DNA methylation (Figure 2C). Collectively, these results indicate that oncogenic EGFR is sufficient to induce epigenetic silencing of NDRG4 by increasing promoter DNA methylation in lung cancer cells.
Oncogenic EGFR-Mediated Epigenetic Silencing Occurs via MAPK Pathways
Oncogenic EGFR constitutively activates phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways. To determine which pathway mediates oncogenic EGFR epigenetic repression of TSGs, we used pharmacological inhibitors to individually block each of these three signaling pathways in HCC827/Del cells [Wortmannin (PI3K inhibitor), U0126 (MEK inhibitor), or ruxolitinib (JAK inhibitor)]. Inhibition of the PI3K or JAK pathways did not induce re-expression of TSGs (Figures 2D and 2E). However, inhibition of the MAPK pathway was sufficient to re-express TSGs that were epigenetically silenced by oncogenic EGFR (Figures 2D and 2E). Consistent with these results, we observed reduced CpG island DNA methylation (Figure 2F) and enhanced RNA pol II enrichment (Figure 2G) on TSG promoters when MEK inhibitor was used. Similar results were obtained using two different U0126 concentrations at two different time points (Figures S2A–S2F). Collectively, these results indicate that oncogenic EGFR mediates epigenetic silencing of TSGs by promoting MAPK pathway activity.
Oncogenic EGFR Transcriptionally Represses DNA Demethylase TET1
By definition, epigenetic gene silencing is a reversible process. Therefore, we tested whether oncogenic EGFR-mediated epigenetic silencing of TSGs is reversible by performing an oncogenic EGFR inhibitor washout experiment. HCC827/Del cells were treated with gefitinib for 24 h, gefitinib was removed by washing cells with fresh media lacking the drug, and samples were collected at 24 and 48 h after drug washout. Untreated controls and gefitinib-treated cells were included in the analysis. The results showed that gefitinib-induced inhibition of oncogenic EGFR in HCC827/Del cells caused the re-expression of oncogenic EGFR-repressed TSGs (Figure 3A). Consistent with the activation of oncogenic EGFR signaling, TSGs were repressed 48 h after drug washout (Figure 3A). TSG re-expression correlated with reduced DNA methylation at CpG islands in TSG promoters (Figure 3B). Collectively, these results indicate that oncogenic EGFR-mediated epigenetic TSG silencing requires continuous activity of the oncogenic EGFR pathway.
Figure 3. EGFR transcriptionally represses the active DNA demethylase TET1 (See also Figure S2).

(A and B) HCC827/Del cells were treated with gefitinib (0.1 μM) for 48 h and then washed out. DMSO-treated samples, gefitinib-treated samples, and samples harvested 24 and 48 h after washout of gefitinib were collected. (A) Samples were analyzed for the indicated proteins via immunoblot analyses (Left), or for the transcripts of the indicated TSGs via RT-qPCR (Right). (B) DNA methylation of the CpG islands in the promoters for indicated TSGs were analyzed via Me-DIP analyses. The percent of CpG island DNA methylation for HCC827/Del cells under the indicated conditions relative to that of DMSO-treated cells is shown. (C) HCC827/Del cells were treated with aphidicolin (5 μM) and gefitinib (0.1 μM) for 48 h, and the expression levels of the indicated TSGs were analyzed via RT-qPCR. Relative mRNA expression levels for the indicated TSGs in drug-treated cells were compared with those in DMSO-treated cells. (D) The indicated EGFR-mutant lung cancer cell lines were treated with afatinib, and the expression of TET1 mRNA was measured via RT-qPCR. Gene expression was normalized with respect to GAPDH mRNA expression. (E) The indicated EGFR-mutant lung cancer cell lines were treated with afatinib, and the expression levels of TET1 and other indicated proteins were measured via immunoblot analyses. GAPDH expression was used as a loading control. (F) The indicated lung cancer cell lines were analyzed for TET1 expression via RT-qPCR. TET1 expression in lung cancer cell lines relative to that of MRC5 cells is shown. *p < 0.05.
CpG island DNA demethylation can occur via passive or active DNA demethylation (Kohli and Zhang, 2013; Pastor et al., 2013). To distinguish between these mechanisms, we treated HCC827/Del cells with the DNA replication inhibitor aphidicolin in conjunction with the EGFR inhibitor gefitinib. Combined aphidicolin and gefitinib treatment resulted in TSG re-expression even in the absence of DNA replication (Figure 3C), suggesting that active DNA demethylation is involved in this process.
Recent studies reported that TET family proteins can induce active DNA demethylation (Kohli and Zhang, 2013; Pastor et al., 2013). Therefore, we investigated whether TET proteins mediated oncogenic EGFR-induced epigenetic silencing of TSGs by measuring TET1, TET2, and TET3 expression levels in a panel of EGFR-mutant lung cancer cell lines. Afatinib inhibition of oncogenic EGFR significantly enhanced TET1 expression in all EGFR-mutant lung cancer cell lines (Figures 3D and 3E). We assessed basal levels of TET1 mRNA and protein in a panel of control and EGFR-mutant lung cancer cell lines. The results indicated that TET2 and TET3 expression did not significantly differ in control and mutant cells, whereas TET1 expression was generally lower in EGFR-mutant cells than in wild-type EGFR lung cancer cell lines (Figure 3F). Additional control experiments measuring the expression of TET2, TET3, and DNA methyltransferases (DNMTs) did not detect any significant differences in the expression of these genes in response to inhibition of oncogenic EGFR signaling (Figures S2G and S2H). Similar results were obtained using immortalized human airway epithelial cells (HAE/SV40-ER+hTERT) expressing either the EGFR Del mutant (EGFR-Del747-752) alone or the EGFR-Del and T790M mutants (EGFR-Del747-752+T790M) (Figure S3). These results further confirm a role for oncogenic EGFR signaling in epigenetic silencing of TSGs.
EGFR Regulates TET1 Expression via the C/EBPα Transcription Factor
To determine the mechanism of TET1 upregulation following oncogenic EGFR inhibition, we analyzed the TET1 promoter sequence and identified potential DNA binding sites for several transcription factors (TFs) (Table S2). To determine which of these TFs regulate TET1 transcription after oncogenic EGFR inhibition, we checked the expression of these TFs in a panel of EGFR-mutant lung cancer cell lines after treatment with afatinib. Oncogenic EGFR inhibition induced the expression of GATA2, C/EBPα, and C/EBPβ in five out of six EGFR-mutant lung cancer cell lines (Figures 4A and S4A), which correlated with the TET1 re-expression pattern in these EGFR-mutant lung cancer cells.
Figure 4. EGFR regulates TET1 expression via the transcription factor C/EBPα (See also Figures S3 and S4).

(A) The indicated lung cancer cell lines were treated with afatinib (0.1 μM) for 48 h, and the mRNA and protein expression levels of the transcription factor C/EBPα were analyzed via RT-qPCR (Left) or immunoblot analysis (Right), respectively. (B) The HCC827/Del cell line expressing C/EBPα or non-specific (NS) shRNAs was treated with afatinib (0.1 μM) for 48 h, and analyzed for TET1 mRNA expression or TET1 protein expression via RT-qPCR (Left) or immunoblot analysis (Right), respectively. (C) The HCC827/Del cell line was treated with afatinib (0.1 μM) for 48 h, and the occupancy of C/EBPα on the TET1 promoter was analyzed via ChIP. The relative enrichment of C/EBPα in afatinib-treated cells was compared with that of an IgG control. is shown at two predicted C/EBPα. (D and E) HCC827/Del cells expressing TET1 or NS shRNAs were treated with afatinib (0.1 μM) for 48 h, and analyzed for TET1 expression and TSG mRNA and protein levels via RT-qPCR (D) and immunoblot analysis (E), respectively. (F) HCC827/Del cells were treated with afatinib (0.1 μM) for 48 h, and analyzed for TET1 occupancy on the promoters of the indicated TSGs via ChIP. *p < 0.05 and **p < 0.005.
To determine which TFs regulate TET1 transcription, we knocked down the expression of each TF using shRNAs (Figures 4B, S4B, and S4C). We found that C/EBPα knockdown blocked TET1 re-expression after oncogenic EGFR inhibition (Figure 4B), whereas GATA2 and C/EBPβ knockdown did not affect TET1 expression (Figure S4D). Similar results were obtained in other EGFR-mutant lung cancer cell lines (Figure S4E).
To confirm that C/EBPα directly regulates TET1 expression, we performed a chromatin immunoprecipitation (ChIP) experiment. ChIP results showed that C/EBPα directly bound to the TET1 promoter after EGFR signaling inhibition (Figure 4C). Next, we determined the mechanism of oncogenic EGFR pathway inhibition upregulation of C/EBPα by analyzing the C/EBPα promoter sequence and identifying several TF binding sites. We selected YY1 for further analysis because it repressed transcription (Gordon et al., 2006) and was previously reported to be regulated by the MAP kinase pathway (Stoeckius et al., 2012). We tested whether YY1 was regulated by EGFR in a MAP kinase pathway-dependent manner in EGFR-mutant lung cancer cells. The results showed that treatment of HCC827/DEL cells with afatinib or U0126 reduced the YY1 protein level (Figure S4F), and YY1 knockdown upregulated C/EBPα (Figure S4G). We also performed ChIP assays to test whether YY1 directly repressed C/EBPα expression. We found that YY1 was enriched on the C/EBPα promoter in HCC827/Del cells, which was inhibited by treatment with afatinib or U0126 (Figure S4H). Collectively, these results indicate that oncogenic EGFR pathway inhibition upregulates the C/EBPα TF via YY1, which, in turn, stimulates the expression of the active DNA demethylase TET1.
TET1 Upregulation after Oncogenic EGFR Signaling Inhibition is Necessary for TSG Re-Expression
We also evaluated the functional association between TET1 upregulation and TSG re-expression after oncogenic EGFR inhibition. We find that shRNA-induced TET1 knockdown prevented re-expression of repressed TSGs after afatinib treatment (Figures 4D and 4E). Similar to afatinib treatment, U0126 was able to re-express C/EBPα (Figures S5A and S5B), However, ectopic expression of oncogenic KRASG12D prevented afatinib-induced C/EBPα and TSG re-expression (Figures S5C and S5D). Collectively, these results indicate that TET1 activation is required for TSG re-expression after oncogenic EGFR pathway inhibition.
TET1 catalyzes the hydroxylation of 5-methyl cytosine (5mC) to 5-hydroxymethyl cytosine (5hmC) and thereby facilitates active DNA demethylation (Pastor et al., 2013). Therefore, we analyzed TET1 enrichment on TSG promoters by performing ChIP analysis. We found that TET1 proteins were significantly enriched on TSG promoters in afatinib treated HCC827/Del cells (Figure 4F). Collectively, these results indicate that TET1 re-expression after oncogenic EGFR signaling inhibition is necessary for TSG re-expression in EGFR-mutant lung cancer cells.
Oncogenic EGFR-Mediated Epigenetic Silencing of TSGs is Conserved in GBM Cells
Oncogenic EGFR mutations are known to be present in GBM cells (Furnari et al., 2015). Therefore, we analyzed the mechanism of TSG silencing in GBM cells. We used an isogenic GBM cell line that was generated by ectopic expression of the EGFRvIII mutation in U87 cells (U87/EGFRvIII), and analyzed TET1 expression in U87 and U87/EGFRvIII cells. Ectopic expression of EGFRvIII in U87 cells repressed TET1 expression (Figure 5A) in an oncogenic EGFR-dependent manner, as gefitinib treatment of U87/EGFRvIII cells and two patient-derived xenograft GBM cell lines (GBM6 and GBM313) resulted in TET1 re-expression (Figures 5B and 5C). Similar results were obtained using the MEK inhibitor U0126 (Figure S5E). Collectively, these results indicate that TET1 is regulated by MAP kinase pathway activity in U87/EGFRvIII, and additional GBM cell lines.
Figure 5. EGFR-driven TET1 repression is conserved in EGFR-mutant GBM (See also Figure S5).

(A) The mRNA expression level of TET1 was measured via RT-qPCR. Relative TET1 mRNA levels in U87/EGFRvIII cells were compared with those in U87 cells (Left). TET1 protein expression level as measured via immunoblot analysis. TET1 and GAPDH expression in U87/EGFRvIII cells was compared with that in U87 cells (Right). (B) The indicated GBM cells were treated with the indicated gefitinib concentrations for 48 h, and analyzed for TET1 mRNA expression via RT-qPCR. The relative mRNA expression levels for TET1 and the indicated TSGs in drug-treated cells were compared with those in DMSO-treated GBM cells. (C) The indicated GBM cells were treated with the indicated gefitinib concentrations for 48 h, and analyzed for TET1 expression level via immunoblotting. GAPDH expression was used as an internal control. (D) U87/EGFRvIII cells were treated with gefitinib (10 μM) and analyzed for the expression of the indicated TSGs via RT-qPCR. The relative expression levels of the TSGs in U87/EGFRvIII cells were compared with those in DMSO-treated cells. (E) U87/EGFRvIII cells were treated with DMSO or gefitinib (10 μM) for 48 h, and analyzed for the expression of TET1 and the indicated TSG proteins via immunoblot analyses. (F) U87/EGFRvIII cells were treated with gefitinib and analyzed for DNA methylation at CpG islands in the promoters of the indicated TSGs via Me-DIP. The relative DNA methylation levels for the indicated TSGs in drug-treated cells were compared with those observed in U87/EGFRvIII cells treated with DMSO. (G) U87/EGFRvIII cells expressing either a non-specific (NS) shRNA or shRNAs targeting TET1 were treated with gefitinib (10 μM) and analyzed for the expression of the indicated genes via RT-qPCR. *p < 0.05 and **p < 0.005.
We then analyzed the expression of five TSGs (ANGPTL4, ARNT2, ATF3, NDRG1, and NDRG4) that were tested in lung cancer cells to determine if oncogenic EGFR epigenetically repressed TSG expression in GBM cells. We found that oncogenic EGFR inhibition was sufficient to induce re-expression of four of these TSGs (Figures 5D and 5E). Consistent with epigenetic TSG silencing, we also found that the TSG re-expression correlated with reduced CpG island methylation in TSG promoters (Figure 5F). TET1 knockdown in U87/EGFRvIII cells prevented TSG re-expression after gefitinib treatment (Figure 5G). Collectively, these results indicate that oncogenic EGFR epigenetically represses the same TSGs in different tumor types via a conserved TET1-dependent mechanism.
TET1 Expression is Necessary for EGFR-Mutant Lung Cancer Cell Response to EGFR Inhibitors
Previous studies reported that TET1 can function as a tumor suppressor (Cimmino et al., 2015; Neri et al., 2014). Therefore, we analyzed whether TET1 repression was necessary for tumor formation in EGFR-mutant lung cancers and glioblastomas by ectopically expressing TET1 in lung cancer and GBM cell lines. We found that ectopic expression of TET1 reduced tumor growth in soft-agar assays, indicating that tumor growth was inhibited (Figure 6A), and significantly reduced tumor formation in athymic nude mice (Figure 6B). To confirm these in vivo results, we evaluated the effect of TET1 expression in a patient-derived xenograft (PDX) of EGFR-mutant lung adenocarcinoma. EGFR-mutant lung cancer PDX-bearing mice were intratumorally injected with either control adenovirus expressing GFP or adenovirus expressing TET1 (Ad-TET1) every 3 days for 2 weeks. Ectopic expression of TET1 in this EGFR-mutant lung cancer PDX model inhibited tumor growth in vivo (Figure 6C).
Figure 6. TET1 confers tumor suppressive activity in lung cancer and GBM cells and is necessary for the response to EGFR TKIs (See also Figure S5).

(A) (Left) The indicated lung cancer and GBM cell lines expressing either empty vector or TET1 were grown on soft agar. Representative wells are shown. (Right) Relative colony sizes for the indicated lung cancer and GBM cell lines are shown. (B) The indicated lung cancer and GBM cell lines expressing either empty vector or TET1 were injected into athymic nude mice. Average tumor volumes for the indicated conditions are shown at the indicated time points. (C) EGFR-mutant lung cancer PDX-bearing mice were injected with either control (Ad-LacZ) or TET1 (Ad-TET1) adenovirus. Average tumor volumes for the indicated conditions are shown at the indicated time points. (D) HCC827/Del cells expressing either TET1 or non-specific (NS) shRNAs were treated with gefitinib (0.1 μM). The relative percent of cell viability and the percent of annexin V-positive cells of the indicated cell lines expressing TET1 shRNA relative to cells expressing NS shRNAs are shown after 48 h of treatment. (E) H3255 cells expressing either TET1 or NS shRNAs were treated with gefitinib (0.1 μM). The percent of cell viability and the percent of annexin V-positive cells of the indicated cell lines expressing TET1 shRNA relative to that of cells expressing NS shRNAs are shown after 48 h of treatment. *p < 0.05 and **p < 0.0005.
EGFR represses TET1 expression, so we evaluated whether TET1 played a role in determining the response to EGFR inhibitors by knockdown of TET1 expression in EGFR-mutant cell lines treated with gefitinib. Loss of TET1 in the EGFR-mutant HCC827/Del and H3255 lines increased cellular resistance to gefitinib (Figures 6D, 6E, S5F, and S5G). Next, we evaluated whether this mechanism of resistance operated in vivo by analyzing tumor samples derived from a previously described EGFR-driven mouse model of lung tumorigenesis (Politi et al., 2006). Specifically, we compared normal lung samples with lung tumor samples that were either untreated, treated for 5 days with erlotinib and presumed to be responding to the drug (erlotinib responsive), or had acquired resistance to erlotinib (erlotinib resistant). Consistent with our cell culture studies, erlotinib-responsive lung tumors exhibited increased TET1 expression, whereas erlotinib-resistant lung tumors did not (Figure 7A). TET1 expression inversely correlated with MAP kinase target gene expression, and erlotinib-resistant samples continued to display higher MAP kinase target gene expression (Figure S6A).
Figure 7. TET1 is downregulated or mislocalized in lung cancer samples and in EGFR TKI resistant samples (See also Figure S6).

(A) Normal lung samples, EGFR-driven untreated lung cancer samples, and EGFR-driven erlotinib-treated lung cancer samples that were either sensitive or resistant to erlotinib treatment were analyzed for TET1 mRNA expression. The relative TET1 expression is shown. (B) Erlotinib-sensitive or erlotinib-resistant lung tumor samples were used to isolate genomic DNA, and the number of 5hmC marks was evaluated by dot blot analyses of 100 ng genomic DNA (Left). The membrane was then stained with methylene blue to confirm equal DNA loading (Right). (C) Analysis of patient-derived lung cancer samples shows the loss of TET1 expression and the cytoplasmic localization of TET1, indicating that TET1 expression is reduced in lung cancer. (Left) Representative images for EGFR-mutant lung cancers and the percent of samples across three genotypes with no staining, cytoplasmic staining, or cytoplasmic and nuclear staining are shown. (Right) Summary of the immunohistochemistry (IHC) results. (D) (Upper panel) Representative images for lung cancer samples stained for TET1. (Lower panel) Summary of the IHC results. (E) The proposed model considers that EGFR represses the active DNA demethylase TET1 to induce epigenetic TSG silencing. TET1 repression is required for EGFR-driven tumor growth and determines the response to EGFR TKIs, because TET1 expression inhibits tumor growth and TET1 knockdown inhibits EGFR TKI-mediated inhibition of cell growth.
We analyzed these samples further to determine if TET1 expression correlated with overall 5hmC marks. Consistent with our TET1 expression results, we found that erlotinib-responsive samples exhibited higher levels of 5hmC marks compared with those of erlotinib-resistant samples (Figure 7B). We also analyzed patient-derived samples of lung cancers with various genotypes (36 KRAS mutants, 27 EGFR mutants, and 53 KRAS/EGFR wild-type samples). TET1 was significantly inactivated in all genotypes (Figure 7B). For example, ~44% of EGFR-mutant lung cancers were negative for TET1 expression, whereas ~44% showed cytoplasmic localization of TET1, and only 11% showed both cytoplasmic and nuclear localization of TET1 (Figure 7C). These results demonstrate that a large percentage of lung cancer samples display TET1 inactivation in patient-derived lung cancer samples.
We tested whether TET1 inactivation correlated with EGFR tyrosine kinase inhibitor (TKI) resistance by analyzing 6 EGFR TKI-sensitive and 7 EGFR TKI-resistant human lung cancer samples. Our results show that although none of the EGFR TKI-resistant tumors expressed TET1 (0/7, 0%), half of the EGFR TKI-sensitive samples exhibited nuclear TET1 staining (3/6, 50%). These results further support a role for TET1 in modulating the response to EGFR TKIs in lung cancer cells (Figures 7D and S6B). We also analyzed the expression of several tumor suppressor genes in publicly available datasets and observed a significant association of reduced TSG expression with overall reduced survival in lung cancer patients (Figure S7). Collectively, our data provide evidence that TET1 functions as a tumor suppressor in lung cancer and GBM cells, and oncogenic EGFR inhibition-induced TET1 activation may be necessary for cellular response to EGFR inhibitors (Figure 7E). Therefore, future analysis using a larger cohort of lung cancer samples will be important for further establishing the utility of TET1 expression as a possible biomarker for predicting responses to EGFR inhibitors.
DISCUSSION
Our results allow us to draw several important conclusions. First, we found that oncogenic EGFR epigenetically silenced several unrelated TSGs in lung cancer and GBM cells via a conserved mechanism that involved transcriptional repression of the active DNA demethylase TET1. Second, we determined that oncogenic EGFR-induced epigenetic silencing was rapid and reversible; therefore, it was not truly epigenetic because it required continuous activity of the oncogenic EGFR pathway. Third, we showed that ectopic TET1 expression induced tumor suppressive effects in lung cancer and GBM cells, and TET1 expression was necessary to elicit responses to EGFR TKIs in lung cancer cells. Finally, our study documented a non-redundant function of the TET1 protein within the context of TSG epigenetic silencing; TET1 was necessary for oncogenic EGFR-induced epigenetic gene silencing, but TET2 and TET3 were not.
Oncogenic EGFR-Mediated Epigenetic Silencing of TSGs
Previous studies have established the importance of epigenetic alterations in cancer initiation and progression. Genome-wide and exome-wide cancer sequencing projects identified mutations in genes encoding proteins that regulate either DNA methylation or post-translational histone modifications (Huether et al., 2014). Collectively, these results indicate that the cancer epigenome can induce alterations in human cancers, similar to the cancer genome. However, it is not fully understood whether genetic and epigenetic alterations in cancer cells emerge and evolve independently or cooperatively. It is also not known if an oncogene can direct the epigenetic silencing of a group of TSGs, or the mechanism by which this epigenetic silencing may occur.
Here, we report that oncogenic EGFR can instruct the epigenetic silencing of multiple, unrelated TSGs in lung cancer and GBM cells. The observed epigenetic silencing was rapidly reversed and re-established, and so it does not represent a true epigenetic event that self-propagates once established. We find that continuous activity of oncogenic EGFR is required to maintain TSG silencing.
Our results are consistent with those of a previous study of oncogenic KRAS. However, those studies were performed in mouse cells, and the implications of those results in human cancer are unclear (Gazin et al., 2007; Wajapeyee et al., 2013). Our study extends these initial findings and identifies an instructive pathway for epigenetic silencing of TSGs in human lung cancer and GBM cells.
Role of TET1 in Oncogenic EGFR-Induced TSG Silencing
Epigenetic silencing can be reversed by passive DNA demethylation that involves DNA replication, or active DNA demethylation that occurs without DNA replication. Several proteins have been implicated in active DNA demethylation, such as TET family proteins and the DNA repair enzymes TDG and AID, which mediate the enzymatic conversion of 5mC to cytosine (Pastor et al., 2013).
Here, we found that oncogenic EGFR transcriptionally represses TET1, which is necessary for oncogenic EGFR-mediated epigenetic silencing of TSGs in distinct tumor types such as lung cancer and GBM. TET1 repression is necessary for oncogenic EGFR-induced epigenetic TSG silencing. The results suggest that TET proteins have non-redundant functions, as TET1 repression was necessary for epigenetic silencing of TSGs, but TET2 and TET3 repression was not. Collectively, these results show that oncogenic EGFR represses TET1, which enables epigenetic silencing of TSGs.
TET Regulates EGFR-Induced Tumor Growth and Cellular Response to EGFR Inhibitors
Previous studies show that TET proteins play an important role in cancer; reduced expression of TET1, TET2, and TET3 is associated with poor prognoses for patients with early-stage breast cancer (Yang et al., 2015). TET1 has tumor suppressive activity in colon, breast, and gastric cancers (Fu et al., 2014; Lu et al., 2014; Neri et al., 2015; Sun et al., 2013). Here, we show that TET1 expression in lung cancer and GBM cells inhibits tumor formation, indicating that TET1 confers tumor suppressive activity in lung cancer. We detected the loss of TET1 expression and mislocalized cytoplasmic TET1 in many patient-derived lung cancer samples, suggesting that TET1 was likely inhibited.
Our study suggests that TET1 determines the response to EGFR TKIs because the loss of TET1 expression confers resistance to EGFR TKIs. Collectively, these results demonstrate that oncogenic EGFR-mediated epigenetic silencing and regulation of DNA demethylase is central to the role of EGFR as an oncogene, and determines the cellular response to EGFR TKIs.
EXPERIMENTAL PROCEDURES
Gene Expression Profiling and Data Analysis
The HCC827/Del and HCC827/Del-T790M cell lines were treated with dimethyl sulfoxide (DMSO), decitabine (2.5 μM), vorinostat (1 μM), or a combination of both decitabine and vorinostat for 72 h. Total RNA was prepared from three biological replicates for each sample using TRIzol (Invitrogen), and purified with the Qiagen RNAeasy kit. For hybridization, cDNA was prepared using the Illumina TotalPrep™ RNA amplification kit according to the manufacturer’s protocols. Then, cDNA was hybridized to a HumanHT-12 v4 Expression BeadChip, which was scanned and analyzed using Illumina’s HiScan machine.
Microarray data were generated by GenomeStudio™ (Illumina), log2-transformed, and quantile-normalized using ArrayStudio (Omicsoft). Quality control (QC) was performed by checking various plots as in general microarray analysis methods. All samples passed QC. Differential expression analysis was performed using a one-way analysis of variance test with a Benjamini and Hochberg multiple test correction procedure (Benjamini and Hochberg, 1995) to identify statistically significant differentially expressed probes (adjusted P-value ≤ 0.05). Pathway enrichment analysis was performed for an input gene list (absolute fold-change ≥ 2 and significant adjusted P-value ≤ 0.05) using MetaCore™ (version 6.8, build 29806, GeneGo, Inc.).
Accession Numbers and Availability of Gene Expression Data
Gene expression data were submitted to Gene Expression Omnibus (GEO, Accession Number GSE39292). The data can be downloaded using the GEO Accession Number (http://www.ncbi.nlm.nih.gov/gds).
Drug Treatments
Cells were treated with the indicated concentrations of gefitinib, afatinib, aphidicolin, decitabine, and vorinostat (Table S3). For the aphidicolin experiment, cells were treated with EGFR inhibitor along with aphidicolin. In each case, cells were collected for RNA extraction 48 h after treatment. For EGFR inhibitor washout experiments, HCC827/Del cells were treated with gefitinib. Gefitinib was removed after 24 h of treatment, and cells were washed twice with phosphate-buffered saline (PBS) before suspending in fresh media. For the experiments, cells were collected at indicated time points after washout.
Soft-Agar, Tumor Xenograft, and Patient-Derived Xenograft Experiments
Soft-agar assays were performed as described previously (Gazin et al., 2007). For in vivo experiments, the HCC827/Del, PC9, H3255, and U87/EGFRvIII cell lines expressing either an empty vector or TET1 were injected subcutaneously into the flanks of nude mice (n = 10). Tumors were allowed to grow, and tumor growth was monitored every week. Tumor volume was calculated using the formula Volume = Length × Width2 × 0.5, and average tumor volumes were plotted. For the patient-derived xenograft (PDX) experiments, we obtained 15 NOD/SCID mice bearing EGFR mutant (L858R) PDX (Model ID: TM00199) from Jackson laboratory. Mice were assigned to three groups of five mice each, and after the average tumor volume reached 100 mm, tumors were injected with 50 μL (titer = 1 × 1011) of control adenovirus (Ad-LacZ) or TET1 adenovirus (Ad-TET1).
EGFR Mouse Tumor Samples
Mice were bred and housed in a pathogen-free environment under guidelines approved by the Yale University Institutional Animal Care & Use Committee (IACUC). CCSP-rtTA; TetO-EGFRL858R mice were described previously (Politi et al., 2006). Doxycycline was administered by feeding mice with doxycycline-impregnated food pellets (625 ppm; Harlan-Tekland). Erlotinib [Organic Synthesis Core Facility of Memorial Sloan Kettering Cancer Center (MSKCC)] was suspended in 0.5% (w/v) methylcellulose and administered intraperitoneally, 25 mg/kg, 5 days a week. Mice bearing untreated tumors, tumors treated for 5 days (erlotinib-responsive), or erlotinib-resistant tumors (CCSP-rtTA; TetO-EGFRL858R) were enrolled in the study. Erlotinib resistance was assessed by continuous or intermittent drug treatment as described previously (Pirazzoli et al., 2015). For molecular studies, freshly harvested tumors and adjacent normal lung tissue were pulverized in liquid nitrogen. RNA was extracted using the RNeasy platform (Qiagen), and then treated with DNase I (RNase-Free DNA Set, Qiagen). Genomic DNA was extracted using the Wizard genomic purification kit (Promega).
Tissue Microarray Analysis, EGFR TKI-Sensitive and TKI-Resistant Patient Sample Analysis, and TET1 Staining Protocol
Yale tissue microarray (YTMA) 310 contains 139 tumor cores from a non-serial collection of non-small cell lung cancer (NSCLC) patients with different mutation status. For YTMA 310 construction, pathology reports from 2011–2013 were retrieved. NSCLC patient cases that had surgical resection of primary tumor and molecular testing for common NSCLC mutations were selected for further review. The cases that had adequate residual tumor from the primary site were collected and cored. YTMA 310 includes 30 EGFR mutants, 43 KRAS mutants, and 66 non-mutant EGFR and KRAS patient cases.
Six EGFR TKI-sensitive and seven EGFR TKI-resistant samples from either the patients or the patient-derived xenografts were analyzed for TET1 expression using TET1 immunohistochemistry (IHC). All samples selected for this analysis lacked the EGFR T790M mutation. TET1 antibody was used at 1:1,000 dilution for the IHC experiments, and the antigen was retrieved using sodium citrate buffer (pH = 6.0). Secondary anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Envision+ system-HRP labeled polymer anti-rabbit) was obtained from DAKO. The slides were developed using the liquid DAB+ substrate chromogen system from DAKO.
Statistical Analyses
All experiments were performed at least three times, and data were expressed as the mean ± standard error of the mean (SEM). The area under the curve (AUC) values were calculated using GraphPad Prism version 6.02 for Macintosh (GraphPad Software, San Diego, California; www.graphpad.com). The P-values were computed with Student’s t-test in Microsoft Excel.
Supplementary Material
HIGHLIGHTS.
EGFR induces silencing of tumor suppressor genes in cancer cells.
EGFR transcriptionally represses TET1 via the transcription factor C/EBPα.
TET1 repression is necessary for EGFR-induced silencing.
TET1 blocks tumor growth and modulates the response to EGFR inhibitors.
Acknowledgments
We gratefully acknowledge the following grants from the National Institutes of Health: R21CA197758-01 (N.W.), R21CA191364-01 (N.W.), R21CA195077-01A1 (N.W.), R01CA200919-01 (N.W.), and R41 (1R41CA195908-01A1) (N.W.). N.W. is also supported by the Research Scholar Grant from the American Cancer Society (128347-RSG-15-212-01-TBG).
Footnotes
AUTHOR CONTRIBUTIONS
M.F., R.G., and A.N. performed the majority of the experiments, with the help of L.S and Y.D. S.K.D. analyzed the microarray data and helped with the statistical analysis. S.K. provided the cell lines used in this study. K.P. and V.P. provided the EGFR mouse tumor samples. M.T. and D.L.R. collected the lung cancer samples and generated the lung cancer tissue array. R.J.H. scored the TET1 staining and interpreted the results. M.F., R.G., R.J.H., and N.W. analyzed and interpreted the data. M.F., R.G., and N.W. co-authored the manuscript. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H, Lobry C, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015;16:653–662. doi: 10.1038/ni.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley J, Nickerson NK, Nam S, Allen KT, Gilmore JL, Nephew KP, Riese DJ., 2nd EGFR signaling in breast cancer: bad to the bone. Semin Cell Dev Biol. 2010;21:951–960. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu HL, Ma Y, Lu LG, Hou P, Li BJ, Jin WL, Cui DX. TET1 exerts its tumor suppressor function by interacting with p53-EZH2 pathway in gastric cancer. J Biomed Nanotechnol. 2014;10:1217–1230. doi: 10.1166/jbn.2014.1861. [DOI] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–1077. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ, Lowell AM, Golub T, Meyerson M, Tenen DG, Shapiro GI, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006;66:11389–11398. doi: 10.1158/0008-5472.CAN-06-2318. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HG, Zhan W, Yan L, Qin RY, Yan YP, Yang ZJ, Liu GC, Li GQ, Wang HF, Li XL, et al. TET1 partially mediates HDAC inhibitor-induced suppression of breast cancer invasion. Mol Med Rep. 2014;10:2595–2600. doi: 10.3892/mmr.2014.2517. [DOI] [PubMed] [Google Scholar]
- Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77:400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, Maldotti M, Parlato C, Paliogiannis P, Oliviero S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene. 2014 doi: 10.1038/onc.2014.356. [DOI] [PubMed] [Google Scholar]
- Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, Maldotti M, Parlato C, Paliogiannis P, Oliviero S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene. 2015;34:4168–4176. doi: 10.1038/onc.2014.356. [DOI] [PubMed] [Google Scholar]
- Palakurthy RK, Wajapeyee N, Santra MK, Gazin C, Lin L, Gobeil S, Green MR. Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol Cell. 2009;36:219–230. doi: 10.1016/j.molcel.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirazzoli V, Ayeni D, Meador CB, Sanganahalli B, Hyder F, de Stanchina E, Goldberg SB, Pao W, Politi K. Afatinib plus cetuximab delays resistance compared to single agent erlotinib or afatinib in mouse models of TKI-naive EGFR L858R-induced lung adenocarcinoma. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K, Ayeni D, Lynch T. The Next Wave of EGFR Tyrosine Kinase Inhibitors Enter the Clinic. Cancer Cell. 2015;27:751–753. doi: 10.1016/j.ccell.2015.05.012. [DOI] [PubMed] [Google Scholar]
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra RW, Fang M, Park SM, Hutchinson L, Green MR. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife. 2014;3:e02313. doi: 10.7554/eLife.02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Q, Liu J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br J Cancer. 2011;104:1241–1245. doi: 10.1038/bjc.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Erat A, Fujikawa T, Hiromura M, Koulova A, Otterbein L, Bianchi C, Tobiasch E, Dagon Y, Sellke FW, et al. Essential roles of Raf/extracellular signal-regulated kinase/mitogen-activated protein kinase pathway, YY1, and Ca2+ influx in growth arrest of human vascular smooth muscle cells by bilirubin. J Biol Chem. 2012;287:15418–15426. doi: 10.1074/jbc.M111.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, Chen P, Chen J, Chada KK, He C, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013;110:9920–9925. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traish AM, Morgentaler A. Epidermal growth factor receptor expression escapes androgen regulation in prostate cancer: a potential molecular switch for tumour growth. Br J Cancer. 2009;101:1949–1956. doi: 10.1038/sj.bjc.6605376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, Ciardiello F. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13:802–810. doi: 10.2174/138945012800564158. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Malonia SK, Palakurthy RK, Green MR. Oncogenic RAS directs silencing of tumor suppressor genes through ordered recruitment of transcriptional repressors. Genes Dev. 2013;27:2221–2226. doi: 10.1101/gad.227413.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yu SJ, Hong Q, Yang Y, Shao ZM. Reduced Expression of TET1, TET2, TET3 and TDG mRNAs Are Associated with Poor Prognosis of Patients with Early Breast Cancer. PLoS One. 2015;10:e0133896. doi: 10.1371/journal.pone.0133896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.