

Abstract
Quinoline antimalarials target hemozoin formation causing a cytotoxic accumulation of ferriprotoporphyrin IX (Fe(III)PPIX). Well-developed SAR models exist for β-hematin inhibition, parasite activity and cellular mechanisms for this compound class, but no comparably detailed investigations exist for other hemozoin inhibiting chemotypes. Here, benzamide analogues based on previous HTS hits have been purchased or synthesized. Only derivatives containing an electron deficient aromatic ring and capable of adopting flat conformations, optimal for π-π interactions with Fe(III)PPIX, inhibited β-hematin formation. The two most potent analogues showed nanomolar parasite activity, with little CQ cross-resistance, low cytotoxicity and high in vitro microsomal stability. Selected analogues inhibited hemozoin formation in Plasmodium falciparum causing high levels of free heme. In contrast to quinolines, introduction of amine side chains did not lead to benzamide accumulation in the parasite. These data reveal complex relationships between heme binding, free heme levels, cellular accumulation and in vitro activity of potential novel antimalarials.
Graphical Abstract
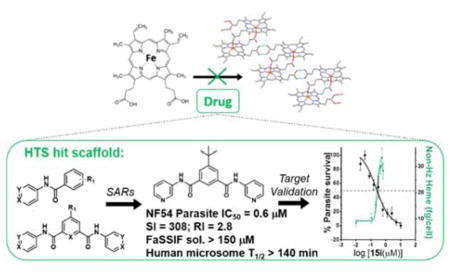
1. INTRODUCTION
Despite encouraging recent developments, malaria remains a major global health burden.1–3 Cross-resistance has been reported among the still extensively used quinolines, chloroquine (CQ), mefloquine (MQ), amodiaquine (AQ) and quinine (QN),4, 5 probably because of their similar structures and modes of action. These drugs are believed to target the hemozoin formation process in the digestive vacuole (DV) of the malaria parasite Plasmodium falciparum.6, 7 Hemozoin (Hz) is an inert crystalline form of ferriprotoporphyrin IX (Fe(III)PPIX), a cytotoxic by-product formed by autoxidation of heme released during parasite digestion of host hemoglobin.
Though there is well established resistance to the quinoline antimalarials, the process of Hz formation is unaltered within the parasite. Rather, mutations in digestive vacuole membrane proteins, for example PfCRT or PfMDR1, are responsible for decreasing drug accumulation at the site of action. Thus, the inhibition of Hz remains a viable drug target.5, 8 The quinolines are known to accumulate in P. falciparum, where CQ and AQ have been directly shown to pH trap in the DV.9 Moreover, these weakly dibasic compounds accumulate several thousand-fold so that they inhibit the target in the micromolar range,10 despite their nanomolar activities against parasites. Quinolines have been thought to inhibit the formation of Hz either via complexation with hematin or interaction with the surface of the Hz crystal in the DV.11–13
Synthetic Hz, known as β-hematin (βH), can be formed efficiently and reliably under abiotic conditions which mimic the lipid-mediated process in the DV. Several assays to identify βH inhibitors have been developed. Early examples did not include lipid mediators and employed IR spectroscopy or radiolabeled heme for detection of inhibition.14–17 More recently, neutral lipid blends (NLBs) have been used to mimic the natural process.18, 19 Huy et al.20 described the use of a detergent, TWEEN 20, which is a cheap and convenient neutral lipid mimetic, to mediate βH formation. Later Carter et al.,21 demonstrated that the detergent Nonidet P-40 (NP-40) is the best available imitator of the NLB, resulting in the highest yield of βH crystals and giving similar IC50 values for the inhibitors CQ and AQ. More importantly this well-established assay, which relies on UV-vis absorbance measurements for detection of a bis-pyridyl-heme complex,22 gave a ten-fold higher hit rate of parasite-active compounds among the βH hits, than any previous methods used in screening efforts.23–25 In addition, the NP40 assay procedure lends itself well to HTS, as it incorporates non-interfering aqueous acetone at pH 7.4 in the final plate development stage to handle poorly soluble compounds, which had previously resulted in false negatives using the pyridine detection method.22 HTS was conducted using this approach at Vanderbilt University with AQ and CQ as standards (βH IC50s of 21 and 53 μM respectively) to discover novel βH inhibitors, active against the D6 strain of P. falciparum.25 Of the 530 βH inhibiting hits, 73 showed parasite IC50s ≤ 5 μM and fourteen primary scaffolds were identified. Subsequent cell fractionation studies to validate Hz as the target revealed dose-dependent increases in parasite free heme. In particular, several compounds, representing two of the primary scaffolds, caused extraordinarily high levels of free heme at 2.5 times the relevant IC50 (30–50%) compared to the levels seen with CQ and other quinoline-containing scaffolds (8–14%).7, 26 Two of these compounds contained either a pyridylbenzamide or phenylbenzamide moiety. The benzamide chemotype was found to occur 33 times among the βH hits, with 13 compounds displaying parasite growth inhibition. Of the fourteen scaffolds, this subset represented the second highest hit rate (45%) for parasite activity of the βH hits after the quinolines. Phenylbenzamides have been reported to show antimalarial activity against the CQ-sensitive 3D7 strain of P. falciparum in a screen published by GSK in 2012.27 One potent benzamide analogue, a kinase inhibitor, was later optimized for solubility and parasite selectivity by Novartis.28 Moreover, two phenylbenzamides in the MMV Malaria Box have been reported to be βH inhibitors in the NP-40 assay.29
While a number of antimalarial chemotypes have previously been reported to inhibit βH formation,25, 29–39 detailed systematic investigation of the relationships between structure and βH inhibition, between βH inhibition and in vitro antimalarial activity and of cellular mechanisms have so far been confined to quinolines. Previous studies on quinolines suggest that suitable compounds containing electron deficient aromatic ring systems can interact strongly with Fe(III)PPIX resulting in inhibition of βH formation.35, 40 However, recent evidence shows that βH inhibition likely occurs via binding of the inhibitor to the crystal surface, rather than free Fe(III)PPIX in solution.13 This probably explains why no convincing direct correlation has been observed between Fe(III)PPIX binding in solution and either βH inhibition or parasite activity for the quinolines.41 Where Hz-inhibiting compounds can access and accumulate in the parasite DV a build-up of cytotoxic free Fe(III)PPIX occurs, resulting in parasite death.7, 41 Extension of such studies to non-quinolines is a prerequisite for a deeper and more general understanding of this pathway and its potential as a target for drug discovery and development.
In this study, benzamide derivatives, starting from the scaffold core, N-phenylbenzamide, have been purchased or synthesized in order to explore structure activity relationships (SARs). The study has been designed to probe the effect of electron deficient versus electron rich aromatic rings on βH inhibition, the relationship between βH inhibition and in vitro antimalarial activity and the effects of inhibitors on cellular Fe(III)PPIX levels in the parasite. In addition, the hypothesis that introducing basic side chains should drastically improve accumulation and hence increase potency in the parasite has been tested and the possible role of Fe(III)PPIX association in accumulation has also been explored. It must be emphasized that the primary aim of this study was to gain an understanding of the mechanistic aspects of βH inhibitors in a non-quinoline system, rather than to optimize the activity and medicinal properties of the series. Nevertheless, preliminary in silico and in vitro pharmacological studies have also been carried out on the most potent analogues to investigate the potential of this series for drug optimization.
2. RESULTS AND DISCUSSION
2.1 Scaffold selection
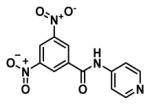
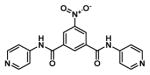
One of the major challenges of drug development is selecting appropriate research routes after HTS, particularly when it comes to identifying potential leads. In our case, HTS revealed a monobenzamide (Figure 1a) and dibenzamide, 5-nitro-N,N′-di(pyridin-4-yl)benzene-1,3-dicarboxamide (Figure 1b), which inhibited βH formation with over three times the potency of CQ. The dibenzamide further showed a promising parasite activity of 0.19 μM against the D6 strain, the third most potent compound of all 171 actives in the screening effort. This compound was shown to be non-cytotoxic against the Chinese hamster ovarian (CHO) cell line with an IC50 >275 μM. Besides potent activity and lack of cytotoxicity, the benzamides were attractive from a synthetic point of view owing to the ease with which derivatives could be prepared, as well as their structural difference from many of the Hz inhibiting antimalarials that contain fused ring systems such as the quinolines and neocryptolepines.30, 31, 42, 43 Structural alerts for potential metabolic stability were confirmed in silico using MetaSite,44 which identified the reduction of the nitro group, N-oxidation of the pyridyl moiety and amide hydroxylation as potential sites of metabolism in the liver. However, VolSurf 45 predicted the benzamide derivatives from the HTS to have suitable metabolic stability (within the 95% confidence boundary) when mapped relative to the chemical space of known drugs. Furthermore, very successful drugs such as Lipitor, Lidoderm and paracetamol contain benzamide moieties and we hypothesized that replacing the nitro and pyridyl moieties with other functionalities could overcome potential metabolic weaknesses. Other models within VolSurf predicted acceptable CACO2 permeation, protein binding, solubility and volume of distribution within the 95% confidence boundary of known drug space.
Figure 1.

Examples of parasite-active hits found in the HTS. a) The mono-pyridylbenzamide, 3,5-dinitro-N-(pyridin-4-yl)benzamide (VU0358764) and b) the di-pyridylbenzamide, 5-nitro-N,N′-di(pyridin-4-yl)benzene-1,3-dicarboxamide (VU0001281).25
2.2 SAR analysis for βH inhibition of the monobenzamide derivatives
The scaffold was deconstructed down to the core, N-phenylbenzamide and this as well as simple derivatives were purchased in order to identify the smallest substructure for which βH activity is observed (IC50 cutoff < 200 μM for activity). The purchased compounds 1 – 6 did not display activity against βH formation (IC50 > 5500 μM). These six compounds contained either an unsubstituted ring A (1) or a substituent in the para position of ring A (2–6) as shown in Table 1. Purchased compounds 7 – 10, all possessing a halogen substituent in meta and para positions demonstrated an interesting phenomenon at very high (mM) concentrations where upon addition of Fe(III)PPIX, a pink precipitate rapidly formed, indicative of low spin Fe(III)PPIX complex formation in a similar manner to that of pyridine-Fe(III)PPIX coordination in the βH assay. Coordination of these compounds to the central Fe atom of Fe(III)PPIX was believed to be via the N of their pyridyl substituent which possesses added nucleophilicity over that of pyridine owing to some amide resonance to the para pyridyl N. The precipitation of this complex was probably a result of the lipophilic influence of the halogen(s) with 9, containing two chloro substituents, showing the effect at the lowest compound concentration and 7, with only one p-chloro, showing the effect at five times higher concentration.
Table 1.
Purchased benzamides for core structure analysis. All the compounds were inactive and pyridylbenzamides 7–10 formed a pink precipitate upon monodentate coordination to the Fe center at the indicated concentration range when added to a solution of 100 μM Fe(III)PPIX and 30.5 μM NP40 at pH 4.8.
| Code | Concentration range at which precipitate forms (μM) |
|---|---|
| 1 – 6 | N/A (Inactive ≤ 5500 μM) |
| 7 | 4500–5500 |
| 8 | 1300–2300 |
| 9 | 700–1300 |
| 10 | 1300–2300 |
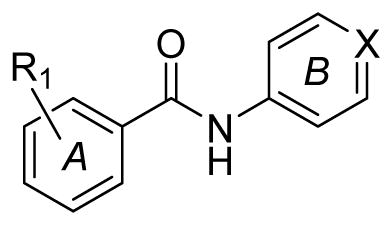
1: R1 = H; X = N
2: R1 = 4-NO2; X = CH
3: R1 = 4-NO2; X = N
4: R1 = 2,4-di-NO2; X = N
5: R1 = 4-CH3; X = N
6: R1 = 4-OCH3; X = N
7: R1 = 4-Cl; X = N
8: R1 = 3-Cl; X = N
9: R1 = 3,4-di-Cl; X = N
10: R1 = 3-CF3; X = N
The strategy for the synthesis of monobenzamides was aimed to determine the effect of an ortho substituent on ring A and whether or not a phenyl ring was tolerated as ring B, leading to generation of compounds 12a–e (Figure 2).
Figure 2.

Synthesis of mono-benzamide derivatives. *Triethylamine (Et3N) was only added in the synthesis of the pyridylbenzamides (12c–e).
The SAR analysis revealed that, for the mono-benzamides two nitro groups in the meta positions, as opposed to ortho or para, were favorable for inhibition of βH formation (Table 2) and that both phenyl or pyridyl for R2 were tolerated. The 4-pyridyl analogue (12c) was most active while the 2-pyridyl (12e) was the least active.
Table 2.
βH inhibition activity for the synthesized monobenzamides
| Compound | R1 | R2 | βH inhibition IC50 (μM) |
|---|---|---|---|
| 12a | 2-nitro | phenyl | >1000 |
| 12b | 3,5-dinitro | phenyl | 22 ± 1 |
| 12c | 3,5-dinitro | 4-pyridyl | 10.9 ± 0.3 |
| 12d | 3,5-dinitro | 3-pyridyl | 13 ± 1 |
| 12e | 3,5-dinitro | 2-pyridyl | 134 ± 8 |
| CQ | - | - | 31.5 ± 0.5 |
The precise mechanism by which βH inhibitors inhibit crystal growth is still a matter of debate. Over the past two decades it has been proposed that inhibition by quinoline antimalarials is via direct drug complexation to hematin in solution,14, 16, 46 by binding to the fastest growing faces of the βH crystal,11, 47, 48 or via a drug-heme complex capping the Hz crystal to block sites of further crystal growth.49 Nevertheless, all the proposed mechanisms involve an interaction between the drug compound and Fe(III)PPIX. Recently, de Villiers et al.12 have reported crystal structures of quinidine-heme (QD-Fe(III)PPIX) and QN-Fe(III)PPIX complexes, which demonstrate that three key interactions are involved in heme binding by these particular drugs, namely coordination, hydrogen bonding and π-π stacking. For QD and QN, hydrogen bonding occurs between the protonated quinuclidine nitrogen and the propionate group of Fe(III)PPIX. Another study showed that the interaction between Fe(III)PPIX and dihydroxyxanthones features hydrogen bonding via their hydroxyl groups, as well as π-π stacking and carbonyl-Fe coordination.50 Most of the benzamides possess features which are capable of binding to Fe(III)PPIX via all three of these interactions, specifically, a pyridyl N for coordination, an amide for hydrogen bonding and aromatic rings for π-π stacking. However, 12b does not possess a pyridyl moiety for coordination to the Fe(III)PPIX center and yet retains a low IC50, ruling out coordination as a key interaction for βH activity. Furthermore, the lack of hydrogen bonding substituents in 12b–e suggests that this interaction can only occur via the amide. Since many of the compounds with amides, including all the purchased benzamides, were inactive, an amide hydrogen bonding interaction was considered very unlikely to be the crucial determinant of βH inhibition activity. By a logical process of elimination, π-π stacking was left as the major interaction leading to inhibitory activity. Although the exact requirements for π-π stacking are still not clear, it was hypothesized that having larger planar aromatic molecular surfaces would increase the strength of the interaction with either hematin or Hz. The ability of the molecule to lie flat would therefore be expected to influence the βH inhibition activity. This hypothesis was considered to be the only feasible explanation for the observed SARs.
The π-π stacking mechanism was consistent with the structure-activity data for pyridyl derivatives 4 with 2,4-dinitro groups (inactive) vs 12c with 3,5-dinitro substituents (active) and phenyl derivatives, 12a with 2-nitro groups (inactive) vs 12b with 3,5-dinitro substituents (active). In silico internal energy calculations carried out using Gaussian software51 demonstrated that the barrier to rotation around the bond linking the carbonyl group to the phenyl ring for 4 possesses minimum energy conformers with torsion angles about the C=C-C=O bond of 70° and −110°. For the molecule to be planar, the torsional angle would be 0° or ±180°. The steric strain energies of these flat conformers are far greater than the thermally accessible energy 3kT indicated in Figure 3a. If the molecule is only able to bind to Fe(III)PPIX when in the planar conformation, this hypothesis would explain the lack of βH inhibition activity for 4. This hypothesis is also consistent with the positive activity data for 12c which is able exist in a planar conformation (±180°) below 3kT (Figure 3b) owing to the low steric constraint caused by meta as opposed to ortho-nitro substituents. It may also explain why 2e with a 2-pyridyl group had lower activity than the 3-pyridyl and 4-pyridyl analogues due to the close proximity of the negative ortho nitrogen lone pair to the electron cloud on the carbonyl oxygen, which is likely to affect the conformational preference of 12e.
Figure 3.

Free energy diagrams for rotation around the indicated bond where 0° or ±180° represents the flat conformation for (a) purchased compound 4 (βH inactive – shown in green) compared to synthesized compound 12c (βH active – shown in blue) and (b) compound 12c (βH active). The value 3kT represents the thermally accessible energy value at 37 °C. Note the difference in scale of the y-axes (a) and (b).
A summary of the SARs for the monobenzamides is given in Figure 4. These results demonstrate the minimum requirements for βH activity of the benzamides, which were then applied in the synthesis of di- and tri-benzamide derivatives. Most importantly, only compounds with substituents in the meta position on ring A were chosen for further investigation, since substituents in the ortho and para positions were not tolerated for βH inhibitory activity. Furthermore, 2-pyridyl was excluded for the R2 substituent since the results for the monobenzamides suggested that the βH inhibition activity was substantially lower in this analogue. The effect of different R2 substituents on the SARs was further investigated for the dibenzamides.
Figure 4.

βH inhibitory SARs for the monobenzamides.
2.3 SAR analysis for βH inhibition of the dibenzamide derivatives
Although two of the monobenzamides were able to inhibit βH inhibition with comparable activity to AQ, the parasite data from the HTS indicated that dibenzamides were more effective at preventing parasite growth than the monobenzamides. For this reason, a focused library of dibenzamides was synthesized. In addition, the 4-pyridyl parent compound was resynthesized (15a) along with 3-pyridyl (15b) and phenyl (15c) derivatives (Table 3). In addition to varying the type of ring B (R2 substituent), the substituent on ring A (R1) was also varied from the parent nitro to cyano, methyl, t-butyl, hydrogen or methoxy, resulting in derivatives 15b – 15n. These substituents were chosen for their range of electron-withdrawing vs releasing as well as hydrophobic vs hydrophilic properties, which excluded the halogens, since the chloro derivative was shown to be inactive in the purchased mono-benzamides. In addition, replacing X = CH with X = N resulted in derivatives with a pyridyl central ring as opposed to a phenyl (15o and 15p) all of which are defined in Figure 5, along with the tribenzamide 20.
Table 3.
βH inhibition activities for the di-benzamides 15a–15p and tri-benzamide 20.
| Compound | R1 substituent | βH inhibition IC50 (μM) |
|---|---|---|
| 15a | Nitro | 4.3 ± 0.1 |
| 15b | 4.7 ± 0.7 | |
| 15c | 7.0 ± 0.2 | |
|
| ||
| 15d | Cyano | 6.34 ± 0.09 |
| 15e | 4.4 ± 0.2 | |
|
| ||
| 15f | Methyl | 8 ± 1 |
| 15g | >1000 | |
|
| ||
| 15h | tert-Butyl | 13.3 ± 0.7 |
| 15i | 6.8 ± 0.1 | |
| 15j | >1000 | |
|
| ||
| 15k | H | 507 ± 33 |
| 15l | >1000 | |
|
| ||
| 15m | Methoxy | 22 ± 1 |
| 15n | > 1000 | |
|
| ||
| 15o | H (X = N) | 260 ± 7 |
| 15p | >1000 | |
|
| ||
| 20 | N/A | 21 ± 1 |
|
| ||
| CQ | N/A | 31.5 ± 0.5 |
Figure 5.

The synthesized dibenzamides, analogues of the hit compound 15a, and the tribenzamide 20. *Triethylamine (Et3N) was only added in the synthesis of the pyridylbenzamides.
Dibenzamides 15a, 15b and 15c displayed similarly potent βH IC50 values (2.9–7.0 μM), four to ten times more active than CQ and at least three times the activity of the monobenzamide analogues (Table 3). This improved activity is in agreement with the hypothesis that larger planar surfaces result in improved βH inhibition due to increased π-π stacking ability. In addition, the IC50 reduction for the dibenzamides parallels the 2-fold improvement in βH inhibition activity seen for the di-quinolines, piperaquine and hydroxypiperaquine compared to mono-quinoline CQ.52 The cyano derivatives (15d and 15e) maintained excellent βH activity about six times that of CQ for both the pyridyl and phenyl rings. However, for the methyl (15f and 15g), t-butyl (15h–j), hydrogen (15k and 15l) and methoxy derivatives (15m and 15n), only those with pyridyl rings at R2 showed activity, while those with a phenyl ring displayed no activity below 1000 μM. Compounds 15o and 15p with a central unsubstituted pyridyl ring and R1 = H showed weak activity for the pyridyl derivative and no activity below 1000 μM for the phenyl derivative. The tribenzamide 20 showed good activity, slighter better than that of CQ.
In general, a compound with a flat, hydrophilic, electron withdrawing group such as NO2 (15a–c), CN (15d, e) or benzamide (20)) at R1 was able to maintain activity with either a pyridyl or phenyl ring at R2. Conversely, when R1 was H or a more electron releasing group, only the pyridyl derivative(s) were active and those with phenyl at R2 showed no ability to inhibit βH formation even at a relatively high concentration. This may indicate that the molecules have two possible orientations of interaction with Fe(III)PPIX giving rise to βH inhibition. The first involves π-π stacking with the central ring, which can only take place with the flat, electron withdrawing nitro and cyano groups. The second involves an interaction with the aromatic substituent R2, which appears not to occur in the case where R2 is phenyl as opposed to pyridyl, similar to that for naphthalene analogues of the 4-aminoquinolines, which also lack βH inhibition activity.53 This may be as a result of the relative electron richness of the phenyl as opposed to the pyridyl ring, with the latter being electron deficient owing to the electronegative nitrogen atom. Interestingly, the high IC50 of 15o, the X = N derivative with R2 = pyridyl, can be explained in a similar manner to that of 12e. Since both these compounds have an ortho-pyridyl ring B, the influence of the ortho nitrogen in close proximity to the amide results in a decreased probability of π-π interaction, either by causing the molecule to be less planar, or by adding too much electron density at that particular site of the molecule. The SARs for the dibenzamides are shown in Figure 6a. Note that the planar and electron withdrawing nitro and cyano substituents at R1 follow a different SAR scheme to that of the methyl, t-butyl, H and methoxy substituents. Although the exact role of the R1 substituent is not clear, it is possible that this substituent forces or directs the benzamide rings into the groove on the crystal face of βH were it binds, accounting for the strong inhibitory activity (Figure 6b). Previous studies on xanthones and porphyrins as βH inhibitors also reported significant interactions between the planar aromatic rings of Fe(III)PPIX and the inhibitor.39, 54 However, additional interactions not present in the benzamides, involving carbonyl or hydroxyl groups to bind the iron of heme for enhanced activity, were also described.
Figure 6.

(a) βH inhibitory SARs for the dibenzamides and (b) a possible mode of interaction to explain the observed βH inhibition activities. In this example, the dibenzamide, 15h interacts with the fastest growing Hz crystal face7 with the R1 t-butyl moiety directed outwards. Graphics were supplied by R. Muller and generated using Materials Studio.55
2.4 Activity of benzamides against P. falciparum in culture
The synthesized benzamide derivatives were tested for inhibitory activity against the CQ-sensitive NF54 strain of P. falciparum. The monobenzamides 12b–e displayed a range of IC50 values from 2.3 to 22 μM and 12a, which was not a βH inhibitor, showed no activity below 41.3 μM, the maximum concentration tested (Table 4). A good correlation was identified between the inverse of the βH inhibition IC50 and inverse parasite growth inhibition IC50, with an r2 of 0.88 (Figure 7a).
Table 4.
CQ-sensitive parasite activities for the synthesized mono-, di- and tri-benzamides.
| Compound | Derivative info | NF54 parasite IC50 (μM) | D10 parasite IC50 (μM) |
|---|---|---|---|
| 12a | Monobenzamides | >41.3 ± ND | ND |
| 12b | 5 ± 1 | ND | |
| 12c | 2.3 ± 0.3 | 2.4 ± ND | |
| 12d | 5 ± 1 | 2.3 ±ND | |
| 12e | 22 ± 4 | 24 ± 5 | |
|
| |||
| 15a | R1 = Nitro | 0.7 ± 0.2 | 0.9 ± 0.1 |
| 15b | 1.32 ± 0.04 | ND | |
| 15c | 1.4 ± 0.2 | ND | |
|
| |||
| 15d | R1 = Cyano | 1.9 ± 0.2 | 2.0 ± 0.2 |
| 15e | ND | 5.2 ± ND | |
|
| |||
| 15f | R1 = Methyl | 2.5 ± 0.6 | ND |
| 15g | 146 ± 1 | ND | |
|
| |||
| 15h | R1 = Tert-butyl | 1.6 ± 0.2 | ND |
| 15i | 0.6 ± 0.1 | 0.7 ± 0.1 | |
| 15j | 8.1 ± 0.5 | ND | |
|
| |||
| 15k | R1 = H | 9 ± 4 | ND |
| 15l | >314 | ND | |
|
| |||
| 15m | R1 = Methoxy | 3.0 ± 0.5 | ND |
| 15n | 37 ± 13 | ND | |
|
| |||
| 15o | R1 = H (X = N) | >315 | ND |
| 15p | >315 | ND | |
|
| |||
| 20 | Tri-phenylbenzamide | 81 ± 12 | >42 |
|
| |||
| CQ | Standard | 0.008 ± 0.002 | 0.021 ± 0.002 |
Figure 7.

A linear correlation between the inverse of the βH and parasite growth IC50 values for the CQ-sensitive NF54 strain for (a) the monobenzamides (r2 = 0.875; P = 0.0195) and (b) the mono-, di- and tri-benzamides combined (r2 = 0.68, P < 0.0001). The statistics improve when derivative 15i (red circle) is excluded (r2 = 0.84, P < 0.0001). Measurements of βH inhibition were in triplicate and parasite growth inhibition in at least duplicate.
The original hit compound, 15a (Figure 1b), was resynthesized and tested in the available CQ-sensitive NF54 strain. The activity was found to be three times weaker than in the D6 strain used in the HTS, with an IC50 of 0.7 μM. The 3-pyridyl and phenyl analogues, 15b and 15c, exhibited a further 2-fold lower activity. The other derivatives were also tested in the NF54 and/or D10 strains of P. falciparum. Where comparisons were made, both strains gave similar IC50 values for tested compounds (Table 4). Replacing the nitro substituent (R1) of 15a with cyano caused a 3-fold increase in IC50 to 1.9 μM for 15d, the pyridyl derivative. A further IC50 increase to 5.2 μM was observed for the phenyl derivative 15e. A similar reduction in activity was noticed when replacing the R2 pyridyl ring with a phenyl group for the derivatives where R1 is methyl (15f and 15g), t-butyl (15i and 5j), hydrogen (15k and 15l) or methoxy (15m and 15n). Both compounds with X = N (15o and 15p) were inactive up to 315 μM in the parasite, most likely because they did not possess sufficient βH inhibition activity. Only compound 15i, the t-butyl derivative, showed a slight improvement in parasite activity relative to the parent compound, with IC50 of 0.6 μM. The trend in activities for the 4-pyridyl vs 3-pyridyl vs phenyl analogues of the t-butyl derivatives (15h–j) was different from that of the nitro derivatives (15a–c), in that the order of potency between the 3-pyridyl vs 4-pyridyl was reversed. In all cases, however, the phenyl derivatives were less active against P. falciparum. The tribenzamide, 20, showed very poor parasite activity despite being a good βH inhibitor. This may have been as a result of its excessive hydrophobicity (predicted logP = 5.21).
As with the monobenzamides alone, inverse activity of the βH and NF54 parasite IC50s were correlated when all the benzamides were taken into account, in contrast to the 4-aminoquinolines which do not show convincing correlations between these activities.33, 35 The relationship was statistically significant with P < 0.0001. Furthermore, a moderately good r2 value of 0.68 was observed for the correlation, which increased to r2 = 0.84 upon removal of the point corresponding to compound 15i, which was an apparent outlier (Figure 7b). This compound showed a better parasite activity than expected based on its βH inhibition IC50. Possible reasons for this could be that its properties differ markedly from the other compounds, that there is favorable uptake in the parasite, or that the βH IC50 measured for this compound in the NP-40 assay does not accurately represent its Hz inhibitory activity in the parasite. Nevertheless, the correlations in Figure 7 suggest that the benzamides do indeed exert their antiparasitic action via inhibition of intracellular Hz formation.
This mechanistic hypothesis was validated using a cellular fractionation assay to convincingly demonstrate that the benzamides cause concentration dependent increases in free heme and corresponding decreases in Hz within whole cell parasite cultures. In this assay, for which full validation and details have been reported previously,26 parasitized red cells are incubated with the test compound for 32 h, the trophozoites are isolated, lysed and spun down to separate the Hb-containing supernatant. The resulting precipitate consists of two fractions; firstly, the non-Hz heme, consisting of free heme or heme associated with drug, lipids or proteins, which is readily solubilized in SDS buffer and pyridine for detection; and secondly, the Hz fraction which remains completely insoluble until dissolved in NaOH and SDS, and which is then neutralized before being independently measured via the pyridine detection procedure.22 Three benzamides were analyzed using this cell fractionation assay. Two of the selected compounds (12d and 15i) showed excellent βH inhibition activity in the NP-40 assay, while one compound was not a βH inhibitor (15j). The latter served as a negative control. The monobenzamide 12d and the dibenzamide 15i were chosen because they have relatively weak and strong parasite growth inhibition activity respectively. This was desirable so as to observe whether free heme levels had an influence on this activity. No significant change in the free heme or Hz levels with increasing concentration was observed for 15j (Figure 8a). This was in agreement with the NP-40 assay for βH inhibition and provided strong support for the technique for finding Hz inhibiting compounds used in this study. It would appear that this compound acts weakly on an alternate parasite target. By contrast, 12d and 15i showed a large increase in the amount of non-Hz heme (Δheme ~40 fg/cell at 2.5 times the IC50) with a corresponding dramatic decrease in Hz (ΔHz ~40 fg/cell at 2.5 times the IC50) within the parasite relative to the control (Figure 8b, c). The significant changes in free heme and Hz levels for these benzamides suggested that the precise manner by which they cause heme-related toxicity differs from that of CQ, since much smaller changes in free heme and Hz levels by ~5 fg/cell and ~15 fg/cell respectively were observed at 2.5 times the CQ IC50.26 Other quinoline antimalarials, such as AQ, QN and mefloquine showed even smaller changes than CQ.7 This finding demonstrates that higher levels of free heme are required for disrupting parasite growth in the case of the benzamides compared to CQ and other quinoline antimalarials.
Figure 8.


Hz and free heme levels from cultured P. falciparum treated with varying concentrations of benzamides a) 15j, a negative control showing no statistically significant difference in Hz or free heme as a function of dose, b) 12d, a monobenzamide and c) 15i, a dibenzamide exhibit strong dose dependent changes. Asterisks indicate statistical significance relative to no drug (2-tailed t-test): *P < 0.05; **P < 0.01; ***P < 0.001, n = 3.
In the case of the βH inhibitors, the free heme curves and the parasite survival curves with varying test compound concentrations crossed over close to the D10 IC50 of 12d (Figure 9a) and 15i (Figure 9b) respectively, strongly supporting the hypothesis that they target the Hz formation process. Direct evidence that these compounds disrupt the process of Hz crystallization in the parasite is provided by the transmission electron microscopy (TEM) images in Figure 10, demonstrating clear morphological changes in parasites treated with 15i relative to the untreated cells. When dosed at 2.5 times the IC50 and incubated for 32 h, intracellular Hz crystals appeared within swollen DVs as irregularly scattered and shapeless clumps surrounded by a distinct electron dense penumbra, in contrast to the well-formed, tightly-packed and sharply delineated crystals observed in the control. Swelling of the DV and a lack of crystal uniformity have previously also been observed in TEM images of CQ treated cells.7, 56, 57
Figure 9.

Dose response curves for the non-Hz heme and the % parasite survival, which cross near the parasite survival IC50 for compounds a) 12d and b) 15i. Scales adjusted so that the IC50 for parasite survival coincides with 50% of the final value for increase in free heme (indicated by the dotted line).
Figure 10.

TEM images of human erythrocytes infected with P. falciparum showing a) an untreated control and b) two examples of cells treated with 15i. The thick white arrows show the swollen DVs of treated cells. The brown arrows indicate the Hz crystal in the DV which has been enlarged and adjusted (20% brightness, 20% contrast in each) for clarity to readily discern the difference between crystal forms in untreated vs treated samples.
2.5 Attempts to improve accumulation through pH trapping
Fitch et al.58 showed that the accumulation of CQ is sensitive to the pH gradient between the parasitized erythrocyte and the medium by varying the pH of the latter. Yayon et al.59 reported high pH-dependent accumulation levels of CQ in the DV, investigated by flow dialysis, in which diffusion across a membrane can be measured60 as well as the tracing of CQ via fluorescence or a 14C labelled analogue.61 Millimolar concentrations of CQ were predicted to exist in the DV at therapeutic doses, accounting for the high activity of the drug. In the 1990s, more sophisticated methods allowed the proton gradients to be varied, by decreasing the extravacuolar pH with the acidifying agent NH4Cl or via inhibitors of vacuolar proton pumps.62,63 Accumulation measurements under these conditions showed that lowering the external pH results in less accumulation and hence these studies concluded that CQ accumulates in the acidic DV as a result of its weak base properties. This pH-dependent accumulation can be explained via the phenomenon of pH trapping, whereby a neutral species passively diffuses into the acidic DV and is protonated to form a less lipophilic (lower logD) charged species, which is then unable to cross the membrane. Designing a compound with greater basicity may be expected to enhance the pH trapping effect and result in improved parasite activity. Indeed, upon addition of basic amino side chains to xanthones, increased activities in the D6 parasite strain as well as heme affinities were previously reported.64 This is also believed to be the case for the 4-aminoquinolines AQ and CQ. It has previously been reported that 4-amino-7-chloro-quinoline is able to inhibit βH formation and shows a D10 parasite IC50 of 3.8 μM.41 Upon attachment of a variety of tertiary amino containing side chains at the 4-amino position, βH inhibition was maintained and parasite activity was greatly enhanced by approximately 100-fold. According to the study, this was probably as a result of improved accumulation in the DV. In addition, it was proposed that rational design could be employed to develop novel antimalarials from scaffolds that form π-π complexes with Fe(III)PPIX and inhibit βH formation.41
A benzamide derivative where the R2 substituents were each N,N-dimethyl-phenylmethanamine (17) was expected to possess suitable properties for this study, namely, a molecular weight below 500 g/mol and a positive clogD value of 1.12 at neutral pH (Figure 11). This compound was synthesized in order to probe the effect of greater basicity and pH trapping for the benzamide series.
Figure 11.

Preparation of 17 via reductive amination from 16.
Compound 17 was tested for both βH inhibition and parasite activity. In the NP40 assay, the compound maintained potent βH activity with an IC50 of 7.9 ± 0.2 μM, appearing to be a stronger inhibitor than AQ or CQ. However, upon assessing the activity against P. falciparum of this more basic derivative, which was expected to accumulate to a greater extent in the parasite DV than 15a and hence possess enhanced activity, no improvement was found. An NF54 parasite IC50 of 1.5 ± 0.2 μM was obtained. This activity was the same as that of the phenyl derivative, 15c, within the error margin, suggesting that no significant accumulation had taken place. This lack of effect was especially striking when compared with the predicted accumulation that would have been expected to take place due to pH trapping assuming achievement of equilibrium. The predicted VAR due to pH trapping alone was calculated using the Henderson–Hasselbach equation (Eq. 1) at an external pH of 7.4 (pHe) and intravacuolar pH of 4.8 (pHv) for a compound (B) with two protonatable sites.
| Eq. 1 |
Using the predicted pKa values of 9.12 and 8.52 for compound 17, the expected VAR was estimated at ~147,000. This indicated that the derivative should have been about 147,000 times more concentrated in the DV compared with 15c, which is not able to pH trap. Since the compounds possessed the same activity, two hypotheses were suggested; either compound 17 does not effectively accumulate, resulting in a higher required concentration to inhibit parasite growth, or compound 17 accumulates but is not able to inhibit Hz formation in the DV. The latter hypothesis was ruled out upon observing an increase in non-Hz heme in the cellular fractionation assay from 7% to 41% at the IC50 of 17, with a simultaneous decrease in the % Hz from 89% for the control to 53% (see supporting information S4.2). With the biological target for the basic analogue strongly supported, the ability of 17 to accumulate in parasitized red cells relative to the hit compound was investigated by measuring the inoculum effect. The effect is only detected when accumulation of the drug occurs in the organism, in this case, the parasitized erythrocyte.65 An apparent decrease in activity with greater parasitemia can be rationalized by the depletion of total drug concentration in the extracellular medium upon incubation with infected red blood cells, due to a significant uptake by the parasite.66, 67 By measuring the changes in IC50 with varying initial parasitemia levels, the amount of drug accumulation can be compared for the pyridyl derivative 15a versus the weak base derivative, 17. An insignificant inoculum effect and cellular accumulation was observed for this derivative, since the IC50 of the compound remained essentially constant over the range of tested parasitemias and the slope was not statistically significant. On the other hand, the pyridyl derivative 15a showed larger accumulation ratios in the cell as demonstrated by the slopes of the linear fits in Figure 12. The lack of correlation between the calculated pH trapping ratio and the observed accumulation ratio for the benzamides suggested that the mechanism of drug accumulation is much more complex, at least in the CQ-sensitive D10 strain, than the simple pH trapping model suggests. These studies further showed that the parasite activity of Hz inhibitors cannot necessarily be improved by adding basic moieties.
Figure 12.

Comparison of inoculum measurements for compounds 15a (r2 = 0.952; P = 0.0045) and 17 (r2 = 0.828; P > 0.05; not significant).
2.6 Possible role of Fe(III)PPIX association in cellular accumulation
Although pH trapping could not directly account for the lower accumulation of the weakly basic benzamide relative to the pyridylbenzamide, some studies have suggested that despite the lack of correlation between Fe(III)PPIX interaction strength (K) and activity, the total cellular uptake of aminoquinolines and bis-quaternary ammonium compounds nonetheless occurs via binding to Fe(III)PPIX.68–71 Furthermore, a correlation between heme affinity and D6 parasite activity was reported for 6-bis-ω-N, N-diethylaminoalkoxyxanthones, showing a dramatic improvement in activity (IC50s from 2.2 to 0.07 μM) with a small change in logK (logKs from 5.9 to 5.5).39 While there is no consensus as to the validity of the conclusions of these studies, the extent of Fe(III)PPIX binding for selected benzamides was investigated in an attempt to determine whether there is a correlation with cellular accumulation. This involved measurement of the association constants via titration of the compounds into a solution of 7.5 μM monomeric hematin in 40% v/v DMSO at pH 7.5. Upon titration of the test compound solution into the hematin solution, a hypochromism or convergent decrease in intensity of the characteristic monomeric hematin spectrum at 401 nm (Soret band) was observed owing to significant Fe(III)PPIX binding. The information gained from this experiment could be extended by monitoring the maximum absorbance at 405 nm as a function of test compound concentration. This allowed the association constant (K) to be calculated from a non-linear least squares fitting model for a 1:1 complex. A representative example is shown in Figure 13. It should be noted that the benzamide solutions were made up in 100% DMSO owing to their low solubility in 40% DMSO. This resulted in a small increase in total percent DMSO in the working solution over the course of the titration. However, this did not appear to affect the absorbance, and isosbestic points were observed as expected, confirming a lack of intermediates. In addition, the fit to the titration curve (Figure 13b) was consistent with 1:1 stoichiometry.
Figure 13.

Measurement of the association constant between compound 15a and Fe(III)PPIX at pH 7.5 in 40% v/v DMSO. Plots of a) the hypochromic effect on the Fe(III)PPIX absorbance spectrum with increasing compound concentration, exhibiting isosbestic points (*) and b) the maximum Fe(III)PPIX absorbance vs the concentration of compound 15a fitted to a 1:1 association isotherm.
Hematin binding experiments were also attempted for compounds 12c, 15f and 17. However, the last precipitated out of solution and useable data could not be obtained owing to its low solubility. The data relating to strength of Fe(III)PPIX binding in solution are shown in Table 5 for synthesized benzamides 12c, 15a and 15f, as well as for selected 4-aminoquinolines. The three benzamides showed similar logK values, on the order of 2-fold lower than those of the known antimalarials and 1 log unit lower than 4-amino-7-chloroquinoline. These values are consistent with the hypothesis that the benzamides associate with Fe(III)PPIX through π-π interactions alone, in accordance with the predicted logK values reported by Kuter et al.72 The linear free energy equation reported by these authors to predict the association constant due to π-π interactions with Fe(III)PPIX (logKπ–calc) incorporates the number of available π electrons (nπ) via logKπ–calc = 0.23 × nπ. Assuming fourteen available π electrons in compound 12c are able to interact with Fe(III)PPIX (six for each aromatic ring and two for the amide), the logKπ–calc is 3.22, remarkably close to the experimentally obtained value. This is despite the fact that the predicted value does not take into account substituent effects on the strength of the interaction, suggesting that they do not play a significant role in this interaction. In addition, the fact that the dibenzamides 15a and 15f do not have significantly larger logK values than the monobenzamide 12c, suggests that association does not take place over the entire molecule, which would result in a logKπ–calc of ~5 for the 22 π electrons in the dibenzamide. This study was also in agreement with the finding that monodentate coordination of protonatable groups to the Fe center is relatively weak compared to π-π interactions,72 owing to competition from protons at the DV pH, since high very concentrations of the pyridylbenzamides were required for coordination of the pyridyl N to the heme Fe at pH 4.8 (Table 1).
Table 5.
Association constants for the interaction of selected benzamides and quinolines with Fe(III)PPIX.
| Compound | logK | Binding constant ratio relative to 12c (K/K12c) |
|---|---|---|
 12c |
3.20 ± 0.36 | 1 |
 15a |
3.23 ± 0.11 | 1.1 |
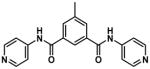 15f |
3.92 ± 0.21 | 5.24 |
| QD | 5.26 ± 0.01 | 115 |
| CQ | 5.52 ± 0.03a | 209 |
| 4-amino-7-chloroquinoline | 4.43 ± 0.01a | 17 |
Data from Egan et al.41
It should be noted that these values were calculated by assuming a 1:1 Fe(III)PPIX binding ratio. CQ has recently been shown to have a binding stoichiometry of 1:2 CQ:Fe(III)PPIX in aqueous solution, resulting in a logKobs of 13.3 ± 0.2.73 However, here the 1:1 complexes were compared in 40% DMSO and the value reported by Egan et al.41 for CQ in this medium was used.
Finally, an analogue expected to π-π stack with heme more effectively, was proposed for synthesis. This required conversion of the R2 pyridyl rings into fused aromatic rings in order to increase the number of π electrons available for interaction. Since it was intended to avoid the inclusion of a quinoline moiety and because a variety of benzimidazoles were shown to be good βH inhibitors in the HTS, a derivative where R2 was benzimidazole was suggested. This group was predicted to add a log unit to the logKπ–calc value since it contains ten π electrons, as opposed to the six of the pyridyl group. Owing to synthetic difficulties relating to the physical properties of a compound with two benzimidazole moieties, the molecule had to be desymmetrized to an analogue containing just one benzimidazole unit (Figure 14). Although this compound (18) could be synthesized and isolated, its solubility in aqueous solution and most organic solvents was poor and it could not be used in heme-binding experiments. In addition, the activities of compound 18 were found to be uninteresting. It was a moderate inhibitor of βH formation with an IC50 of 68 μM but displayed no parasite activity up to the maximum dose concentration of 249 μM.
Figure 14.

Synthesis of the unsymmetrical benzamide 18.
2.7 Preliminary cross-resistance and pharmacological studies
The two most active compounds were tested against the K1 CQ-resistant strain and found to have resistance index (RI) values below 5, suggesting little CQ cross-resistance. In addition, IC50 values for the CHO mammalian cell line showed selectivity indices (SIs) of >300 for both compounds, indicating low cytotoxicity and good selectivity for P. falciparum (see Table 6).
Table 6.
K1 and CHO data for selected benzamides
| Compound | K1 parasite IC50, μM (RI)a | CHO IC50, μM (SI)b |
|---|---|---|
| 15a | 2.86 ± 0.08 (4.1) | >275 (>393) |
| 15i | 1.7 ± 0.4 (2.8) | 185 (308) |
| CQ | 0.222 (27.8) | N/A |
Resistance index, IC50 (K1)/IC50 (NF54);
Selectivity index, IC50 (CHO)/IC50 (NF54)
Kinetic solubility was determined for the hit compound (15a) and the most potent analogue (15i). The results shown in Table 7 indicate a significant improvement in the solubility at pH 6.5 and in a simulating fasting-state biorelevant medium, pH 6.5 (FaSSIF) for 15i containing the non-planar t-butyl group (FaSSIF: 153.6 μM) versus 15a, which instead contains a planar nitro group (FaSSIF: 13 μM). This indicated favorable potential of the benzamide series for further optimization as orally-dosed drugs, given the acceptable solubility at pH 2, the approximate pH of the stomach, and in FaSSIF medium for derivatives with non-planar substituents. In general, these weakly basic compounds were most soluble in the more acidic medium owing to protonation of the pyridyl nitrogen, resulting in a cationic compound. The metabolic stability of the series was verified by in vitro microsomal studies with human, rat and mouse liver microsomes. 15a was shown to be particularly stable in all species up to the limit of detection in this assay; however, this result may not be entirely reliable owing to the incompatibility of this poorly-soluble compound with the assay conditions. Nonetheless, encouragingly the more soluble analogue, 15i, was also shown to be stable in human and rat microsomes. Its stability was diminished in mouse, but this was likely the result of the known variation in levels of CYP isoforms, substrate specificity and enzyme inhibition across species.74 These results demonstrated that the benzamine scaffold has potential for optimization of this important pharmacological parameter. (Table 7)
Table 7.
Kinetic solubility in various media and metabolic stability in human, rat and mouse liver microsomes.
| Compound | Kinetic solubility (μM) pH 2 /6.5 / FaSSIF | Liver microsomal species | Degradation half-life (min) | In vitro clearance (μL/min/mg protein) | Hepatic extraction (EH) |
|---|---|---|---|---|---|
| 15a | 199.7 / <5 / 13 | Human | >150 | <11.6 | <0.42 |
| Rat | >150 | <11.6 | <0.30 | ||
| Mouse | >150 | <11.6 | <0.33 | ||
|
| |||||
| 15i | 197.2 / 92.7/ 153.6 | Human | 141.9 | 14.1 | 0.4 |
| Rat | 93.2 | 33.5 | 0.4 | ||
| Mouse | 52.4 | 130.3 | 0.6 | ||
3. CONCLUSIONS
Screening of diverse compound libraries using the NP-40 detergent based assay allows for efficient identification of novel βH inhibiting compounds. Where previously only series of quinoline-containing compounds have been systematically analyzed to determine relationships between structure and βH inhibition as well as between βH inhibition and in vitro antimalarial activity, this study reports the first rational synthesis and investigation of a non-quinoline-based βH-inhibiting series to probe these effects. As with the quinolines, electron deficient aromatic ring systems were shown favor βH inhibition activity, at least for mono- and di-benzamide analogues capable of adopting flat conformations. However, unlike the quinoline antimalarials QD and QN, and the non-quinoline antimalarial halofantrine, which reportedly require π-π interactions, hydrogen bonds and coordination to Fe(III)PPIX,12, 75 the SARs identified in this study strongly indicate that only π-π interactions between the benzamide analogue and Fe(III)PPIX take place to inhibit βH formation. Specifically, dibenzamides without an electron withdrawing group on the central ring required pyridyl rings attached to the amide nitrogen for βH inhibition activity, while those with an electron withdrawing group tolerated phenyl rings. The lack of Fe(III)PPIX coordination and hydrogen bonding capabilities for the benzamides does not adversely affect their βH inhibition activities, however the overall strength of heme binding was generally weaker for selected mono- and dibenzamide analogues compared to QD, CQ and the hydroxyxanthones. Furthermore, direct linear correlations between the inverse of βH inhibition and the inverse of in vitro antimalarial activity were clearly observed for both mono- and dibenzamides, indicating that other factors are less important than βH inhibition for determining biological activity, as opposed to the 4-aminoquinolines, where activities are greatly influenced by substituent basicity for pH trapping and accumulation. This hypotheses was further supported via inoculum measurements of an analogue with two basic tertiary amino side chains, which demonstrated that accumulation and hence activity cannot necessarily be improved by introducing basic substituents to βH inhibiting compounds, as originally proposed.35 Nonetheless, two pyridylbenzamides showed nanomolar antimalarial activity with little CQ-cross resistance, high in vitro microsomal stability and low toxicity to mammalian cells. In addition, cellular fractionation studies showed increases in intracellular free heme, at remarkably higher levels than the known quinoline antimalarials, AQ, MQ, QN and CQ. This suggests that larger concentrations of free heme, or possibly of a free heme-drug complex are required to cause P. falciparum toxicity leading to cell destruction, compared with the quinolines. Ongoing studies to validate a hypothesis which explains the differences in free heme levels, heme binding, accumulation and activity between the benzamides, other HTS hits and the standard quinoline antimalarials are being pursued.
4. EXPERIMENTAL
General methods
Commercially obtained chemicals (AR or higher grade) were purchased from Sigma-Aldrich or Kimix Chemicals. Nonidet P-40 was acquired from Pierce Biotechnology, Rockford, IL, USA. Compounds 1–10 were purchased from Vitas-M Laboratory. All reactions were monitored by thin layer chromatography (TLC) using silica gel plates (Merck F254 aluminum-backed). TLC plates were visualized with ultraviolet light (254 nm) and, where an amine was present, plates were stained with ninhydrin spray (0.2% w/w in EtOH). Silica gel flash column chromatography was carried out with Fluka 60: 70-230 mesh on either a Biotage Isolera One Flash Chromatography System or using a combination of a D-star DVW-10 variable wavelength detector with a Teledyne ISCO fraction collector. Melting points were measured using a Reichert-Jung Thermovar hot stage microscope. Proton (1H) and carbon (13C) NMR spectra were recorded using a Varian Mercury spectrometer (300 MHz for 1H), a Bruker 300 (at 300.08 MHz for 1H) or a Bruker Ultrashield 400 Plus (at 399.95 MHz for 1H and 100.64 MHz for 13C) spectrometer. NMR experiments were performed in deuterodimethyl sulfoxide (DMSO-d6), deuterochloroform (CDCl3), deuteromethanol (MeOD) or deuteroacetone (acetone-d6) with internal standards at δH = 2.50, 7.26, 3.31 or 2.05 ppm respectively for 1H NMR and δC = 40.05, 77.16, 49.00 or 29.8 and 206.26 ppm respectively for 13C NMR. All chemical shifts were reported in ppm and J coupling values in Hz. Electron ionization mass spectrometry was recorded using a JEOL GC mate II single magnetic mass spectrometer. High resolution mass spectrometry (HRMS) was performed on a Time-of-flight (TOF) Waters Synapt G2 instrument using leucine encephalin as a standard. All mass spectra for the benzamide series were recorded using the electrospray negative (ES−) technique and introduction of the sample was via an ESI probe injected into a stream of MeOH. Purity of final compounds (≥ 95%) was confirmed using high performance liquid chromatography (HPLC) carried out on an Agilent Technologies 1220 Infinity LC (G4288C) in UV mode at 250 nm with a C18 reverse phase column in HPLC grade acetonitrile (ACN) and double distilled deionized Millipore® Direct-Q water.
General procedure for the synthesis of the monobenzamides 12a–b
Aniline and the relevant acid chloride (11a or 11b) were stirred in dry pyridine under N2 at room temperature (RT). Upon completion of the reaction, indicated by TLC analysis, the organic components were extracted in EtOAc (3 × 20 ml) and then washed with 1M HCl to remove excess pyridine. The organic fractions were combined, stirred with MgSO4 and concentrated by evaporation under reduced pressure. The product was dried under high vacuum for 2 h to yield the desired product.
2-Nitro-N-phenylbenzamide (12a).76
Scale: aniline (186 mg, 2 mmols) and 2-nitrobenzoyl chloride (371 mg, 2 mmols), stirred for 1 h. Yield of 12a: 477 mg, 99%. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 12a as off-white crystals: Mp 149–151 °C; Lit Mp76 155–156 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 10.62 (br. s, 1H), 8.13 (td, J = 0.70, 7.89 Hz, 1H), 7.86 (m, 1H), 7.76 (m, 2H), 7.66 (m, 2H), 7.35 (m, 2H), 7.11 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 164.6, 147.0, 139.3, 134.5, 133.2, 131.4, 129.8, 129.3, 124.7, 124.4, 120.2; HRMS-ES−: Observed 241.0604 (M-H)−; Calculated 241.0613 for C13H9N2O3; HPLC: 96.9%.
3,5-Dinitro-N-phenylbenzamide (12b).77
Scale: aniline (200 mg, 2.1 mmols) and 3,5-dinitrobenzoyl chloride (606 mg, 2.6 mmols), stirred for 2 h. Yield of 12b: 505 mg, 84% as a pale-yellow solid. Recrystallization from absolute EtOH afforded 12b as white needles: Mp 236–238 °C; Lit Mp77 236 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 10.80 (s, 1H), 9.15 (d, J = 2 Hz, 2H), 8.98 (t, J = 2 Hz, 1H), 7.76 (m, 2H), 7.39 (m, 2H), 7.16 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 161.7, 148.6, 138.8, 138.0, 129.3, 128.5, 125.0, 121.6, 121.2; HRMS-ES−: Observed 286.0450 (M-H)−; Calculated 286.0464 for C13H8N3O5; HPLC: 97.3%.
General procedure for the synthesis of the monobenzamides 12c–e
The relevant aminopyridine and acid chloride were added to dry pyridine, followed by the addition of Et3N (1 eq.). The mixture was stirred under N2 at RT. Upon completion of the reaction, indicated by TLC analysis, a saturated solution of Na2CO3 (20 ml) was added and the organic components were extracted into EtOAc (3 × 20 ml). The fractions were combined, washed with brine and then stirred with MgSO4 before being concentrated by evaporation under reduced pressure. Excess pyridine was removed by an azeotropic evaporation with toluene to give a solid, which was dried under high vacuum for 2 h to yield the desired product.
3,5-Dinitro-N-(pyridin-4-yl)benzamide (12c).78
Scale: 4-aminopyridine (94 mg, 1 mmol) and 3,5-dinitrobenzoyl chloride (230 mg, 1 mmol), stirred for 2 h. Yield of 12c: 268 mg, 93% as an off-white solid. Recrystallization from AR grade acetone afforded 12c as a white powder: Mp 267–268 °C; Lit Mp78 240–250 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.11 (br. s., 1H), 9.15 (d, J = 2 Hz, 2H), 9.02 (t, J = 2 Hz, 1H), 8.54 (d, J = 6.4 Hz, 2H), 7.78 (d, J = 6.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ ppm 162.8, 151.0, 148.7, 145.7, 137.3, 128.7, 122.0, 114.8; HRMS-ES−: Observed 287.0411 (M-H)−; Calculated 287.0416 for C12H7N4O5; HPLC: 99.7%.
3,5-Dinitro-N-(pyridin-3-yl)benzamide (12d).79
Scale: 3-aminopyridine (94 mg, 1 mmol) and 3,5-dinitrobenzoyl chloride (230 mg, 1 mmol), stirred for 3 h. Yield of 12d: 234 mg, 81% as an off-white solid. Recrystallization from AR-grade acetone afforded 12d as a white powder: Mp 236–238 °C; Lit Mp79 231–232 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.02 (s, 1H), 9.17 (d, J = 2.0 Hz, 2H), 9.02 (t, J = 2.0 Hz, 1H), 8.94 (d, J = 2.4 Hz, 1H), 8.38 (m, 1H), 8.20 (m 1H), 7.45 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 162.3, 148.7, 145.9, 142.8, 137.4, 135.5, 128.6, 128.4, 124.2, 121.8; HRMS-ES−: Observed 287.0403 (M-H)−; Calculated 287.0416 for C12H7N4O5; HPLC: 99.3%.
3,5-Dinitro-N-(pyridin-2-yl)benzamide (12e).80
Scale: 2-aminopyridine (94 mg, 1 mmol) and 3,5-dinitrobenzoyl chloride (230 mg, 1 mmol), stirred for 3 h. Yield of 12e: 286 mg, 99% as an off-white solid. Recrystallization from AR-grade MeOH afforded 12e as a white powder: Mp 196–198 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.57 (s, 1H), 9.18 (d, J = 2.2 Hz, 2H), 8.98 (t, J = 2.2 Hz, 1H), 8.42 (m, 1H), 8.19 (m, 1H), 7.89 (m, 1H), 7.23 (m, 1H); 13C NMR (151 MHz, DMSO-d6) δ ppm 162.7, 152.1, 148.6, 148.5, 138.8, 137.4, 129.0, 121.8, 121.0, 115.4; HRMS-ES−: Observed 287.0402 (M-H)−; Calculated 287.0416 for C12H7N4O5; HPLC: 99.7%.
General procedure for acid chlorides (14a–n) from the corresponding isophthalic acid (13a–n)
Thionyl chloride (10 ml) was added to the appropriate isophthalic acid (13a–n) and refluxed at 80 °C for 4 h and then cooled and stirred for 1 h or overnight. The excess thionyl chloride was evaporated under reduced pressure. After drying the residue for 1 h on a high vacuum pump, the product (14a–n) was not isolated but used immediately in the next step from the same flask.
General procedure for dibenzamides (15a–15p) from the corresponding acid chloride (14a–p)
The appropriate freshly prepared acid chloride (14a–n) or purchased acid chloride (14o–p) was dissolved in 100% dry pyridine unless otherwise specified, followed by the addition of the relevant amine (aniline or aminopyridine) and Et3N (2 equiv.). The reaction was stirred under N2 at RT for the required time. Upon completion of the reaction as indicated by TLC analysis, one of three procedures was followed for the isolation. i) For the pyridylbenzamides (15a, 15b, 15d, 15f, 15h, 15i and 15m), a saturated solution of Na2CO3 (20 ml) was added and the organic components were extracted into EtOAc (3 × 20 ml). The organic fractions were combined, washed with brine and then dried with MgSO4. The solutions were concentrated by evaporation under reduced pressure and excess pyridine was removed by an azeotropic evaporation with toluene to give a solid. ii) For the phenylbenzamides (15c, 15e, 15g, 15j and 15n), the organic components were extracted into EtOAc (3 × 20 ml) and then washed with 1M HCl to remove excess pyridine. The organic fractions were then combined, dried with MgSO4 and concentrated by evaporation under reduced pressure. iii) For the compounds 15k, 15l, 15o and 15p, precipitation of the compounds from the reaction mixture allowed for isolation of the white solid by filtration. All compounds 15a–15p, were dried under high vacuum for 2 h to yield the desired product.
5-Nitro-N,N-di(pyridin-4-yl)isophthalamide (15a)
Scale: 14a (818 mg, 3.3 mmols) and 4-aminopyridine (627 mg, 6.7 mmols), stirred for 3 h. Yield of 15a: 795 mg, 92% as an off-white solid. Recrystallization from AR-grade acetone afforded 15a as small off-white crystals: Mp 303–306 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.93 (br. s, 2H), 9.33 (t, J = 1.5 Hz, 1H), 8.95 (d, J = 1.5 Hz, 2H), 8.69 (d, J = 6.9 Hz, 4H), 8.26 (d, J = 6.9 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ ppm 164.7, 150.6, 148.6, 146.0, 135.5, 134.2, 127.1, 115.5; HRMS-ES−: Observed 362.0887 (M-H)−; Calculated 362.0889 for C18H12N5O4; HPLC: 96.6%.
5-Nitro-N,N-di(pyridin-3-yl)isophthalamide (15b)
Scale: 14b (496 mg, 2 mmols) and 3-aminopyridine (394 mg, 4.2 mmols), stirred for 4 h. Yield of 15b: 576 mg, 79% as an off-white solid. Recrystallization from AR-grade acetone afforded 15b as small off-white crystals: Mp 134–136 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 2H), 9.00 (br.s, 3H), 8.96 (d, J = 2.0 Hz, 2H), 8.36 (m, 2H), 8.21 (m, 2H), 7.44 (m, 8.3 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 163.7, 148.4, 145.7, 142.7, 136.6, 135.8, 133.7, 128.2, 125.8, 124.1 HRMS-ES−: Observed 362.0880 (M-H)−; Calculated 362.0889 for C18H12N5O4; HPLC: 98.4%.
5-Nitro-N,N-diphenylisophthalamide (15c)81
Scale: 14c (496 mg, 2 mmols) and aniline (376 mg, 4 mmols), stirred for 3 h. Yield of 15c: 675 mg, 93% as an off-white solid. Recrystallization from AR-grade acetone afforded 15c as white crystals: Mp 280–283 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 2H), 8.79 – 9.12 (m, 3H), 7.78 (m, 4H), 7.41 (m, 4H), 7.13 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 163.3, 148.4, 139.1, 137.1, 133.6, 129.2, 125.5, 124.8, 121.1; HRMS-ES−: Observed 360.0970 (M-H)−; Calculated 360.0984 for C20H14N3O4; HPLC: 98.8%.
5-Cyano-N,N-di(pyridin-4-yl)isophthalamide (15d)
Scale: 14d (114 mg, 0.5 mmols) and 4-aminopyridine (108 mg, 1.1 mmol), stirred for 3 h in dry DCM (20 ml) and pyridine (0.5 ml). Purification by column chromatography (gravity) with 5–10% MeOH/DCM was carried out. Yield of 5d: 45 mg, 26%. Recrystallization from AR-grade acetone afforded 15d as small off-white crystals: Mp 285–288 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.58 (br. s, 2H), 8.99 (t, J = 1.6 Hz, 1H), 8.68 (d, J = 1.6 Hz, 2H), 8.65 (d, J = 6.6 Hz, 4H), 8.12 (d, J = 6.6 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ ppm 164.5, 151.0, 145.9, 136.2, 135.0, 132.4, 118.1, 114.6, 112.5; HRMS-ES−: Observed 342.0987 (M-H)−; Calculated 342.0991 for C19H12N5O2; HPLC: 99.0%.
5-Cyano-N,N-diphenylisophthalamide (15e)
Scale: 14e (228 mg, 1 mmol) and aniline (201 mg, 2.1 mmols), stirred for 2 h. Yield of 15e: 308 mg, 90% as an off-white solid. Recrystallization from AR-grade DCM gave 15e as a white powder: Mp 300–303 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.55 (br. s, 2H), 8.78 (t, J = 1.7 Hz, 1H), 8.60 (d, J = 1.7 Hz, 2H), 7.80 (m, 4H), 7.42 (m, 4H), 7.17 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ ppm 163.7, 139.1, 136.8, 134.3, 132.1, 129.2, 124.7, 120.9, 118.4, 112.3; MS-EI+ Observed 341.1129 M+; Calculated 341.1164 for C21H15N3O2; HPLC: 96.7%.
5-Methyl-N,N-di(pyridin-4-yl)isophthalamide (15f)
Scale: 14f (434 mg, 2 mmols) and 4-aminopyridine (376 mg, 4 mmol), stirred for 1 h. Yield of 15f: 557 mg, 84% as an off-white solid. Recrystallization from absolute EtOH afforded 15f as a white powder: Mp 209–212 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.70 (s, 2H) 8.49 (d, J = 6.4 Hz, 4H) 8.34 (s, 1H) 8.00 (m, 2H) 7.79 (d, J = 6.4, 4H) 2.49 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.4, 150.8, 146.3, 139.0, 135.1, 132.2, 125.1, 114.6, 21.3; HRMS-ES−: Observed 331.1191 (M-H); Calculated 331.1195 for C19H15N4O2; HPLC: 99.3%.
5-Methyl-N,N-diphenylisophthalamide (15g)
Scale: 14g (434 mg, 2 mmols) and aniline (372 mg, 4 mmol), stirred for 2 h. Yield of 15g: 557 mg, 84% as an off-white solid. Recrystallization from MeOH and AR-grade DCM afforded 15g as small white crystals: Mp 257–258 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 2H), 8.31 (s, 1H), 7.95 (m, 2H), 7.79 (m, 4H), 7.36 (m, 4H), 7.10 (m, 2H), 2.48 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.7, 139.6, 138.6, 135.8, 131.6, 129.1, 124.8, 124.3, 120.9, 21.4; HRMS-ES−: Observed 329.1288 (M-H)−; Calculated 329.1290 for C21H17N2O2; HPLC: 99.0%.
5-(tert-Butyl)-N,N-di(pyridin-4-yl)isophthalamide (15h)
Scale: 14h (518 mg, 2 mmols) and 4-aminopyridine (376 mg, 4 mmol), stirred for 3 h. Yield of 15h: 467 mg, 62% as an off-white solid. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 15h as a white powder: Mp 245–247 °C; 1H NMR (300 MHz, DMSO-d6) δ 10.74 (s, 2H), 8.52 (d, J = 6.2 Hz, 4H), 8.41 (t, J = 1.6 Hz, 1H), 8.17 (d, J = 1.6 Hz, 2H), 7.82 (d, J = 6.2 Hz, 4H), 1.42 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ ppm 166.7, 152.2, 150.8, 146.3, 134.9, 128.6, 125.1, 114.7, 35.4, 31.4; HRMS-ES−: Observed 373.1657 (M-H)−; Calculated 373.1665 for C22H21N4O2; HPLC: 99.1%.
5-(tert-Butyl)-N,N-di(pyridin-3-yl)isophthalamide (15i)
Scale: 14i (259 mg, 1 mmol) and 3-aminopyridine (188 mg, 2 mmols), stirred for 3 h in pyridine (2 ml) and dry DCM (2 ml). Yield of 15i: 294 mg, 79% as an off-white solid. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 15i as a white powder Mp 227–229 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.57 (s, 2H), 8.94 (t, J = 1.3 Hz, 2H), 8.41 (t, J = 1.3 Hz, 1H), 8.33 (d, J = 4.7 Hz, 2H), 8.18 (m, 4H), 7.41 (d, J = 4.7, 2H), 1.4 (m, 9H); 13C NMR (151 MHz, DMSO-d6) δ ppm 166.2, 152.1, 145.3, 142.7, 136.1, 135.0, 128.3, 128.1, 125.0, 124.1, 35.4, 31.5; HRMS-ES−: Observed 373.1653 (M-H)−; Calculated 373.1665 for C22H21N4O2; HPLC: 99.4%.
5-(tert-Butyl)-N,N-diphenylisophthalamide (15j)
Scale: 14j (518 mg, 2 mmols) and aniline (372 mg, 4 mmols), stirred for 2 h. Yield of 15j: 603 mg, 81% as an off-white solid. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 15j as white needles: Mp 285–286 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 11.26 (s, 2H) 9.26 (t, J = 1.6, 1H) 9.00 (d, J = 1.6 Hz, 2H) 8.67 (m, 4H) 8.25 (m, 4H) 7.99 (m, 2H); 13C NMR (151 MHz, DMSO-d6) δ ppm 165.8, 151.8, 139.5, 135.5, 129.1, 127.9, 124.8, 124.3, 121.1, 35.4, 31.5; HRMS-ES−: Observed 371.1749 (M-H)−; Calculated 371.1760 for C24H23N2O2; HPLC: 99.3%.
N,N-Di(pyridin-4-yl)isophthalamide (15k).82
Scale: 14k (406 mg, 2 mmols) and 4-aminopyridine (376 mg, 4 mmols), stirred for 2 h in pyridine (4 ml) and DCM (10 ml). The product was isolated by filtration of the white reaction precipitate. Yield of 15k: 422 mg, 66%. Recrystallization from AR-grade DCM afforded 15k as a white powder: Mp 334–335 °C; Lit Mp83 >350 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.74 (s, 2H), 8.54 (t, J = 1.8 Hz, 1H), 8.49 (d, J = 6.3 Hz, 4H), 8.18 (dd, J = 1.8, 7.9 Hz, 2H), 77.80 (d, J = 6.3 Hz, 4H), 7.73 (t, J = 7.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 166.3, 150.9, 146.3, 135.1, 131.8, 129.3, 127.8, 114.6; HRMS-ES−: Observed 317.1033 (M-H)−; Calculated 317.1039 for C18H13N4O2; HPLC: 95.6%.
N,N-Diphenylisophthalamide (15l).84
Scale: 14l (406 mg, 2 mmols) and aniline (372 mg, 4 mmols), stirred for 2 h. Precipitation of the white product was assisted with DCM. The product was isolated by filtration of the reaction precipitate. Yield of 15l: 455 mg, 72%. Recrystallization from AR-grade DCM afforded 15l as a white powder: Mp 383–384 °C; Lit. Mp84 >300 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.39 (s, 2H), 8.53 (t, J = 1.6 Hz, 1H), 8.13 (dd, J = 1.6, 7.8 Hz, 2H), 7.82 (m, 4H), 7.68 (t, J = 7.8 Hz, 1H), 7.27 – 7.46 (m, 4H), 7.11 (m, 7.5 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ ppm 165.5, 139.5, 135.7, 131.1, 129.1, 129.1 127.5, 124.3, 120.9; HRMS-ES−: Observed 315.1135 (M-H)−; Calculated 315.1134 for C20H15N2O2; HPLC: 98.3%.
5-Methoxy-N,N-di(pyridin-4-yl)isophthalamide (15m)
Scale: 14m (466 mg, 2 mmols) and 4-aminopyridine (376 mg, 4 mmols), stirred for 4 h. Yield of 15m: 483 mg, 69% as an off-white solid. Recrystallization from AR-grade MeOH afforded 15m as a white powder: Mp 144–145 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 10.74 (s, 2H), 8.52 (d, J = 6.4 Hz, 4H), 8.16 (t, J = 1.5 Hz, 1H), 7.82 (d, J = 6.4 Hz, 4H), 7.74 (d, J = 1.5 Hz, 2H), 3.95 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm 166.1, 159.8, 150.8, 146.3, 136.4, 120.1, 117.3, 114.6, 56.4; HRMS-ES−: Observed 347.1132 (M-H)−; Calculated 347.1144 for C19H15N4O3; HPLC: 95.6%.
5-Methoxy-N,N-diphenylisophthalamide (15n)
Scale: 14n (466 mg, 2 mmols) and aniline (372 mg, 4 mmols), stirred for 2 h. Yield of 15n: 637 mg, 92% as an off-white solid. Recrystallization from AR-grade MeOH afforded 15n as white needles: Mp 223–224 °C; 1H NMR (300 MHz, DMSO-d6) δ ppm 10.37 (s, 2H), 8.14 (t, J = 1.5 Hz, 1H), 7.81 (m, 4H), 7.70 (d, J = 1.5 Hz, 2H), 7.39 (m, 4H), 7.12 (m, 2H), 3.94 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ ppm 165.3, 159.7, 139.5, 137.1, 129.1, 124.3, 121.0, 119.8, 116.6, 56.3; HRMS-ES−: Observed 345.1225 (M-H)−; Calculated 345.1239 for C21H17N2O3; HPLC: 99.3%.
N,N-Di(pyridin-4-yl)pyridine-2,6-dicarboxamide (15o).83
Scale: 14o (204 mg, 1 mmol) and 4-aminopyridine (188 mg, 2 mmols), stirred for 3 h. The product was isolated by filtration of the white reaction precipitate. Yield of 15o: 307 mg, 92%. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 15o as a white powder: Mp 327–328 °C; Lit. Mp83 >300 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 12.17 (s, 2H), 8.71 (d, J = 6.6 Hz, 4H), 8.49 (m, 2H), 8.46 (d, J = 6.6 Hz, 4H), 8.39 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 164.2, 149.7, 148.6, 146.5, 140.9, 127.3, 115.8; HRMS-ES−: Observed 318.0986 (M-H)−; Calculated 318.0991 for C17H12N5O2; HPLC: 97.0%.
N,N-Diphenylpyridine-2,6-dicarboxamide (15p).85
Scale: 14p (204 mg, 1 mmol) and aniline (186 mg, 2 mmols), stirred for 3 h. The product was isolated by filtration of the white reaction precipitate. Yield of 15p: 302 mg, 91%. Recrystallization from AR-grade MeOH and AR-grade DCM afforded 15p as a white powder: Mp 271–272 °C; Lit. Mp85 278 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.00 (s, 2H), 8.39 (m, 2H), 8.29 (m, 1H), 7.91 (m, 4H), 7.43 (m, 4H), 7.18 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ ppm 162.2, 149.4, 140.4, 138.5, 129.3, 125.8, 124.9, 121.7; HRMS-ES−: Observed 316.1088 (M-H)−; Calculated 316.1086 for C19H14N3O2; HPLC: 98.1%.
N,N-bis(4-((Dimethylamino)methyl)phenyl)-5-nitroisophthalamide (17)
5-Nitroisophthaloyl dichloride (173 mg, 0.7 mmols) was prepared as per 14a and then dissolved in pyridine (1 ml) and DCM (4 ml). 4-[(N-Boc)aminomethyl]aniline (318 mg, 1.4 mmols) was added and the reaction stirred at RT for 2 h. Following completion of the reaction indicated by TLC, EtOAc and 1 M HCl were added to the flask. The product 16 was extracted into EtOAc (3 × 20 ml) and washed with cold 1 M HCl (3 × 20 ml) to remove the pyridine. The combined organic components were dried with MgSO4 and the solvent removed under reduced pressure before being dissolved in dry DCM (5 ml) under inert atmosphere. TFA (1 ml) was slowly added at 0 °C and the mixture was stirred for 2.5 h. The excess TFA and DCM were removed on a rotoevaporator, followed by the addition of MeOH (20 ml) and K2CO3 (200 mg). The solution was stirred for 30 mins, then filtered through Celite and the MeOH evaporated off under reduced pressure. The resulting solid was dissolved in DMF (~10 ml) and formaldehyde (1.5 ml, 37% in water). Sodium cyanoborohydride (315 mg, 5 mmols) was added to the reaction stirred at RT for 5 h. Water (50 ml) was added and the product was extracted with EtOAc (3 × 50 ml), dried with MgSO4 and the solvents evaporated under reduced pressure. The residual DMF was removed by heating under high vacuum on the pump for 12 h, and the resulting crude solid was recrystallized from MeOH to yield 17 as pale yellow crystals (111 mg, 53%); Mp 202–204 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.00 (s, 2H), 9.03 (t, J = 1.7 Hz, 1H), 8.61 (d, J = 1.7 Hz, 2H), 7.97 (d, J = 8.6 Hz, 4H), 7.55 (d, J = 8.6 Hz, 4H), 4.14 (s, 4H), 2.63 (s, 12H); 13C NMR (101 MHz, DMSO-d6) δ ppm 163.5, 148.4, 140.0, 137.0, 133.6, 129.6, 126.5, 125.7, 120.5, 65.2, 49.0; MS-EI+ Observed 475.1728 M+; Calculated 475.2220 for C26H29N5O4; HPLC: 97.1%
N1-(1H-Benzo[d]imidazol-2-yl)-5-nitro-N3-phenylisophthalamide (18)
5-Nitroisophthaloyl dichloride (1.49 g, 6 mmols) was prepared as per 14a and then dissolved in DCM (10 ml) under N2 and cooled to −40 °C in an acetonitrile/liquid N2 cooling bath. 2-Aminobenzimidazole (133 mg, 1 mmol) was added and the reaction allowed to come to RT before the addition of pyridine (2 drops) whereupon the reaction went from cloudy white to clear yellow. Once TLC monitoring indicated the disappearance of the amine starting material, the reaction was again cooled to −40 °C and another portion of 2-aminobenzimidazole (4 mmols) was slowly added. The reaction was left to come to RT and then heated to 40 °C for 30 mins, after which TLC indicated no remaining benzimidazole starting material, after which it was then brought back to RT. Aniline (500 mg, 5.4 mmols) was added and the solution stirred for a further 30 mins before commencing the work-up. The organic product was extracted into EtOAc (3 × 50 ml) from a solution of brine and sodium carbonate and then the combined EtOAc fractions dried with MgSO4. The EtOAc was removed on a rotoevaporator and then DCM:MeOH (1:1) was added to the yellow solid and briefly heated. The undissolved solid was filtered off and the process repeated twice using acetone to remove the dissolved impurities. The product was then recrystallized in hot pyridine to yield 18 as yellow crystals (200 mg, 10%); Mp 339–342 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.63 (br. s, 2H), 10.72 (s, 1H), 9.02 – 9.18 (m, 2H), 8.91 (t, J = 1.9 Hz, 1H), 7.80 (dd, J = 1.1, 8.6 Hz, 2H), 7.42 – 7.48 (m, 2H), 7.35 – 7.41 (m, 2H), 7.18 – 7.23 (m, 2H), 7.11 – 7.17 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 163.5 (2), 147.8, 139.7, 138.7 (2), 136.5, 133.9, 130.1, 128.8, 126.0, 124.5, 124.3, 122.6, 120.5, 112.2; HRMS-ES−: Observed 400.1052 (M-H)−; Calculated 400.1046 for C21H14N5O4; HPLC: 93.2%.
N,N,N-Triphenyl-benzene-1,3,5-tricarboxamide (20).86
Benzene-1,3,5-tricarbonyl trichloride (19, 265 mg, 1 mmol) was dissolved in dry DCM:pyridine (4:1) and then aniline (279 mg, 3 mmols) was added. The reaction was stirred at RT for 1 h after which the precipitated product was filtered off and dried. The white solid was recrystallized in MeOH and DCM to give 20 as white crystals (240 mg, 55%); Mp 314–315 °C; Lit Mp86 327–329 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 10.55 (br. s, 3H) 8.69 (s, 3H) 7.81 (m, 6H) 7.40 (m, 6H) 7.11 (m, 3H); 13C NMR (151 MHz, DMSO-d6) δ ppm 160.7, 133.1, 130.2, 129.2, 128.1, 123.4, 121.4; HRMS-ES−: Observed 434.1484 (M-H)−; Calculated 434.1505 for C27H20N3O3; HPLC: 96.7%.
Supplementary Material
Acknowledgments
We wish to acknowledge and thank Nina Lawrence and Sylva Schwager from the Division of Pharmacology, UCT Medical School for conducting the ADMET assays as well as Ronel Muller and Katherine A. de Villiers from the Department of Chemistry at the University of Stellenbosch for graphics generated in Accelrys Materials Studio. The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI110329. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- AQ
Amodiaquine
- AR
Analytical reagent
- βH
β-hematin
- CHO
Chinese hamster ovarian
- CQ
Chloroquine
- DV
Digestive vacuole
- Fe(III)PPIX
Ferriprotoporphyrin IX
- Hz
Hemozoin
- EH
Hepatic extraction
- MQ
Mefloquine
- NLB
Neutral lipid blend
- NP-40
Nonidet P-40
- QD-Fe(III)PPIX
Quinidine-heme complex
- QN
Quinine
- QN-Fe(III)PPIX
Quinine-heme complex
- RI
Resistance index
- RT
Room temperature
- SI
Selectivity index
- TEM
Transmission electron microscopy
Footnotes
The Supporting Information is available free of charge on the … website at DOI: ….
Detergent mediated assay for β-hematin inhibition, heme binding procedure, P. falciparum culturing and assays, LDH malaria parasite survival assay, cell fractionation, TEM images, inoculum measurements and in vitro ADMET assays (PDF).
SMILES strings, β-hematin inhibition IC50 data, parasite IC50 data, selected CHO IC50 data and ADME data (CSV).
References
- 1.World Health Organisation. World Malaria Report. 2014 Jan http://www.who.int/malaria/publications/world_malaria_report_2014/wmr-2014-no-profiles.pdf?ua=1.
- 2.Kappe SHI, Vaughan AM, Boddey JA, Cowman AF. That Was Then but This Is Now: Malaria Research in the Time of an Eradication Agenda. Science. 2010;328:862–866. doi: 10.1126/science.1184785. [DOI] [PubMed] [Google Scholar]
- 3.Sutherland CJ, Polley D. Genomic Insights into the Past, Current and Future Evolution of Human Parasites of the Genus Plasmodium. In: Tibayrenc M, editor. Genetics and Evolution of Infectious Diseases. Elsevier; London: 2011. pp. 607–608. [Google Scholar]
- 4.Smrkovski LL, Buck RL, Alcantara AK, Rodriguez CS, Uylangco CV. Studies of Resistance to Chloroquine, Quinine, Amodiaquine and Mefloquine Among Philippine Strains of Plasmodium falciparum. Trans R Soc Trop Med Hyg. 1985;79:37–41. doi: 10.1016/0035-9203(85)90228-7. [DOI] [PubMed] [Google Scholar]
- 5.Cowman AF, Galatis D, Thomson JK. Selection for Mefloquine Resistance in Plasmodium falciparum is Linked to Amplification of the Pfmdrl Gene and Cross-Resistance to Halofantrine and Quinine. Proc Nati Acad Sci USA. 1994;91:1143–1147. doi: 10.1073/pnas.91.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. Piperaquine: a Resurgent Antimalarial Drug. Drugs. 2005;65:75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 7.Combrinck JM, Mabotha TE, Ncokazi KK, Ambele MA, Taylor D, Smith PJ, Hoppe HC, Egan TJ. Insights into the Role of Heme in the Mechanism of Action of Antimalarials. ACS Chem Biol. 2013;8:133–137. doi: 10.1021/cb300454t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LMB, Sidhu AS, Naude B, Deitsch KW, Su X, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum Digestive Vacuole Transmembrane Protein PfCRT and Evidence for Their Role in Chloroquine Resistance. Molecular Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foley M, Tilley L. Quinoline Antimalarials: Mechanisms of Action and Resistance and Prospects for New Agents. Pharmacol Therapeut. 1998;79:55–87. doi: 10.1016/s0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 10.Hawley SR, Bray PG, Mungthin M, Atkinson JD, O’Neill PM, Ward SA. Relationship between Antimalarial Drug Activity, Accumulation, and Inhibition of Heme Polymerization in Plasmodium falciparum In Vitro. Antimicrob Agents Chemother. 1998;42:682–686. doi: 10.1128/aac.42.3.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buller R, Peterson ML, Almarsson O, Leiserowitz L. Quinoline Binding Site on Malaria Pigment Crystal: A Rational Pathway for Antimalaria Drug Design. Cryst Growth Des. 2002;2:553–562. [Google Scholar]
- 12.de Villiers KA, Gildenhuys J, Roex Tl. Iron(III) Protoporphyrin IX Complexes of the Antimalarial Cinchona Alkaloids Quinine and Quinidine. ACS Chem Biol. 2012;7:666–671. doi: 10.1021/cb200528z. [DOI] [PubMed] [Google Scholar]
- 13.Olafson KN, Ketchum MA, Rimer JD, Vekilov PG. Mechanisms of Hematin Crystallization and Inhibition by the Antimalarial Drug Chloroquine. Proc Natl Acad Sci. 2015;112:4946–4951. doi: 10.1073/pnas.1501023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan TJ, Ross DC, Adams PA. Quinoline Anti-Malarial Drugs Inhibit Spontaneous Formation of Beta-Haematin (Malaria Pigment) FEBS Lett. 1994;352:54–57. doi: 10.1016/0014-5793(94)00921-x. [DOI] [PubMed] [Google Scholar]
- 15.Parapini S, Basilico N, Pasini E, Egan TJ, Olliaro P, Taramellia D, Monti D. Standardization of the Physicochemical Parameters to Assess in Vitro the Beta-Hematin Inhibitory Activity of Antimalarial Drugs. Experimental Parasitology. 2000;96:249–256. doi: 10.1006/expr.2000.4583. [DOI] [PubMed] [Google Scholar]
- 16.Dorn A, Vippagunta S, Matile H, Bubendorf A, Vennerstrom J, Ridley R. A Comparison and Analysis of Several Ways to Promote Haematin (Haem) Polymerisation and an Assessment of Its Initiation In Vitro. Biochem Pharmacol. 1998;55:737–747. doi: 10.1016/s0006-2952(97)00509-1. [DOI] [PubMed] [Google Scholar]
- 17.Kurosawa Y, Satoh T, Dorn A, Matile H, Kitsuji-Shirane M, Hofheinz W, Kansy M, Ridley RG. Haematin Polymerisation Assay as a High-Throughput Screen for Identification of New Antimalarial Pharmacophores. Antimicrob Agents Chemother. 2000;44:2638–2644. doi: 10.1128/aac.44.10.2638-2644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitch CD, Cai GZ, Chen YF, Shoemaker JD. Involvement of Lipids in Ferriprotoporphyrin IX Polymerization in Malaria. Biochim Biophys Acta. 1999;1454:31–37. doi: 10.1016/s0925-4439(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 19.Pisciotta JM, Coppens I, Tripathi AK, Scholl PF, Shuman J, Bajad S, Shulaev V, Sullivan DJ. The Role of Neutral Lipid Nanospheres in Plasmodium falciparum Haem Crystallization. Biochem J. 2007;402:197–204. doi: 10.1042/BJ20060986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huy NT, Uyen DT, Maeda A, Trang DTX, Oida T, Harada S, Kamei K. Simple Colorimetric Inhibition Assay of Heme Crystallization for High-Throughput Screening of Antimalarial Compounds. Antimicrob Agents Chemother. 2007;51:350–353. doi: 10.1128/AAC.00985-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter MD, Phelan VV, Sandlin RD, Bachmann BO, Wright DW. Lipophilic Mediated Assays for β-Hematin Inhibitors. Comb Chem High T Scr. 2010;3:285–292. doi: 10.2174/138620710790980496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ncokazi KK, Egan TJ. A Colorimetric High-Throughput B-Hematin Inhibition Screening Assay for Use in the Search for Antimalarial Compounds. Anal Biochem. 2005;338:306–319. doi: 10.1016/j.ab.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Rush MA, Baniecki ML, Mazitschek R, Cortese JF, Wiegand R, Clardy J, Wirth DF. Colorimetric High-Throughput Screen for Detection of Heme Crystallization Inhibitors. Antimicrob Agents Chemother. 2009;53:2564–2568. doi: 10.1128/AAC.01466-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandlin RD, Carter MD, Lee PJ, Auschwitz JM, Leed SE, Johnson JD, Wright DW. Use of the NP-40 Detergent-Mediated Assay in Discovery of Inhibitors of Beta-Hematin Crystallization. Antimicrob Agents Chemother. 2011;55:3363–3369. doi: 10.1128/AAC.00121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandlin RD, Fong KY, Wicht KJ, Carrell HM, Egan TJ, Wright DW. Identification of Beta-Hematin Inhibitors in a High-Throughput Screening Effort Reveals Scaffolds with in vitro Antimalarial Activity. Int J Parasitol. 2014;4:316–325. doi: 10.1016/j.ijpddr.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Combrinck JM, Fong KY, Gibhard L, Smith PJ, Wright DW, Egan TJ. Optimization of a Multi-Well Colorimetric Assay to Determine Haem Species in Plasmodium falciparum in the Presence of Antimalarials. Malaria J. 2015;14:1–14. doi: 10.1186/s12936-015-0729-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Jr, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. Chemical Genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu T, Nagle A, Sakata T, Henson K, Borboa R, Chen Z, Kuhen K, Plouffe D, Winzeler E, Adrian F, Tuntland T, Chang J, Simerson S, Howard S, Ek J, Isbell J, Deng X, Gray NS, Tully DC, Chatterjee AK. Cell-Based Optimization of Novel Benzamides as Potential Antimalarial Leads. Bioorg Med Chem Lett. 2009;19:6970–6974. doi: 10.1016/j.bmcl.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fong KY, Sandlin RD, Wright DW. Identification of β-Hematin Inhibitors in the MMV Malaria Box. Int J Parasitol Drugs Drug Resist. 2015;5:84–91. doi: 10.1016/j.ijpddr.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu WJ, Wicht KJ, Wang L, Imai K, Mei ZW, Kaiser M, Sayed IETE, Egan TJ, Inokuchi T. Synthesis and Antimalarial Testing of Neocryptolepine Analogues: Addition of Ester Function in Sar Study of 2,11-Disubstituted Indolo[2,3-B]quinolines. Eur J Med Chem. 2013;64:498–511. doi: 10.1016/j.ejmech.2013.03.072. [DOI] [PubMed] [Google Scholar]
- 31.Wang N, Wicht KJ, Imai K, Ngoc TA, Wang MQ, Kaiser M, Egan TJ, Inokuchi T. Synthesis, β-haematin Inhibition, and In Vitro Antimalarial Testing of Isocryptolepine Analogues: SAR Study of Indolo[3,2-c]quinolines with Various Substituents at C2, C6, and N11. Bioorg Med Chem. 2014;22:2629–2642. doi: 10.1016/j.bmc.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Wicht KJ, Combrinck JM, Smith PJ, Egan TJ. Bayesian Models Trained with HTS Data for Predicting β-Haematin Inhibition and In Vitro Antimalarial Activity. Bioorg Med Chem Lett. 2015;23:5210–5217. doi: 10.1016/j.bmc.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ongarora DSB, Strydom N, Wicht K, Njoroge M, Wiesner L, Egan TJ, Wittlin S, Jurva U, Masirembwa CM, Chibale K. Antimalarial Benzoxaquines: Structure Activity Relationship, In Vivo Evaluation, Mechanistic and Bioactivation Studies. Bioorg Med Chem. 2015;23:5419–5432. doi: 10.1016/j.bmc.2015.07.051. [DOI] [PubMed] [Google Scholar]
- 34.Joshi MC, Wicht KJ, Taylor D, Hunter R, Smith PJ, Egan TJ. In Vitro Antimalarial Activity, β-Haematin Inhibition and Structure-Activity Relationships in a Series of Quinoline Triazoles. Eur J Med Chem. 2013;69:338–347. doi: 10.1016/j.ejmech.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 35.Kaschula CH, Egan TJ, Hunter R, Basilico N, Parapini S, Taramelli D, Pasini E, Monti D. Structure-Activity Relationships in 4-Aminoquinoline Antiplasmodials. The Role of the Group at the 7-Position. J Med Chem. 2002;45:3531–3539. doi: 10.1021/jm020858u. [DOI] [PubMed] [Google Scholar]
- 36.Omodeo-Salè F, Cortelezzi L, Basilico N, Casagrande M, Sparatore A, Taramelli D. Novel Antimalarial Aminoquinolines: Heme Binding and Effects on Normal or Plasmodium falciparum-Parasitized Human Erythrocytes. Antimicrob Agents Chemother. 2009;53:4339–4344. doi: 10.1128/AAC.00536-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vargas S, Ioset KN, Hay AE, Ioset JR, Wittlin S, Hostettmann K. Screening Medicinal Plants for the Detection of Novel Antimalarial Products Applying the Inhibition of β-Hematin Formation. J Pharm Biomed Anal. 2011;56:880–886. doi: 10.1016/j.jpba.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 38.Ignatushchenkoa MV, Winterb RW, Bächingerd HP, Hinrichsb DJ, Riscoe MK. Xanthones as Antimalarial Agents; Studies of a Possible Mode of Action. FEBS Lett. 1997;409:67–73. doi: 10.1016/s0014-5793(97)00405-5. [DOI] [PubMed] [Google Scholar]
- 39.Kelly JX, Winter R, Peyton DH, Hinrichs DJ, Riscoe M. Optimization of Xanthones for Antimalarial Activity: the 3,6-Bis--Diethylaminoalkoxyxanthone Series. Antimicrob Agents Chemother. 2002;46:144–150. doi: 10.1128/AAC.46.1.144-150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vippagunta SR, Dorn A, Matile H, Bhattacharjee AK, Karle JM, Ellis WY, Ridley RG, Vennerstrom JL. Structural Specificity of Chloroquine-Hematin Binding Related to Inhibition of Hematin Polymerization and Parasite Growth. J Med Chem. 1999;42:4630–4639. doi: 10.1021/jm9902180. [DOI] [PubMed] [Google Scholar]
- 41.Egan TJ, Hunter R, Kaschula CH, Marques HM, Misplon A, Walden J. Structure-Function Relationships in Aminoquinolines: Effect of Amino and Chloro Groups on Quinoline-Hematin Complex Formation, Inhibition of B-Hematin Formation, and Antiplasmodial Activity. J Med Chem. 2000;43:283–291. doi: 10.1021/jm990437l. [DOI] [PubMed] [Google Scholar]
- 42.Wang N, Wicht KJ, Wang L, Lu WJ, Misumi R, Wang MQ, Gokha AAAE, Kaiser M, Sayed IETE, Egan TJ, Inokuchi T. Synthesis and In Vitro Testing of Antimalarial Activity of Non-Natural-Type Neocryptolepines: Structure-Activity Relationship (SAR) Study of 2,11- and 9,11-Disubstituted 6-Methylindolo[2,3-b]quinolines. Chem Pharm Bull. 2013;61:1282–1290. doi: 10.1248/cpb.c13-00639. [DOI] [PubMed] [Google Scholar]
- 43.Shaban E, Wicht KJ, Wang N, Mei ZW, Hayashi I, Aleem AA, Gokha E, Kaiser M, Tantawy IE, Sayed E, Egan TJ, Inokuchi T. Synthesis and Antimalarial Activity of Some Neocryptolepine Analogues Carrying a Multifunctional Linear and Branched Carbon-Side Chains. Heterocycles. 2014;89:1055–1064. [Google Scholar]
- 44.MetaSite, v20. Molecular Discovery; Italy: 2012. [Google Scholar]
- 45.VolSurf+, v1.0.7.1. Molecular Discovery; Italy: 2012. [Google Scholar]
- 46.Egan TJ. Interactions of Quinoline Antimalarials with Hematin in Solution. J Inorg Biochem. 2006;100:916–926. doi: 10.1016/j.jinorgbio.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Egan TJ, Mavuso WW, Ncokazi KK. The Mechanism of β-Hematin Formation in Acetate Solution. Parallels between Hemozoin Formation and Biomineralization Processes. Biochem. 2001;40:204–213. doi: 10.1021/bi0013501. [DOI] [PubMed] [Google Scholar]
- 48.Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. The Structure of Malaria Pigment Beta-Haematin. Nature. 2000;404:307–310. doi: 10.1038/35005132. [DOI] [PubMed] [Google Scholar]
- 49.Sullivan DJ, Gluzman IY, Russell DG, Goldberg DE. On the Molecular Mechanism of Chloroquine’s Antimalarial Action. Proc Natl Acad Sci USA. 1996;93:11865–11870. doi: 10.1073/pnas.93.21.11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly JX, Winter R, Riscoe M, Peyton DH. A Spectroscopic Investigation of the Binding Interactions Between 4,5-Dihydroxyxanthone and Heme. J Inorg Biochem. 2001;86:617–625. doi: 10.1016/s0162-0134(01)00217-3. [DOI] [PubMed] [Google Scholar]
- 51.Frisch MJ, Trucks GW. Gaussian Software, v30. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 52.Warhurst DC, Craig JC, Adagu IS, Guy RK, Madrid PB, Fivelman QL. Activity of Piperaquine and Other 4-Aminoquinoline Antiplasmodial Drugs Against Chloroquine-Sensitive and Resistant Blood-Stages of Plasmodium falciparum: Role of β-haematin Inhibition and Drug Concentration in Vacuolar Water- and Lipid-Phases. Biochem Pharma. 2007;73:1910–1926. doi: 10.1016/j.bcp.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 53.Chou AC, Fitch CD. Control of Heme Polymerase by Chloroquine and Other Quinoline Derivatives. Biochem Biophys Res Comm. 1993;195:422–427. doi: 10.1006/bbrc.1993.2060. [DOI] [PubMed] [Google Scholar]
- 54.Basilico N, Monti D, Olliaro P, Taramelli D. Non-Iron Porphyrins Inhibit Beta-Haematin (Malaria Pigment) Polymerisation. FEBS Lett. 1997;409:297–299. doi: 10.1016/s0014-5793(97)00533-4. [DOI] [PubMed] [Google Scholar]
- 55.Materials Studio, v4.0. Accelrys Software Inc; San Diego: 2013. [Google Scholar]
- 56.Warhurst DC, Hockley DJ. Mode of Action of Chloroquine on Plasmodium berghei and P. cynomolgi. Nature. 1967;214:935–936. doi: 10.1038/214935a0. [DOI] [PubMed] [Google Scholar]
- 57.Macomber PB, Sprinz H, Tousimis AJ. Morphological Effects of Chloroquine on Plasmodium berghei in Mice. Nature. 1967;214:937–939. doi: 10.1038/214937a0. [DOI] [PubMed] [Google Scholar]
- 58.Fitch CD, Yunis NG, Chevli R, Gonzales Y. High-Affinity Accumulation of Chloroquine by Mouse Erythrocytes Infected with Plasmodium berghii. J Clin Invest. 1974;54:24–33. doi: 10.1172/JCI107747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yayon A, Cabantchik ZI, Ginsburg H. Identification of the Acidic Compartment of Plasmodium falciparum Infected Human Erythrocytes as the Target of the Antimalarial Drug Chloroquine. EMBO J. 1984;3:2695–2700. doi: 10.1002/j.1460-2075.1984.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colowick SP, Womack FC. Binding of Diffusible Molecules by Macromolecules: Rapid Measurement by Rate of Dialysis. J Biol Chem. 1969;244:774–777. [PubMed] [Google Scholar]
- 61.Yayon A, Cabantchik ZI, Ginsburg H. Susceptibility of Human Malaria Parasites to Chloroquine is pH Dependent. Proc Natl Acad Sci USA. 1985;82:2784–2788. doi: 10.1073/pnas.82.9.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bray PG, Howells RE, Ward SA. Vacuolar Acidification and Chloroquine Sensitivity in Plasmodium falciparum. Biochem Pharmacol. 1992;43:1219–1227. doi: 10.1016/0006-2952(92)90495-5. [DOI] [PubMed] [Google Scholar]
- 63.MacIntyre AC, Cutler DJ. Kinetics of Chloroquine Uptake into Isolated Rat Hepatocytes. J Pharm Sci. 1993;82:592–600. doi: 10.1002/jps.2600820610. [DOI] [PubMed] [Google Scholar]
- 64.Kumar Sanjay, Guha Mithu, Choubey Vinay, Maity Pallab, Bandyopadhyay U. Antimalarial Drugs Inhibiting Hemozoin (B-Hematin) Formation: A Mechanistic Update. Life Sci. 2007;80:813–828. doi: 10.1016/j.lfs.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 65.Brook I. Inoculum Effect. Clin Infect Dis. 1989;11:361–368. doi: 10.1093/clinids/11.3.361. [DOI] [PubMed] [Google Scholar]
- 66.Gluzman IY, Schlesinger PH, Krogstad DJ. Inoculum Effect with Chloroquine and Plasmodium falciparum. Antimicrob Agents Chemother. 1987;31:32–36. doi: 10.1128/aac.31.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Neill PM, Willock DJ, Hawley SR, Bray PG, Storr RC, Ward SA, Park BK. Synthesis, Antimalarial Activity, and Molecular Modeling of Tebuquine Analogues. J Med Chem. 1997;40:437–448. doi: 10.1021/jm960370r. [DOI] [PubMed] [Google Scholar]
- 68.Bray PG, Janneh O, Raynes KJ, Mungthin M, Ginsburg H, Ward aSA. Cellular Uptake of Chloroquine Is Dependent on Binding to Ferriprotoporphyrin IX and Is Independent of NHE Activity in Plasmodium falciparum. J Cell Biol. 1999;145:363–376. doi: 10.1083/jcb.145.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bray PG, Mungthin M, Ridley RG, Ward SA. Access to Hematin: The Basis of Chloroquine Resistance. Mol Pharm. 1998;54:170–179. doi: 10.1124/mol.54.1.170. [DOI] [PubMed] [Google Scholar]
- 70.Biagini GA, Richier E, Bray PG, Calas M, Vial H, Ward SA. Heme Binding Contributes to Antimalarial Activity of Bis-Quaternary Ammoniums. Antimicrob Agents Chemother. 2003;47:2584–2589. doi: 10.1128/AAC.47.8.2584-2589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Omodeo-Sale F, Cortelezzi L, Basilico N, Casagrande M, Sparatore A, Taramelli D. Novel Antimalarial Aminoquinolines: Heme Binding and Effects on Normal or Plasmodium falciparum-Parasitized Human Erythrocytes. Antimicrob Agents Chemother. 2009;53:4339–4344. doi: 10.1128/AAC.00536-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuter D, Chibale K, Egan TJ. Linear Free Energy Relationships Predict Coordination and π-Stacking Interactions of Small Molecules with Ferriprotoporphyrin IX. J Inorg Biochem. 2011;105:684–692. doi: 10.1016/j.jinorgbio.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 73.Kuter D, Benjamin SJ, Egan TJ. Multiple Spectroscopic and Magnetic Techniques Show that Chloroquine Induces Formation of The μ-Oxo Dimer of Ferriprotoporphyrin IX. J Inorg Biochem. 2014;133:40–49. doi: 10.1016/j.jinorgbio.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 74.Jia L, Liu X. The Conduct of Drug Metabolism Studies Considered Good Practice (II): In Vitro Experiments. Curr Drug Metab. 2007;8:822–829. doi: 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Villiers KA, Marques HM, Egan TJ. The Crystal Structure of Halofantrine–Ferriprotoporphyrin IX and the Mechanism of Action of Arylmethanol Antimalarials. J Inorg Biochem. 2008;102:1660–1667. doi: 10.1016/j.jinorgbio.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 76.Kakuta H, Zheng X, Oda H, Harada S, Sugimoto Y, Sasaki K, Tai A. Cyclooxygenase-1-Selective Inhibitors Are Attractive Candidates for Analgesics That Do Not Cause Gastric Damage. Design and in Vitro/in Vivo Evaluation of a Benzamide-Type Cyclooxygenase-1 Selective Inhibitor. J Med Chem. 2008;51:2400–2411. doi: 10.1021/jm701191z. [DOI] [PubMed] [Google Scholar]
- 77.Ahn KH, Chung YM, Jun M, Kim DS. Selective Colorimetric Sensing of Anions in Aqueous Media through Reversible Covalent Bonding. J Org Chem. 2009;74:4849–4854. doi: 10.1021/jo900573v. [DOI] [PubMed] [Google Scholar]
- 78.Il’ina IG, Mikhalev OV. Design, Synthesis, and Spectral Parameters of Transition Metal Coordination Compounds Based on Organic Autocomplexes. Russ J Org Chem. 2010;47:1488–1493. [Google Scholar]
- 79.Mallakpour S, Zarei M. Novel, Thermally Stable and Chiral Poly(Amide-Imide)S Derived from a New Diamine Containing Pyridine Ring and Various Amino Acid-Based Diacids: Fabrication and Characterization. High Perform Polym. 2013;25:245–253. [Google Scholar]
- 80.George S, Shanmugapandiyan P. Synthesis and Antimicrobial Evaluation Of 2-(5-(Substituted Phenyl-1h-Tetrazol-1-Yl) Pyridines. Int J Pharm Pharm Sci. 2011;4:104–106. [Google Scholar]
- 81.Miller TM, Neenan TX. Convergent Synthesis of Monodisperse Dendrimers Based Upon 1,3,5-Trisubstituted Benzenes. Chem Mater. 1990;2:346–349. [Google Scholar]
- 82.Odago MO, Hoffman AE, Carpenter RL, Tse DCT, Sun SS, Lees AJ. Thioamide, Urea and Thiourea Bridged Rhenium(I) Complexes as Luminescent Anion Receptors. Inorg Chim Acta. 2011;374:558–565. [Google Scholar]
- 83.Qin Z, Jennings MC, Puddephatt RJ. Self-Assembly in Palladium(II) and Platinum(II) Chemistry: The Biomimetic Approach. Inorg Chem. 2003;42:1956–1965. doi: 10.1021/ic020322z. [DOI] [PubMed] [Google Scholar]
- 84.Lukin O, Kubota T, Okamoto Y, Schelhase F, Yoneva A, Müller WM, Müller U, Vögtle F. Knotaxanes-Rotaxanes with Knots as Stoppers. Angew Chem Int Ed. 2003;42:4542–4545. doi: 10.1002/anie.200351981. [DOI] [PubMed] [Google Scholar]
- 85.Horino H, Sakaba H, Arai M. Facile Preparation of 6-Bromopyridine-2-Carboxamide and Pyridine-2,6-Dicarboxamide: Partial Aminocarbonylation of 2,6-Dibromopyridine. Synthesis. 1989;9:715–718. [Google Scholar]
- 86.Palmans ARA, Vekemans JAJM, Fischer H, Hikmet RA, Meijer EW. Extended-Core Discotic Liquid Crystals Based on the Intramolecular H-Bonding in N-Acylated 2,2′-Bipyridine-3,3′-diamine Moieties. Chem Eur J. 1997;3:300–307. doi: 10.1002/chem.19970030220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.