

INTRODUCTION
The renin-angiotensin system (RAS) is an essential regulator of blood pressure homeostasis. The primary effector molecule of the RAS is angiotensin (Ang) II, an eight-amino acid peptide that initiates tissue-specific processes via binding to the type 1 or 2 angiotensin II receptors (AT1 or AT2). The AT1 receptor is a G-protein coupled receptor expressed in most tissues throughout the body. While the rodent expresses 2 AT1 receptor isoforms, AT1A and AT1B, the closest functional homolog to the human AT1 receptor in most rodent tissues is AT1A. The most widely appreciated functions of Ang II signaling via the AT1 receptor are to augment global vascular tone and promote sodium retention in the kidney, thus playing a crucial role in acute and long-term maintenance of blood pressure.1, 2 Ang II elicits these effects by increasing the contractility of vascular smooth muscle, promoting the release of aldosterone from the adrenal cortex, and directly increasing the expression and/or activity of solute transporters in the renal tubules that promote sodium reabsorption. As such, blockade of the renin-angiotensin system (RAS) has been a mainstay in the treatment for hypertension and its complications, including cardiovascular and kidney disease.2, 3 Although the etiology of essential hypertension is unclear, inhibition of the RAS is effective in reducing blood pressure in a wide variety of patients, which suggests that inappropriate RAS activation is common in the pathogenesis of this idiopathic condition. Global RAS inhibitors ameliorate hypertension by blocking the production of Ang II or by preventing activation of the AT1 receptor. These medications are “global” inhibitors in the sense that they disrupt AT1 receptor signaling concomitantly in all tissues.
The ongoing effort to uncover novel pathogenic mechanisms of hypertension has revealed a significant role for the immune system.4 Although augmentation of hypertension and associated organ damage by the immune system has been appreciated for over 5 decades, intense experimental investigation of this phenomenon commenced roughly 15 years ago. Over this period, leukocytes have been shown to modulate the hypertensive response in a plethora of experimental models.5 Immune cells infiltrate cardiovascular control centers that regulate blood pressure, such as the kidney, blood vessels, and brain. There they produce factors that promote local inflammation and sodium retention and mediate tissue injury, ultimately leading to elevated blood pressure. Human data regarding this phenomenon are currently sparse, yet indicative of an immune-related component to hypertension.6, 7
The dominant angiotensin receptor expressed on immune cells is the AT1 receptor, and their inflammatory functions are altered by its activation.8–10 Furthermore activation of the AT1 receptor on distinct cell populations within the immune compartment has effects on blood pressure and/or renal damage that contrast starkly with AT1 receptor actions in the kidney and vasculature.11 These emerging data suggest that global inhibition of the RAS may not be optimal in the treatment of hypertension, and that understanding cell-specific actions of the AT1 receptor in the setting of hypertension may guide novel therapeutics that manipulate the RAS via tissue-specific strategies. The aim of this review is to describe the interplay between the renin-angiotensin and immune systems in hypertension.
THE IMMUNE SYSTEM, THE AT1 RECEPTOR, AND HYPERTENSION
The immune system is composed of two arms: the innate immune system, which mounts a rapid, non-specific response to a wide array of pathogens, and the adaptive immune system, which executes an antigen-specific response subsequent to the innate response. Myeloid cells largely comprise the innate arm and trigger engagement of the adaptive immune response. By contrast, lymphoid cells contribute primarily to adaptive immunity. The myeloid compartment contains a vast collection of immune cells, including monocytes, macrophages, and dendritic cells, with varying capacities to fight invading pathogens, while the lymphoid compartment is largely comprised of two cell types, T and B lymphocytes.
A link between the immune system and hypertension has long been recognized, as inflammatory cells were shown to traffick to organs that regulate blood pressure decades ago. For example, Sommers et al. recognized a positive correlation between lymphocyte aggregation and the degree of arteriolar nephrosclerosis in kidney biopsies from hypertensive patients.12 Experimentally in 1967, monocytes were shown to accumulate in the aortic intima as early as 4 hours after hypertension was induced in rats by aortic constriction.13 Moreover in 1969, studies of kidney glomeruli in the hypertensive rat revealed a marked accrual of monocytes compared to normotensive controls.14 Olsen then showed that transfer of splenocytes from a hypertensive rat could recapitulate hypertension in a normotensive rat.15 Inversely, Svendsen reported that athymic mice were protected from DOCA hypertension.16 Since these initial observations and in several hypertensive models, immune cells have been implicated in vascular injury, kidney damage, and inappropriate sodium retention – all pathological amplifiers of blood pressure elevation. Moreover, hypertension emerges in autoimmune disorders such as systemic lupus erythematosus (SLE), and blocking inflammatory responses in SLE can dramatically reduce blood pressure.17, 18
As RAS stimulation and immune system activation both contribute to the pathogenesis of hypertension, several investigators have explored whether ligation of AT1 receptors on immune cells can modulate blood pressure and/or target organ damage in hypertension. Bone marrow transplant (BMT) experiments, utilizing donor mice that are genetically deficient of the dominant rodent AT1 receptor isoform, AT1A (Agtr1a−/−), have demonstrated context-specific effects of the immune AT1 receptor in hypertension and cardiovascular disease. One such study employed the Tsukuba mouse, which carries the human renin and angiotensinogen transgenes. This mouse is a model of RAS-mediated, persistent hypertension and atherosclerosis precipitated by an atherogenic diet.19 Tsukuba mice underwent BMT using Agtr1a−/− or Agtr1a+/+ donors and were placed on the atherogenic diet 6 weeks later. The level of hypertension was similar between Tsukuba mice receiving Agtr1a−/− or Agtr1a+/+ BMCs, but the absence of the AT1A receptor on BM cells increased the severity of atherosclerotic lesions in the aorta.20 Our lab utilized BMT to explore the actions of AT1A receptors on immune cells in Ang II-induced hypertension.21 Although the absence of AT1A on bone marrow cells did not affect baseline blood pressure, the mice that received Atgr1a−/− BM cells had an augmented hypertensive response to chronic Ang II infusion compared to mice receiving Atgr1a+/+ BM cells, along with exaggerated accumulation of macrophages and T cells in the hypertensive kidney. Together, these studies pointed to a protective effect of the immune cell AT1 receptor in hypertension and associated complications.
AT1A receptors on bone marrow-derived cells can similarly ameliorate kidney damage even in the absence of hypertension. For example, in a RAS-dependent model of kidney fibrosis induced by unilateral ureteral obstruction (UUO), transplantation of Agtr1a−/− bone marrow into WT mice induced greater renal expression of pro-fibrotic genes and more severe interstitial fibrosis compared to WT transplant controls.22 Ma and Fogo similarly found that AT1A receptor-deficient mice had exaggerated obesity-dependent kidney damage, attributed to enhanced pro-inflammatory macrophage responses.23 By contrast, in the apolipoprotein E deficient mouse model of atherosclerosis, reconstitution with Agtr1a−/− BM cells significantly blunted the formation of atherosclerotic lesions in the aorta and kidney,24, 25 consistent with pathogenic actions of AT1 receptors on bone marrow-derived cells.
We speculate that these conflicting actions of the immune cell AT1 receptor in hypertension and cardiovascular disease may relate to the specific subsets of immune cells involved in mediating tissue pathology in the respective models. To this point, a variety of immune cells have been implicated in regulating blood pressure and the extent of tissue injury in hypertension. These include both pro- and anti-inflammatory subsets that are proposed to have deleterious and salutary functions, respectively, in target organ pathology.26 Because the bone marrow transplant studies mentioned above abrogated AT1A receptor signaling on all immune cell populations, more precise approaches are required to determine in vivo how AT1 receptor signaling influences the function of particular immune cell subsets in the setting of hypertension. We have pursued cell-specific, conditional deletion strategies to confront this problem, and others have utilized adoptive transfer approaches. Below we will review the current understanding of how particular myeloid and lymphoid subsets participate in the pathogenesis of hypertension and discuss recent findings regarding the effects of AT1A receptor activation on immune cell function in this setting. We will focus predominantly on monocytes, macrophages, and T cells, as the majority of studies in the field of hypertension to date have generated data regarding these immune cell subsets.
MONOCYTES/MACROPHAGES, THE AT1 RECEPTOR, AND HYPERTENSION
Monocytes are a subset of circulating myeloid cells that originate in the bone marrow and play a pivotal role in immunity during homeostasis and disease.27 The stratification of murine monocytes into functionally distinct populations is continually revised, but one schema parses monocytes based on their expression of the surface marker Ly6C.28 Robust expression of Ly6C (Ly6Chi) designates a pro-inflammatory subset of monocytes that are heavily recruited to sites of inflammation and produce reactive oxygen species (ROS), interleukin 1β (IL-1β), and tumor necrosis factor-α (TNF). The human counterpart is the pro-inflammatory CD14hiCD16− monocyte population. Minimal or no expression of Ly6C (Ly6Clo) identifies an anti-inflammatory subset of murine monocytes said to “patrol” the luminal endothelium under homeostatic conditions in order to rapidly detect vascular insult.29 In humans, these patrolling monocytes are identified by CD14loCD16hi double positivity. Besides their direct inflammatory capacity, monocytes have long been thought to maintain appropriate levels of resident tissue macrophages via differentiation, although recent evidence suggests a more complex relationship between circulating and resident tissue myeloid cell pools.30, 31
Macrophages are resident tissue phagocytes that play a critical role in innate immunity. Apart from their phagocytic capacity, macrophages release inflammatory mediators and recruit and program other immune cells. In one paradigm, macrophages are divided into two subsets, M1 and M2.31 M1, or classically activated, macrophages are pro-inflammatory and, like their Ly6Chi monocyte precursors, release the cytokines IL-1β and TNF and produce ROS. M2, or alternatively activated, macrophages display an ant-inflammatory phenotype, are involved in tissue repair, and elaborate regulatory cytokines such as interleukin-10 (IL-10) and transforming growth factor β (TGFβ).32 Although monocytes and macrophages are essential for proper immune homeostasis, their dysregulation contributes to diverse pathologies ranging from classic autoimmune diseases to hypertension.4, 33
Both experimental and clinical data point to a key role for monocytes and macrophages in hypertension. The monocyte chemokine CCL2 (MCP-1) is highly expressed in the kidney and vasculature during hypertension,34, 35 leading to the infiltration of monocytes expressing CCR2, the receptor for MCP-1, into these tissues.36, 37 Accordingly, depletion of LysM-expressing monocytes blunts the chronic hypertensive response to Ang II,38 whereas inhibition of MCP-1 or CCR2 blunts renal and aortic myeloid cell invasion and attenuates both hypertension and consequent tissue injury.37, 39 Within the target organ, monocytes and macrophages instigate and sustain inflammation that causes tissue damage and augments blood pressure elevation. In the Ang II hypertension model, pro-inflammatory monocytes and macrophages expressing M1 markers infiltrate the aorta by day 7,40 whereas later, at day 14, these aortic macrophages favor an M2 phenotype featuring CD206 expression.34 Whether this phenotypic shift reflects temporal plasticity of the initial M1 macrophage infiltrate or its replacement by M2 macrophages is unclear, but there is precedent for plasticity of macrophages in tissue injury and repair.32, 41 In clinical hypertension, histological analysis of post-mortem kidneys show a significant increase in cortical density of CD68+ monocytes/macrophages compared to normotensive controls.42 Moreover, circulating monocytes from hypertensive patients have an enhanced pro-inflammatory phenotype when stimulated ex-vivo,43 and circulating levels of inflammatory chemokines are elevated in hypertensive patients.44–46
Pro-inflammatory monocytes/macrophages have the capacity to potentiate hypertension through multiple mechanisms. As part of the innate immune response, myeloid cells can promote remodeling in the vasculature and epithelial cell injury in the kidney, prompting endothelial dysfunction and impairing appropriate excretion of sodium in the setting of hypertension. However, even in the absence of renal injury, M1 macrophages can promote sodium retention in the kidney through the elaboration of ROS and/or inflammatory cytokines including TNF or IL-1β. Regarding the role of ROS, ablation of monocytes/macrophages significantly reduces aortic superoxide production, vascular dysfunction, and blood pressure during Ang II-induced hypertension.38 Inversely, adoptive transfer of WT, but not NADPH oxidase-null monocytes, can restore vascular oxidative stress and hypertension.47 Complementing their intrinsic production of ROS, infiltrating monocytes in the aorta can uncouple NOS3, thus amplifying vascular oxidative stress in hypertension.40
Regarding the role of macrophage cytokines, TNF augments salt appetite and promotes sodium reabsorption in the thick ascending limb of the nephron by suppressing NOS3 expression.48–50 TNF deficiency therefore ameliorates hypertension during RAS activation.47, 48 Our recent studies suggest that macrophages may similarly potentiate hypertension through their secretion of IL-1.51 Accordingly, mice lacking the receptor for IL-1α and β have enhanced accumulation of nitric oxide (NO)-expressing macrophages in the kidney early in the course of hypertension and are partially protected from Ang II-induced blood pressure elevation. In our model, IL-1 receptor stimulation releases the NKCC2 sodium co-transporter from tonic inhibition by nitric oxide (NO) leading to enhanced salt retention during RAS activation. IL-1 receptor activation appears to attenuate NO secretion from intra-renal macrophages by curtailing NOS2 expression. Thus, monocytes/macrophages can promote hypertension via the interlocking actions of ROS and M1 cytokines. Nevertheless, the effects of these M1 cytokines on blood pressure may depend on their absolute levels and localization as pharmacologic infusions of TNF or IL-1 can promote natriuresis.49, 52, 53
Two important recent findings further confound the simplistic notion that myeloid cells uniformly promote blood pressure elevation. First, mononuclear phagocytes in the dermis exert anti-hypertensive actions by allowing the mobilization and removal of non-osmotically stored sodium and by driving vasodilatory prostanoid production.54, 55 Second, a unique population of Gr-1-expressing myeloid derived-suppressor cells can attenuate chronic elevations in blood pressure, at least in part by curtailing the pro-hypertensive effects of activated T lymphocytes in the vasculature.56 Accordingly, the actions of myeloid cell populations in hypertension depend on the inflammatory mediators they elaborate and their effects on oxidant stress in cardiovascular control centers, analogous to the actions of T lymphocyte subsets discussed below.
As monocytes and macrophages express AT1 receptors, the hypothesis that Ang II promotes hypertension in part by stimulating the macrophage AT1 receptor would be consistent with the pathogenic actions of AT1 receptors in the target organ. Indeed, several in vitro experiments support this possibility. For example, incubation of a human monocyte cell line with Ang II elicits activation of NF-κB signaling and promotes the release of TNF via an AT1 receptor-dependent pathway.57–59 Moreover, phagocytic capacity is reduced in rodent macrophages lacking the AT1A receptor.22 The reported effects of AT1 receptor activation of macrophage migration have been inconsistent. Whereas AT1 receptor inhibition attenuated CCR2-dependent migration of monocytes isolated from hypertensive rats,60 we were not able to appreciate alterations in CCL2-induced migration of macrophages from AT1A receptor-deficient mice. Collectively the in vitro studies above would suggest that disrupting AT1A receptor signals may interfere with several important monocyte/macrophage functions. Nevertheless, approaches using pharmacologic antagonists may be confounded by off-target effects, and the development or phenotype of a macrophage from a mouse that is globally deficient of the AT1A receptor may be altered by the absence of AT1A receptor signaling in other immune and even non-immune cell lineages.
To circumvent these potential confounders, we used a conditional gene targeting strategy to selectively delete the coding exon from the Agtr1a gene in LysM-expressing myeloid cells of mice (LysM Cre+ Agtr1aflox/flox = “Macro KO”). We then compared renal injury and blood pressure elevation in these animals and wild-type controls (LysM Cre− Agtr1aflox/flox = “WT”) during RAS activation.10 We found that the Macro KO animals had a chronic hypertensive response to Ang II similar to that of the WTs but had exaggerated levels of tubular and interstitial injury in the kidney. These findings were consistent with the protective actions of AT1A receptors on immune cells detected in our earlier BMT study21 but could not explain the exaggerated levels of hypertension seen in our AT1A receptor-deficient bone marrow chimeras. In particular, our conditional gene targeting approach revealed a capacity for the macrophage AT1A receptor to mitigate fibrosis in the kidney,10 confirming earlier studies from the Ichikawa group using bone marrow chimeras.22 In our hands, activating the AT1A receptor on macrophages in vitro suppressed the emergence of the pro-inflammatory M1 phenotype, which was reflected in vivo by attenuated expression of M1 cytokines including TNF and IL-1β in the Macro KO kidney during RAS activation,10 consistent with the findings of Ma and Fogo in their model of obesity related glomerulopathy.23 One caveat with our LysM Cre deletion strategy is that this approach may have also deleted AT1A receptors from neutrophils, and neutrophils are now receiving attention for their potential role in blood pressure regulation.61 Notwithstanding this limitation, our studies would indicate that activating the AT1 receptor directly on macrophages actually suppresses target organ damage in hypertension, particularly within the kidney, raising the possibility that the effects of global RAS activation to promote inflammation accrue from the activation of AT1 receptors in the target organ or, at least, in non-myeloid hematopoietic cells (Figure 1).
Figure 1.
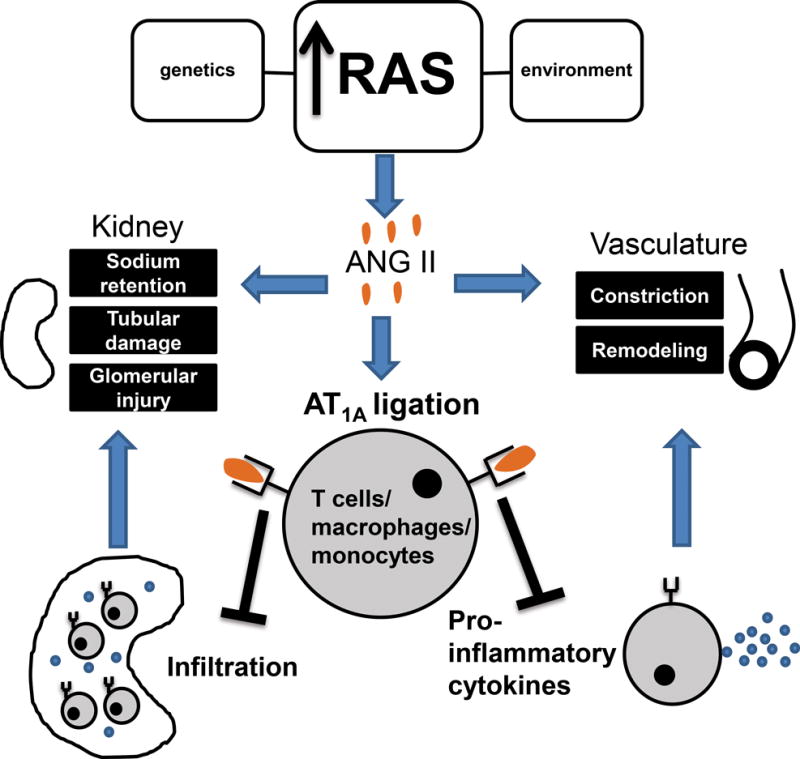
Interactions between the renin angiotensin system (RAS) and immune system during the pathogenesis of hypertension. Inappropriate RAS activation increases circulating Ang II. Elevated Ang II directly promotes sodium retention in the kidney and systemic vascular constriction. During hypertension, T cells and monocytes/macrophages accumulate in the kidney and vasculature where they mediate tissue injury and deleterious remodeling. Immune cells harbor the type 1 Ang II receptor, AT1A. In an apparent feedback mechanism, ligation of the AT1A receptor constrains T cell accumulation around the renal blood vessels, and activation of the AT1A receptor on T cells and macrophages limits their differentiation toward the pro-inflammatory Th1 and M1 subsets, respectively. This inhibition of Th1 and M1 polarization attenuates damage to the kidney during hypertension and mitigates the emergence of renal fibrosis that culminates in organ failure.
T LYMPHOCYTES, THE AT1 RECEPTOR, AND HYPERTENSION
T lymphocytes are key constituents of the adaptive immune system. As such, they are recruited by innate immune cells to aide in the specific response against pathogens. Innate immune cells present pathogen-derived antigens to T cells in peripheral lymphoid tissue. T cells express specific antigen receptors and respond to a cognate antigen by clonal expansion. Following expansion, T cells travel to the site of inflammation and perform their effector functions, which vary depending on their sub-classification. CD4+ T cells can release a wide range of cytokines and are often referred to as T helper (Th) cells. CD8+ T cells, on the other hand, directly kill cells by the release of cytotoxic factors. CD4+ T cells are generally divided into four subtypes depending on their cytokine profile: Th1, Th2, Th17, and Treg. Although additional T helper subsets have recently been defined, such as Th9 and Th22, our understanding of their role in cardiovascular physiology is limited.62–64 Pro-inflammatory Th1 cells release cytokines such as interferon-γ (IFN), TNF, and IL-12, and stimulate macrophages and CD8+ T cells.65 Th2 cells stimulate B cell antibody production by secreting the cytokines IL-4, IL-9, and IL-13. Th17 cells are pro-inflammatory and have been implicated in the pathogenesis of several autoimmune diseases.66 Th17 cells produce IL-17, a potent pro-inflammatory cytokine. Finally T regulatory cells, or Tregs, are anti-inflammatory and release cytokines that inhibit other immune cell types, including IL-10 and TGFβ.67 As with myeloid cells, the distinction among different T cell subsets in vivo is imprecise, since they share many of the same surface receptors. However, each subset is driven by unique transcription factors and produce distinct cytokines that can, with some difficulty, be measured ex vivo.66, 68–70
T lymphocytes were experimentally linked to hypertension many years ago. However, a landmark study by Guzik et al. in 2007 sparked a recent upsurge of studies examining the actions of T cells in hypertension.47 In the Guzik studies, lymphocyte-deficient (Rag1−/−) mice had attenuated Ang II-induced hypertension, and adoptive transfer of WT T cells, but not B cells, restored the hypertensive response. Since then, T cells have been implicated in experimental models of preeclampsia,71 pulmonary hypertension,72 salt-sensitive hypertension,73 DOCA-salt hypertension47, and others. The mechanisms by which T cells intensify tissue injury and elevate blood pressure during hypertension are areas of intense investigation but include contributions to endothelial dysfunction in the vasculature47 and inappropriate sodium retention in the kidney,74 both of which accrue at least in part from local generation of reactive oxygen species (ROS).75
Canonical T cell activation occurs in peripheral lymphoid tissue and requires recognition of peptide antigens on the surface of antigen presenting cells (APCs) via the T cell receptor (TCR). Also required for T cell activation are APC co-stimulatory signals via receptors proximal to the TCR.76 Whereas the proportion of activated circulating CD4+ T cells increases during Ang II-induced or DOCA-salt hypertension, genetic or pharmacological inhibition of T cell co-stimulation blocks T cell activation and attenuates blood pressure elevation in these models.77 With regard to the antigen(s) responsible for eliciting T cell activation in hypertension, little is known. A novel hypothesis from Harrison and colleagues suggests that intrinsic proteins within APCs are structurally modified via byproducts of NADPH oxidase activity during hypertension, and thus are presented as antigens to T cells.78 Pharmacological prevention of this protein modification in vivo attenuated hypertension. In separate studies, Dr. Rodriguez-Iturbe’s group implicated heat shock proteins, particularly HSP70, as a renal autoantigen in salt-sensitive hypertension.79 These studies would suggest that T cell immunity in hypertension is driven by antigen presentation.
Both CD4+ and CD8+ T cells infiltrate the kidney and vasculature during hypertension and contribute to tissue injury and dysfunction by a variety of putative mechanisms, including the production of ROS and the release of pro-inflammatory cytokines. CD8+ T cells are capable of elaborating ROS and appear to play a more critical role than CD4+ T cells in potentiating blood pressure elevation.80 ROS drive endothelial dysfunction, sodium retention, and tissue injury. The amount of ROS in T cells increases upon activation81 and adoptive transfer of NADPH oxidase-null T cells into Rag1−/− mice failed to fully restore the Ang II-induced hypertension and vascular dysfunction seen in Rag1−/− mice repopulated with WT T cells.47 Moreover, CD4+ Th1 cells elaborate TNF, a potent pro-inflammatory cytokine that induces ROS production, activates other immune cells,82 and potentiates sodium retention, as mentioned above. Inhibition of TNF signaling attenuates hypertension and reduces end organ damage.47, 83 Nevertheless, genetic deletion of pro-inflammatory Th1 cells ameliorates glomerular damage without impacting blood pressure11, suggesting that the actions of TNF produced by non-lymphoid cells in the CNS and the kidney to promote salt appetite and reabsorption, respectively,48–50 may figure more prominently in the pathogenesis of hypertension than T cell TNF. The release of IL-17 by tissue-infiltrating Th17 cells exacerbates hypertension during RAS activation.84 By contrast, repeated adoptive transfer of T regulatory cells diminishes vascular injury and Ang II-induced blood pressure elevation.85 Thus, T cell subsets can have drastically varying effects on the hypertensive response depending on their capacities to generate ROS and/or their effects on the cytokine milieu in the kidney and vasculature.
As with other immune cells, expression of the AT1 receptor on T cells guides their differentiation and function with surprising consequences in the setting of hypertension. In our hands, a conditional gene targeting strategy revealed protective actions of the T cell AT1A receptor in hypertension similar to our findings with the macrophage AT1A receptor.10, 11 Specifically, deleting the AT1A receptor from T cells in mice yielded exaggerated perivascular T cell infiltration in the kidney and augmented albuminuria, recapitulating our earlier findings with the AT1A receptor-deficient bone marrow chimeras.21 Consistent with these findings, the T cell AT1A receptor limited damage to the kidney glomerulus and renal expression of the injury marker Ngal (Lcn2) during RAS-mediated hypertension. However, in contrast to the macrophage AT1A receptor, the actions of the T cell AT1A receptor did not influence the extent of tubular injury or interstitial fibrosis in our model. Congruent with the capacity of AT1A receptor activation on T cells to limit glomerular injury in hypertension, CD4+ T cells isolated from the kidney and spleen of the animals lacking the T cell AT1A receptor had enhanced expression of the Th1 cytokines IFN and TNF. Further studies revealed that activating the T cell AT1A receptor limits Th1 differentiation of the T cell and thereby attenuates hypertensive kidney damage by suppressing the Th1 transcription factor T-bet (Figure 1).11, 50, 68
Studies using T lymphocytes isolated from mice that are globally deficient of the AT1A receptor are more consistent with the view that the T cell AT1A receptor promotes rather than suppresses inflammation. For example, splenic lymphocytes from Atgr1a−/− animals showed blunted proliferative responses following allogeneic stimulation.8 In vitro, these Agtr1a−/− T cells had impaired cytokine generation,9 and adoptive transfer of these cells into Rag1−/− mice only partly restored the hypertension and vascular dysfunction observed following adoptive transfer of wild-type T cells.47 One interpretation to reconcile the discrepant findings with the global versus conditional Agtr1a mutants is that activation of the AT1A receptors on non-immune cell lineages indirectly influences AT1A receptor functions in immune cells. The protective effect of the immune cell AT1A receptor in our bone marrow chimera studies would not conflict with this interpretation as bone marrow cells from the globally Agtr1a−/− donors in those studies were permitted to differentiate and repopulate a niche in an environment of non-immune cells with a full complement of AT1A receptors. Another intriguing hypothesis is that AT1A receptor activation on immune cells suppresses hypertensive kidney damage in our conditional mutant and bone marrow chimera studies by triggering apoptosis in leukocytes due to sustained overactivation.86 In this scenario, the direct effects of AT1 receptors on the differentiation of immune cells become less physiologically relevant in vivo.
CONCLUSIONS AND FUTURE DIRECTIONS
Innate and adaptive immune cells can potentiate hypertension and end organ damage by mediating chronic inflammation in cardiovascular control centers including the vasculature and the kidney. After infiltrating these tissues, mononuclear cells release ROS and inflammatory cytokines that regulate tissue injury and alter the release of vasoactive mediators, leading to endothelial dysfunction and/or sodium retention. Ongoing studies continue to parse actions of myeloid and T lymphocyte subpopulations in hypertension, and recent data indicate that distinct pro-inflammatory subsets within these broad populations, for example effector memory T cells,87, 88 play important roles in blood pressure elevation and consequent target organ damage. By contrast, unique anti-inflammatory immune cell populations, including myeloid-derived suppressor cells and Tregs, can attenuate hypertension.56, 85 Further investigation into these pro-and anti-inflammatory subsets should identify novel therapeutic targets for preventing the catastrophic complications of hypertension including cardiovascular and end stage kidney disease. Further exploration of the proximal determinants leading to T cell activation in hypertension, particularly if it accrues from antigen presentation and clonal T cell expansion will allow us to more precisely confront essential hypertension as an autoimmune disease. Another important emerging area of research in hypertension will be to elucidate the role of B lymphocytes in hypertension. Although adoptive transfer of B cells into an empty lymphocyte niche could not recapitulate the hypertensive response,47 B cell deficiency (BAFF-R−/−) in an otherwise intact immune system ameliorates hypertension,89 which suggests that B cells can promote blood pressure elevation through interactions with other immune cell populations. Thus, studies to date implicate several major myeloid and lymphoid populations in promoting hypertension. Despite important actions of some small mononuclear cell subsets to limit the hypertensive response, one might speculate that the broad thrust of pro-inflammatory innate and adaptive immune responses to elevate blood pressure have evolved to prevent circulatory collapse in the wake of overwhelming sepsis. The predilection for oxidant and even environmental stress to enhance susceptibility to hypertension through immune-mediated mechanisms is consistent with this hypothesis.90
The interactions between the immune system and the RAS add yet another layer of complexity to re-interpreting essential hypertension as an autoimmune disease. We acknowledge that our studies highlighting a protective effect of AT1 receptors on macrophages and T cells in hypertension are controversial and await corroboration. Nevertheless, our results with bone marrow chimera studies and conditional mutant approaches are directionally consistent, and the notion that nature may have evolved a protective function for immune cell AT1 receptors to temper the pathogenic actions of AT1 receptors in the vasculature and kidney during hypertension offers an attractive “feedback” paradigm. Moreover, we find evidence that activating AT1 receptors on immune cells also attenuates normotensive kidney fibrosis10 and acute kidney injury.91 Given the well-documented detrimental actions of renal and vascular AT1 receptors in hypertension, confirmation that AT1 receptors on immune cells are protective would not diminish the utility of ARBs for the treatment of hypertension, but, rather, would suggest that adjunct immunomodulatory therapy may be warranted to mitigate the off-target, pro-inflammatory effects of blocking the AT1 receptors in myeloid cells and lymphocytes.
Acknowledgments
SOURCES OF FUNDING
NIH grants DK087893, HL128355, P30DK096493; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893; Edna and Fred L. Mandel Center for Hypertension and Atherosclerosis Research.
Footnotes
DISCLOSURES
None.
References
- 1.Hall JE, Brands MW, Henegar JR. Angiotensin ii and long-term arterial pressure regulation: The overriding dominance of the kidney. J Am Soc Nephrol. 1999;10(Suppl 12):S258–265. [PubMed] [Google Scholar]
- 2.Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: A specific target for hypertension management. Am J Hypertens. 1999;12:205S–213S. doi: 10.1016/s0895-7061(99)00103-x. [DOI] [PubMed] [Google Scholar]
- 3.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, Group LS. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (life): A randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 4.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–1033. doi: 10.1161/CIRCRESAHA.116.303697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Iturbe B, Pons H, Quiroz Y, Johnson RJ. The immunological basis of hypertension. Am J Hypertens. 2014;27:1327–1337. doi: 10.1093/ajh/hpu142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17:S218–225. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida S, Takeuchi T, Kotani T, Yamamoto N, Hata K, Nagai K, Shoda T, Takai S, Makino S, Hanafusa T. Infliximab, a tnf-alpha inhibitor, reduces 24-h ambulatory blood pressure in rheumatoid arthritis patients. J Hum Hypertens. 2014;28:165–169. doi: 10.1038/jhh.2013.80. [DOI] [PubMed] [Google Scholar]
- 8.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM. Angiotensin ii regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104:1693–1701. doi: 10.1172/JCI7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of t-cell function by endogenously produced angiotensin ii. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, Lefler WS, Chen D, Crowley SD. Type 1 angiotensin receptors on macrophages ameliorate il-1 receptor-mediated kidney fibrosis. J Clin Invest. 2014;124:2198–2203. doi: 10.1172/JCI61368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on t lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110:1604–1617. doi: 10.1161/CIRCRESAHA.111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol. 1958;34:685–715. [PMC free article] [PubMed] [Google Scholar]
- 13.Still WJ. The early effect of hypertension on the aortic intima of the rat. An electron microscopic study. Am J Pathol. 1967;51:721–734. [PMC free article] [PubMed] [Google Scholar]
- 14.Takebayashi S. Ultrastructural studies on glomerular lesions in experimental hypertension. Acta Pathol Jpn. 1969;19:179–200. doi: 10.1111/j.1440-1827.1969.tb00701.x. [DOI] [PubMed] [Google Scholar]
- 15.Olsen F. Transfer of arterial hypertension by splenic cells from doca-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C. 1980;88:1–5. doi: 10.1111/j.1699-0463.1980.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 16.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of doca and salt hypertension in mice. Acta Pathol Microbiol Scand A. 1976;84:523–528. doi: 10.1111/j.1699-0463.1976.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 17.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56:643–649. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension. 2014;64:792–800. doi: 10.1161/HYPERTENSIONAHA.114.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugiyama F, Haraoka S, Watanabe T, Shiota N, Taniguchi K, Ueno Y, Tanimoto K, Murakami K, Fukamizu A, Yagami K. Acceleration of atherosclerotic lesions in transgenic mice with hypertension by the activated renin-angiotensin system. Lab Invest. 1997;76:835–842. [PubMed] [Google Scholar]
- 20.Kato H, Ishida J, Nagano K, Honjo K, Sugaya T, Takeda N, Sugiyama F, Yagami K, Fujita T, Nangaku M, Fukamizu A. Deterioration of atherosclerosis in mice lacking angiotensin ii type 1a receptor in bone marrow-derived cells. Lab Invest. 2008;88:731–739. doi: 10.1038/labinvest.2008.42. [DOI] [PubMed] [Google Scholar]
- 21.Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux PL, Ruiz P. A role for angiotensin ii type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin ii-dependent hypertension. Hypertension. 2010;55:99–108. doi: 10.1161/HYPERTENSIONAHA.109.144964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida M, Fujinaka H, Matsusaka T, Price J, Kon V, Fogo AB, Davidson JM, Linton MF, Fazio S, Homma T, Yoshida H, Ichikawa I. Absence of angiotensin ii type 1 receptor in bone marrow-derived cells is detrimental in the evolution of renal fibrosis. J Clin Invest. 2002;110:1859–1868. doi: 10.1172/JCI200215045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma LJ, Corsa BA, Zhou J, Yang H, Li H, Tang YW, Babaev VR, Major AS, Linton MF, Fazio S, Hunley TE, Kon V, Fogo AB. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am J Physiol Renal Physiol. 2011;300:F1203–1213. doi: 10.1152/ajprenal.00468.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuda D, Sata M, Ishizaka N, Nagai R. Critical role of bone marrow angiotensin ii type 1 receptor in the pathogenesis of atherosclerosis in apolipoprotein e deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:90–96. doi: 10.1161/ATVBAHA.107.152363. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto S, Yancey PG, Zuo Y, Ma LJ, Kaseda R, Fogo AB, Ichikawa I, Linton MF, Fazio S, Kon V. Macrophage polarization by angiotensin ii-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2856–2864. doi: 10.1161/ATVBAHA.111.237198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Idris-Khodja N, Mian MO, Paradis P, Schiffrin EL. Dual opposing roles of adaptive immunity in hypertension. Eur Heart J. 2014;35:1238–1244. doi: 10.1093/eurheartj/ehu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 29.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 30.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Italiani P, Boraschi D. From monocytes to m1/m2 macrophages: Phenotypical vs. Functional differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT, Lieu M, Samuel CS, Diep H, Kemp-Harper BK, Tare M, Ricardo SD, Guzik TJ, Sobey CG, Drummond GR. M2 macrophage accumulation in the aortic wall during angiotensin ii infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol. 2015;309:H906–917. doi: 10.1152/ajpheart.00821.2014. [DOI] [PubMed] [Google Scholar]
- 35.Capers Qt, Alexander RW, Lou P, De Leon H, Wilcox JN, Ishizaka N, Howard AB, Taylor WR. Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension. 1997;30:1397–1402. doi: 10.1161/01.hyp.30.6.1397. [DOI] [PubMed] [Google Scholar]
- 36.Elmarakby AA, Quigley JE, Olearczyk JJ, Sridhar A, Cook AK, Inscho EW, Pollock DM, Imig JD. Chemokine receptor 2b inhibition provides renal protection in angiotensin ii - salt hypertension. Hypertension. 2007;50:1069–1076. doi: 10.1161/HYPERTENSIONAHA.107.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, Egashira K. Critical role of monocyte chemoattractant protein-1 receptor ccr2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res. 2004;94:1203–1210. doi: 10.1161/01.RES.0000126924.23467.A3. [DOI] [PubMed] [Google Scholar]
- 38.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme m-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 39.Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A, Jones ES, Widdop RE, Armitage JA, Sakkal S, Ricardo SD, Sobey CG, Drummond GR. Reversal of vascular macrophage accumulation and hypertension by a ccr2 antagonist in deoxycorticosterone/salt-treated mice. Hypertension. 2012;60:1207–1212. doi: 10.1161/HYPERTENSIONAHA.112.201251. [DOI] [PubMed] [Google Scholar]
- 40.Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, Brahler M, Knorr M, Brandt M, Karbach SH, Becker C, Oelze M, Bauersachs J, Widder J, Munzel T, Daiber A, Wenzel P. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin ii. J Biol Chem. 2014;289:27540–27550. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hughson MD, Gobe GC, Hoy WE, Manning RD, Jr, Douglas-Denton R, Bertram JF. Associations of glomerular number and birth weight with clinicopathological features of african americans and whites. Am J Kidney Dis. 2008;52:18–28. doi: 10.1053/j.ajkd.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 43.Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, Scholze J. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–117. doi: 10.1161/01.hyp.34.1.113. [DOI] [PubMed] [Google Scholar]
- 44.Parissis JT, Korovesis S, Giazitzoglou E, Kalivas P, Katritsis D. Plasma profiles of peripheral monocyte-related inflammatory markers in patients with arterial hypertension. Correlations with plasma endothelin-1. Int J Cardiol. 2002;83:13–21. doi: 10.1016/s0167-5273(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 45.Palomo I, Marin P, Alarcon M, Gubelin G, Vinambre X, Mora E, Icaza G. Patients with essential hypertension present higher levels of se-selectin and svcam-1 than normotensive volunteers. Clin Exp Hypertens. 2003;25:517–523. doi: 10.1081/ceh-120025335. [DOI] [PubMed] [Google Scholar]
- 46.Madej A, Okopien B, Kowalski J, Haberka M, Herman ZS. Plasma concentrations of adhesion molecules and chemokines in patients with essential hypertension. Pharmacol Rep. 2005;57:878–881. [PubMed] [Google Scholar]
- 47.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin ii-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramseyer VD, Garvin JL. Tumor necrosis factor-alpha: Regulation of renal function and blood pressure. Am J Physiol Renal Physiol. 2013;304:F1231–1242. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin ii-dependent hypertension. Hypertension. 2014;64:1275–1281. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the nkcc2 co-transporter in the nephron. Cell Metab. 2016;23:360–368. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shahid M, Francis J, Majid DS. Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am J Physiol Renal Physiol. 2008;295:F1836–1844. doi: 10.1152/ajprenal.90297.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohan DE, Merli CA, Simon EE. Micropuncture localization of the natriuretic effect of interleukin 1. Am J Physiol. 1989;256:F810–813. doi: 10.1152/ajprenal.1989.256.5.F810. [DOI] [PubMed] [Google Scholar]
- 54.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Muller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-c-dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 55.Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. 2015;125:4281–4294. doi: 10.1172/JCI81550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE, Shen XZ. Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res. 2015;117:858–869. doi: 10.1161/CIRCRESAHA.115.306539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JA, Berliner JA, Nadler JL. Angiotensin ii increases monocyte binding to endothelial cells. Biochem Biophys Res Commun. 1996;226:862–868. doi: 10.1006/bbrc.1996.1441. [DOI] [PubMed] [Google Scholar]
- 58.Kranzhofer R, Browatzki M, Schmidt J, Kubler W. Angiotensin ii activates the proinflammatory transcription factor nuclear factor-kappab in human monocytes. Biochem Biophys Res Commun. 1999;257:826–828. doi: 10.1006/bbrc.1999.0543. [DOI] [PubMed] [Google Scholar]
- 59.Hahn AW, Jonas U, Buhler FR, Resink TJ. Activation of human peripheral monocytes by angiotensin ii. FEBS Lett. 1994;347:178–180. doi: 10.1016/0014-5793(94)00531-1. [DOI] [PubMed] [Google Scholar]
- 60.Dai Q, Xu M, Yao M, Sun B. Angiotensin at1 receptor antagonists exert anti-inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol. 2007;152:1042–1048. doi: 10.1038/sj.bjp.0707454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fabbiano S, Menacho-Marquez M, Robles-Valero J, Pericacho M, Matesanz-Marin A, Garcia-Macias C, Sevilla MA, Montero MJ, Alarcon B, Lopez-Novoa JM, Martin P, Bustelo XR. Immunosuppression-independent role of regulatory t cells against hypertension-driven renal dysfunctions. Mol Cell Biol. 2015;35:3528–3546. doi: 10.1128/MCB.00518-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugita S, Kawazoe Y, Imai A, Kawaguchi T, Horie S, Keino H, Takahashi M, Mochizuki M. Role of il-22- and tnf-alpha-producing th22 cells in uveitis patients with behcet’s disease. J Immunol. 2013;190:5799–5808. doi: 10.4049/jimmunol.1202677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of t helper 9 cells. Nat Rev Immunol. 2015;15:295–307. doi: 10.1038/nri3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta ‘reprograms’ the differentiation of t helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 65.Constant SL, Bottomly K. Induction of th1 and th2 cd4+ t cell responses: The alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 66.Korn T, Bettelli E, Oukka M, Kuchroo VK. Il-17 and th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 67.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory t cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 68.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, t-bet, directs th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 69.Hori S, Nomura T, Sakaguchi S. Control of regulatory t cell development by the transcription factor foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 70.Zheng W, Flavell RA. The transcription factor gata-3 is necessary and sufficient for th2 cytokine gene expression in cd4 t cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 71.Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW, Jr, Wallace K, LaMarca B. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond) 2016;130:409–419. doi: 10.1042/CS20150702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kherbeck N, Tamby MC, Bussone G, Dib H, Perros F, Humbert M, Mouthon L. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin Rev Allergy Immunol. 2013;44:31–38. doi: 10.1007/s12016-011-8265-z. [DOI] [PubMed] [Google Scholar]
- 73.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol. 2014;307:F499–508. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin ii-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J, 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. J Clin Invest. 2014;124:4642–4656. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol. 2013;304:F289–299. doi: 10.1152/ajprenal.00517.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med. 2004;37:1144–1151. doi: 10.1016/j.freeradbiomed.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 82.Blaser H, Dostert C, Mak TW, Brenner D. Tnf and ros crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Huang B, Cheng Y, Usa K, Liu Y, Baker MA, Mattson DL, He Y, Wang N, Liang M. Renal tumor necrosis factor alpha contributes to hypertension in dahl salt-sensitive rats. Sci Rep. 2016;6:21960. doi: 10.1038/srep21960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin ii-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin ii-induced hypertension and vascular injury. Hypertension. 2011;57:469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 86.Harrison DG, Guzik TJ. Studies of the t-cell angiotensin receptor using cre-lox technology: An unan-t-cellpated result. Circ Res. 2012;110:1543–1545. doi: 10.1161/CIRCRESAHA.112.271411. [DOI] [PubMed] [Google Scholar]
- 87.Itani HA, Harrison DG. Memories that last in hypertension. Am J Physiol Renal Physiol. 2015;308:F1197–1199. doi: 10.1152/ajprenal.00633.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118(8):1233–1243. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, Tipping P, Vinh A, Samuel CS, Peter K, Guzik TJ, Kyaw TS, Toh BH, Bobik A, Drummond GR. Obligatory role for b cells in the development of angiotensin ii-dependent hypertension. Hypertension. 2015;66:1023–1033. doi: 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 90.Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin ii-induced hypertension and vascular inflammation in adult life. Hypertension. 2010;55:494–499. doi: 10.1161/HYPERTENSIONAHA.109.145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang J, Rudemiller NP, Patel MB, Wei Q, Karlovich NS, Jeffs AD, Wu M, Sparks MA, Privratsky JR, Herrera M, Gurley SB, Nedospasov SA, Crowley SD. Competing actions of type 1 angiotensin ii receptors expressed on t lymphocytes and kidney epithelium during cisplatin-induced aki. J Am Soc Nephrol. 2016 Jan 7; doi: 10.1681/ASN.2015060683. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]