

Summary
The molecular clock relies on a delayed negative feedback loop of transcriptional regulation to generate oscillating gene expression. Although the principal components of the clock are present in all circadian neurons, different neuronal clusters have varying effects on rhythmic behavior, suggesting that the clocks they house are differently regulated. Combining biochemical and genetic techniques in Drosophila, we identify a phosphorylation program native to the master pacemaker neurons that regulates the timing of nuclear accumulation of the Period/Timeless repressor complex. GSK-3/SGG binds and phosphorylates Period-bound Timeless, triggering a CK2-mediated phosphorylation cascade. Mutations that block the hierarchical phosphorylation of Timeless in vitro, also delay nuclear accumulation in both tissue culture and in vivo, and predictably change rhythmic behavior. This two-kinase phosphorylation cascade is anatomically restricted to the eight master pacemaker neurons, distinguishing the regulatory mechanism of the molecular clock within these neurons from the other clocks that cooperate to govern behavioral rhythmicity.
eTOC Blurb
Top et al. identify a mechanism through which nuclear entry of the circadian repressor complex is regulated. A GSK-3 triggered CK2 phosphorylation cascade of Timeless protein mediates nuclear accumulation. This mechanism acts in the master pacemaker neurons and regulates behavioral rhythmicity.
Introduction
Circadian behavior is the anticipation and response to cyclic environmental changes, such as light/dark cycles, that recur with a period of ~24 h. Genetic studies have revealed oscillating cell-autonomous molecular clocks that regulate a wide range of genetic programs that underlie rhythmic behavior and control various physiological processes (Abruzzi et al., 2011; Allen et al., 2015; Boothroyd et al., 2007; Wijnen et al., 2005). In both insects and vertebrates, the master regulatory clock resides in specialized circadian centers. In Drosophila, these consist of ~150 circadian neurons, of which 8 small ventral lateral neurons (s-LNvs) are the dominant master pacemakers that govern circadian rhythmicity (Helfrich-Förster, 1998; Renn et al., 1999). The molecular mechanism underlying the dominance of s-LNvs remains unclear, since the same core clock components are expressed in all circadian neurons. It is likely that the clock components in the s-LNvs undergo a specialized regulation distinct from other circadian neurons to account for their dominance in circadian circuitry.
The genetic architecture of these clocks is conserved throughout the animal kingdom, and consists of a primary delayed negative feedback loop (Crane and Young, 2014; Young and Kay, 2001; Zheng and Sehgal, 2012). In Drosophila, Period (PER), Timeless (TIM), Clock (CLK), and Cycle (CYC) are the core proteins that compose the central loop of the molecular clock, with CLK/CYC forming a transcriptional activator complex that regulates per and tim transcription, among thousands of other genes (Abruzzi et al., 2011). PER and TIM form the transcriptional repressor complex in the cytoplasm of pacemaker neurons, where it accumulates for ~6–8 h until prompted to enter the nucleus. In the nucleus, PER inhibits CLK/CYC activity before its eventual degradation, closing the auto-regulatory loop (Allada and Chung, 2010; Crane and Young, 2014; Hardin, 2011; Zheng and Sehgal, 2012). This delay between PER/TIM protein synthesis and nuclear translocation is a critical regulatory mechanism in maintaining the 24-hour periodicity of daily rhythmic behavior.
Two serine/threonine kinases, Glycogen Synthase Kinase-3 (also known as Shaggy; SGG) and Casein Kinase II (CK2) have been implicated in nuclear entry in both flies and mammals, suggesting that a parallel regulatory mechanism is more broadly used than has been previously appreciated, and underlies circadian rhythmicity. (Akten et al., 2003; Iitaka et al., 2005; Lin et al., 2002; Maier et al., 2009; Martinek et al., 2001). In flies, SGG has been shown to phosphorylate TIM and PER, and CK2 to phosphorylate PER in vitro, but a lack of evidence for protein interactions between the kinases and their targets have left unanswered how these kinases cooperate or integrate into the molecular clock, if at all (Ko et al., 2010; Lin et al., 2005; Martinek et al., 2001; Stoleru et al., 2007). Mutations that block SGG and CK2 phosphorylation of PER and TIM in vitro, disrupt behavioral rhythmicity modestly, too small to measure an effect on nuclear accumulation (Ko et al., 2010; Lin et al., 2005). Since both SGG and CK2 are promiscuous kinases involved in a wide range of cellular processes, in vivo changes in kinase activity leaves open the possibility that the health or overall function of the neuron is affected, rather than any step within the molecular clock directly. Therefore, although SGG and CK2 influence rhythmic behavior, the nature and extent of their involvement in the molecular clock remains to be explored.
Additionally, anatomically restricted kinase (and mutant kinase) overexpression studies have revealed differing neuronal roles within the circadian network for different aspects of circadian behavior (Grima et al., 2004; Stoleru et al., 2004; 2005; Yao and Shafer, 2014). This would suggest that these kinases alter the unique biochemical environments necessary to differently regulate the common clock components. We therefore set out to test if distinct and direct regulations of the molecular clock underlie the identity and function of specific neurons within the neuronal hierarchy.
In this study, we define the nuclear entry mechanism of the molecular clock that is limited to the master pacemaker neurons, providing a biochemical basis for distinguishing this “master clock” from other clocks. We show that SGG binds and phosphorylates TIM to trigger a CK2-mediated phosphorylation cascade in vitro, which itself is dependent on the formation of the PER-TIM complex, and identify the targeted phosphorylation sites. Mutations that block phosphorylation of these TIM sites in vitro delay nuclear accumulation in cultured cells and in vivo, which tightly correlates with changes in behavioral rhythmicity. TIM mutations that block or mimic phosphorylation also block or override the rhythm-shortening effect of kinase overexpression, illustrating that the hierarchical phosphorylation mechanism determined in vitro, regulates nuclear accumulation in vivo. Strikingly, this nuclear translocation timing mechanism is restricted only to the s-LNvs, suggesting that different biochemical environments regulate molecular clocks differently, and underlie the role of the neuron within the circuitry that regulates rhythmic behavior.
Results
SGG and CK2 overexpression in small ventral lateral neurons shortens rhythmic behavior
While overexpression of GSK-3/Shaggy (SGG) in the LNvs results in shortened behavioral rhythms (Stoleru et al., 2005), and Casein Kinase II (CK2) is expressed only in the ventral lateral neurons (LNvs; small and large) (Figure S1), only the small ventral lateral neurons (s-LNvs) have a highly distinct, unique role as the master regulatory pacemaker neurons (Helfrich-Förster, 1998; Renn et al., 1999). Thus we first set out to determine if SGG and CK2 activity distinguishes the s-LNv clock from other molecular clocks. Overexpression of SGG in all circadian neurons or only in LNvs led to shorter behavioral rhythmicity (Figure 1A), as previously reported (Martinek et al., 2001; Stoleru et al., 2005). Further restriction of SGG overexpression to s-LNvs (using R6 Gal4) resulted in shorter behavioral rhythms, but not l-LNVs (using C929 Gal4) (Figure 1A). Overexpressing SGG in all circadian neurons while simultaneously blocking it only in the LNvs restores wild-type rhythmic behavior (Figure 1B). Together, these data indicate that SGG activity only in the s-LNvs is critical for normal rhythmicity.
Figure 1. SGG and CK2 activity in the small ventral lateral neurons regulates behavioral rhythmicity.

(A) SGG was overexpressed in all clock neurons (tim-UAS-Gal4; tUG), both s-LNvs and l-LNvs (pdf-Gal4), s-LNvs (R6-Gal4), or l-LNvs (C929-Gal4) and behavioral periods measured.
(B) Behavioral period of flies overexpressing SGG in all circadian neurons (tUG>sgg; TS/+) or all neurons other than LNvs (tUG>sgg/pdfGal80; TS/p80) are shown, and compared to a control (+/p80).
(C) Behavioral periods are shown for transgenic flies expressing the tUG promoter, UAS-CK2α (CK2α) and UAS-CK2β (CK2β) in the indicated combinations.
(D) CK2 was overexpressed as described in panel A, and behavioral periods measured. All values used in this figure can be found in Table S1. All error bars represent the mean +/− SEM. CK2-overexpressing flies exhibited significantly shorter behavioral periods shorter than flies carrying only the transgene or only the driver. p<0.001 (***), p<0.05 (*), NS = not significant (p≥0.05).
We next set out to test the role of CK2 in s-LNVs. Earlier studies involving CK2 report that overexpression of CK2αTik (a dominant negative inhibitor of CK2 activity) lengthens behavioral rhythmicity and delays nuclear translocation of the PER/TIM complex (Smith et al., 2008). Counter-intuitively, overexpression of wild-type CK2α also lengthens behavioral rhythmicity (Lin et al., 2005). To include CK2 in our study, we first sought to resolve this apparent conflict. Since CK2 is a tetrameric holoenzyme composed of two different subunits, overexpressing a single subunit in a non-stoichiometric manner likely interferes with holoenzyme assembly and reduces kinase activity, leading to long-period rhythmic behavior. We tested this by overexpressing single subunits, which resulted in the expected long-period rhythms, and then overexpressed both subunits to produce the predicted short-period phenotype (Figure 1C). Having resolved the conflict, we overexpressed both CK2 subunits in subsets of circadian neurons to find that CK2 activity in the s-LNvs is responsible for rhythmic activity, similar to our conclusions from SGG overexpression (Figure 1D). Therefore SGG and CK2 overexpression in the s-LNvs is sufficient to shorten behavioral rhythmicity, suggesting that these kinases help distinguish the s-LNv molecular clock from other clocks.
SGG and CK2 physically interact with PER/TIM
Direct physical interaction between SGG and CK2, and components of the molecular clock has previously been unexplored or reported to be absent (Ko et al., 2010; Lin et al., 2005; Stoleru et al., 2007). To determine if SGG and CK2 act directly on the s-LNv clock, we conducted co-immunoprecipitation (co-IP) experiments to systematically test their interactions with the repressor complex (Figure 2). First, epitope-tagged TIM, PER, and SGG were expressed in S2 cells, which do not express endogenous TIM and PER. IP of TIM led to the co-IP of both PER and SGG, when expressed together (Figure 2A, lanes 1–4). The reciprocal IP of PER also led to the co-IP of both TIM and SGG, when expressed together (Figure 2A, lanes 5–8). When PER was expressed without TIM, a reproducibly lower amount of SGG protein was retrieved by co-IP, as compared to cells expressing all three proteins (Figures 2A, S2A). This difference is not observed when TIM is immunoprecipitated in the absence of PER. Therefore all three proteins interact, with SGG preferring to interact with TIM over PER. In addition, TIM and PER levels in cells co-expressing SGG are higher than in those that are not, indicates that SGG stabilizes TIM and PER, possibly through the formation of a PER-TIM-SGG complex (Figure 2B).
Figure 2. SGG and CK2 co-immunoprecipitate with TIM.

C-terminally tagged TIM-HA (T), PER-myc (P), SGG-V5 (S) or CK2α-V5 (C), or empty plasmid (−), was exogenously expressed in cultured S2 cells, in the indicated combinations.
(A) Immunoprecipitation of TIM using HA antibody and of PER using myc antibody as indicated was followed by immunoblotting. TIM, PER and SGG were probed with HA, myc, and V5 antibody, respectively.
(B) Protein load controls used for immunoprecipitation in panel A were similarly immunoblotted.
(C) TIM and PER immunoprecipitation and TIM, PER, and CK2α immunoblotting was conducted similarly to panel A.
(D) Protein load controls used for immunoprecipitation in panel C were immunoblotted similarly.
(E) Protein extracts from wild-type flies collected at 2 h intervals from ZT14 to 22 were pooled and subjected to immunoprecipitation against TIM or rat non-specific (NS) IgG. Co-immunoprecipitated proteins were detected by immunoblotting using the indicated antibodies (left panel). The loading control (LC) was similarly analyzed (right panel).
We similarly tested the interactions between the repressor complex and CK2 by expressing epitope-tagged TIM, PER, and CK2α in S2 cells. Co-IP of CK2α was equivalent with both TIM and PER (Figure 2C, lanes 3–4 and 7–8), suggesting that CK2 does not show a preference between the two in S2 cells. In contrast to the protein-stabilizing effect of TIM/SGG or PER/SGG co-expression, CK2α co-expression with TIM or PER reduces their abundance in whole cell lysates (Figure 2D). This destabilization by CK2 can be countered if PER and TIM are in a complex, suggesting that additional regulatory mechanisms are involved in PER/TIM destabilization.
Finally, we confirmed that physical interactions between TIM, PER, SGG, and CK2 also occur in vivo (Figure 2E). Following IP of endogenous TIM from fly heads collected over an interval of 8 h in the dark, we observed co-IP of PER, SGG, and CK2α. Previously, it was proposed that the TIM-SGG interaction was mediated via the light-sensing protein Cryptochrome (CRY) (Stoleru et al., 2007). To determine the contribution of CRY to TIM-SGG interactions, we repeated this experiment using flies lacking genomic cry. We found that TIM-SGG interactions were not affected by the presence or absence of CRY (Figure S2B). Thus, TIM physically associates with SGG, CK2α and PER in vivo as it does in S2 cells. Therefore TIM, PER, SGG, and CK2 can interact to accommodate a phosphorylation mechanism of the PER/TIM repressor complex.
SGG and CK2 drive a Timeless phosphorylation cascade in vitro
No phosphorylation sites have been identified on TIM, although a nuclear localization signal and a critical proline (and proximate threonine) residue within TIM are involved in nuclear localization in pacemaker cells (Jang et al., 2015; Saez et al., 2011). Since SGG preferentially binds TIM (Figure 2), we first set out to identify the residues it may phosphorylate. A sequence alignment of Drosophila TIM and its homologues in three other insect species revealed potential phosphorylation sites in a region of conserved serines and threonines (Figure 3A). Aligning this region with TIM sequences of other Drosophila species revealed a broader highly conserved serine/threonine-rich region that we termed the TIM ST (Figure 3B). We therefore focused on this TIM ST as a candidate region for SGG- and CK2-mediated regulation of nuclear translocation.
Figure 3. SGG and CK2 cooperate to phosphorylate the TIM ST region.

(A) Drosophila TIM protein is aligned with TIM homologues of the indicated insect species. Residues that share identity are highlighted in grey.
(B) Alignment of D. melanogaster TIM with other Drosophila species reveals a broader region of conserved serine-threonines (highlighted in grey). Serines and threonines that have been mutated to alanines or aspartates are indicated in red font.
(C–E) Purified fragments of TIM were used in an in vitro kinase assay and analyzed by radiography. Coomassie stains (CS) of the analyzed TIM fragments are shown.
(C) TIM fragment spanning amino acids 222–577 phosphorylated by SGG.
(D) TIM fragment spanning amino acids 222–325 phosphorylated by CK2.
(E) TIM fragment spanning amino acids 222–577 phosphorylated by CK2.
wt: wild-type, 2A: TIM S297A/T301A, 2D: TIM S297D/T301D, 3A: T305A/S309A/S313A, 3D: T305D/S309D/S313D, 5A: TIM2A/3A, 5D: TIM2D/3D, 2D3A: TIM2D/3A.
SGG is able to phosphorylate a TIM fragment spanning residues 222–577, a region that harbors the TIM ST, in vitro (Martinek et al., 2001). To determine if the TIM ST is phosphorylated, we mutated five serine/threonine residues that formed consecutive SGG consensus sites (S/T-X-X-X-S/T), and tested these mutants in an in vitro kinase assay. We grouped these serine and threonine residues into two clusters: The N-terminal S297 and T301 residues were mutated to the phospho-null alanine (tim2A) or to the phosphomimetic aspartate (tim2D); the C-terminal T305, S309 and S313 were mutated similarly to generate tim3A and tim3D; and mutations of all five residues generated tim5A or tim5D. Mutation of all five residues (TIM5A or TIM5D) blocked SGG phosphorylation of the TIM fragment, relative to wild-type (Figure 3C). Mutation of the N-terminal TIM ST sites (TIM2A or TIM2D) similarly blocked TIM phosphorylation. In contrast, mutation of the C-terminal three residues (TIM3A or TIM3D) did not impair phosphorylation, with TIM3D yielding similar or modestly augmented phosphorylation. Equivalent amounts of the TIM protein fragments were used in the assay, indicating that changes in phosphorylation are due to intrinsic SGG activity. These data demonstrate that the N-terminal S297/T301 residues are SGG phosphorylation sites, in vitro.
Since CK2 has also been implicated in regulating PER/TIM nuclear accumulation (Akten et al., 2003; Lin et al., 2002), we hypothesized that CK2 targets the C-terminal sites not phosphorylated by SGG. To test this, we repeated the in vitro kinase assay using CK2 and found that all TIM fragments were phosphorylated, with lower levels of phosphorylation of the alanine mutant proteins, and higher levels in the wild type or aspartate mutant proteins, suggesting that regions outside the TIM ST are also phosphorylated by CK2 (Figure S3). To restrict our analysis to the TIM ST, we repeated the experiment using a TIM sub-fragment spanning amino acids 222–325 (Figure 3D). Wild-type and alanine mutant forms of this TIM sub-fragment were not phosphorylated by CK2, while the phosphomimetic TIM5D, TIM2D, and TIM3D sub-fragments were strongly phosphorylated. This suggests that phosphomimetic mutations in the TIM ST can promote CK2-dependent phosphorylation elsewhere within the sub-fragment.
Since wild type TIM, TIM2A and TIM3A are not phosphorylated by CK2, and given that SGG phosphorylates S297 and/or T301 (Figure 3C), we hypothesized that SGG phosphorylation of the two N-terminal residues triggers CK2-mediated phosphorylation of the three C-terminal sites. To test this, we generated a tim2D3A mutant fragment with phosphomimetic substitutions at the SGG sites, and phospho-null substitutions at the C-terminal sites (S297D, T301D, T305A, S309A, S313A). In contrast to TIM2D, which is readily phosphorylated by CK2, alanine substitutions in the C-terminal sites blocked CK2 phosphorylation of TIM (Figure 3E). Therefore SGG phosphorylation of TIM is necessary for the CK2-mediated phosphorylation of the three C-terminal residues in the TIM ST, which in turn promotes further CK2 phosphorylation elsewhere on TIM.
SGG and CK2 phosphorylation of Timeless promotes nuclear accumulation in cultured cells
S2 cells expressing inducible fluorescently tagged PER and TIM exhibit delays in nuclear entry similar to those of pacemaker neurons in vivo, offering a system in which to dissect nuclear entry with high temporal precision (Meyer et al., 2006; Saez and Young, 1996). Using this S2 cell system, we set out to determine the nuclear entry activity of full length TIM mutants informed by our in vitro data (Figure 3). We found that in contrast to wild type PER/TIM, the onset of nuclear accumulation of PER/TIM2A complexes (with SGG sites blocked) were significantly delayed (Figure 4A; compare minute 350 and 425). In the case of PER/TIM3A (with CK2 sites blocked), the onset of PER/TIM nuclear accumulation is not affected (Figure 4A; see minute 325). These results suggest that SGG-mediated phosphorylation of the TIM ST is responsible for initiating nuclear accumulation, while CK2-mediated phosphorylation of the TIM ST regulates its efficiency.
Figure 4. Mutation of TIM phosphorylation sites alters nuclear accumulation in cultured cells.

(A) Wild-type or mutant TIM-YFP, and PER-mCherry expression induced in S2 cells using a heat shock promoter. The change in nuclear fluorescence intensity is plotted in arbitrary units, as a function of time (minutes). Nuclear accumulation of TIM protein (top panels) and PER protein (lower panels) was compared between TIM wt (blue), and TIM2A or TIM3A (red).
(B) RNAi treated cells (red) were analyzed for wild-type TIM and PER nuclear accumulation similarly to panel A, and compared to non-treated cells (in blue). Nuclear accumulation of TIM and PER were plotted in cells depleted of SGG (left panels) or CK2α (right panels). Kinase levels in cells treated (+) or not treated (−) with RNAi were determined by immunoblotting and compared to tubulin (Tub) load control (inset). N = 8–22 cells. All data points are the mean, shaded area represents +/− SEM.
(C) CFP and TIM-mCherry fusion protein were expressed in S2 cells using a constitutively active actin promoter, and imaged. Representative images are shown. Scale bar = 5 μm.
(D) Nuclear accumulation of TIM was analyzed and plotted as a ratio of nuclear and cytoplasmic TIM signal (N/C). The red line is the mean N/C, +/− SEM of Wild-type TIM (wt), TIM2A (2A), TIM2D (2D), TIM3A (3A), TIM3D (3D). The grey dashed line is a reference point for the N/C mean of wild type TIM. p<0.05 (*).
We next wanted to determine the broad activity of SGG and CK2 in PER/TIM nuclear entry. SGG and CK2 are critical for fly development, and null alleles of these genes are lethal (Kuntamalla et al., 2008; Ruel et al., 1993). Therefore we introduced interfering RNA (RNAi) to the S2-nuclear entry assay described above. RNAi-depletion of SGG in S2 cells led to a significant delay of TIM/PER nuclear accumulation, as compared to untreated cells (Figure 4B). This result mirrors the lag in nuclear accumulation observed with cells expressing TIM2A (Figures 4A–B). On the other hand, RNAi against CK2α modestly reduced CK2α protein levels, produced no discernable effect on the timing or rate of nuclear accumulation, and significantly increased the amount of protein accumulating in the nucleus (Figure 4B). These results are consistent with previous observations of TIM and PER destabilization by CK2 (Lin et al., 2002; Meissner et al., 2008; Smith et al., 2008) and our own results in S2 cells (Figure 2). Together, these data suggest that SGG chiefly affects the onset of nuclear accumulation, while CK2 helps regulate nuclear accumulation as well as regulate stability of TIM and PER.
Phosphomimetic TIM mutants accumulate in the nucleus independently of PER
Nuclear translocation of the PER/TIM complex is dependent on the presence of both TIM and PER (Myers et al., 1996; Vosshall et al., 1994), and a TIM-PER interaction was previously proposed as a condition of nuclear entry (Saez and Young, 1996). It is possible that this dimerization initiates the SGG-triggered phosphorylation cascade to promote nuclear accumulation. If so, expression of a TIM phosphomimetic mutant should bypass the need for PER expression, allowing TIM nuclear accumulation in its absence. Using the S2-cell nuclear entry assay, which expresses no endogenous TIM or PER, we expressed wild-type or mutant forms of mCherry-fused TIM, with CFP as a nuclear marker, and monitored TIM nuclear accumulation. Both wild-type TIM and phospho-null mutant forms of TIM accumulated primarily in the cytoplasm. In contrast, phosphomimetic forms of TIM accumulated primarily in the nucleus (Figure 4C). Double-blind analysis revealed a statistically significant increase in nuclear accumulation of phosphomimetic TIM mutant proteins in the absence of PER (Figure 4D). Exogenous over-expression of SGG with TIM or PER alone did not lead to increased nuclear accumulation (Figure S4). These results suggest that SGG-triggered phosphorylation of TIM that regulates nuclear translocation, is PER dependent.
SGG and CK2 phosphorylation sites on Timeless regulate behavioral rhythmicity in vivo
After establishing the TIM phosphorylation mechanism in vitro and its function in nuclear translocation in cultured cells, we next wanted to determine the effect of SGG- and CK2-dependent phosphorylation of TIM in vivo. We therefore assessed the ability of a tim mini-gene (Ousley et al., 1998) carrying mutations blocking or mimicking SGG and CK2 phosphorylation sites, to rescue the tim0 allele (Figures 5A–B). Transgenic flies expressing wild-type TIM exhibited the expected 23.5-h behavioral rhythmicity. In contrast, flies expressing TIM2A or TIM3A produced ~30.5 and 27 h long behavioral rhythms respectively, indicating that these residues are essential for normal TIM function. Flies expressing TIM2D exhibited normal behavioral rhythms (~24 h), indicative of complete rescue of the alanine form, while TIM3D flies exhibited a shorter, 19-h behavioral rhythm. Thus, mutations blocking TIM phosphorylation produce behavioral phenotypes in adult flies that correlate well with the observed delays in nuclear accumulation in cultured cells.
Figure 5. Mutation of TIM phosphorylation sites changes behavioral rhythmicity and timed nuclear accumulation.

(A) Actograms of flies expressing transgenic tim in a tim0 genetic background. Wild-type (wt), tim2A (2A), tim2D (2D), tim3A (3A), tim3D (3D) transgenic flies were monitored in a 5-day light/dark cycle (LD), followed by constant darkness (DD).
(B) The behavioral period of flies described in panel A are plotted (grey bars), and compared to heterozygous flies expressing a single copy (white bars) of the corresponding tim transgene.
(C–D) Behavioral periods of transgenic flies expressing wild-type or mutant tim in either a tim0 genetic background (grey bars), or using tUG (All circadian neurons; C) or pdf-Gal4 (LNvs; D) to overexpress CK2 in a tim0 genetic background (white bars).
(E) Fly heads were collected at the indicated zeitgeber (ZT) or circadian (CT) time points. Representative images of the small ventral lateral neurons stained for PDF (blue), TIM (red), and PER (green) are shown. Scale bar = 5 μm.
(F) All stained fly brains were visually scored to determine whether PER/TIM staining was predominantly in the nucleus or in the cytoplasm of the small ventral lateral neurons, and valued 1 or 0, respectively. 4–10 fly brains were averaged for each mutant (grey line) at each time point, and compared to wild-type (dotted black line). Where data points are absent, protein was not observable, indicating nuclear degradation of the proteins. Times are measured in ZT (black bar) or CT (grey bar), as indicated. Each data point represents the mean +/− SEM.
(G) Nuclear accumulation of wild-type TIM (black line) and TIM2A (grey line) was scored in the large ventral lateral neurons (l-LNv), dorsal lateral neurons (LNd), and the three dorsal neuron clusters (DNs), as described in Panel F (N = 4–8). Each data point represents the mean +/− SEM.
Altering PER levels in flies through transgene copy number changes behavioral rhythmicity, while changes in TIM levels do not (Ashmore et al., 2003; Baylies et al., 1987). Consistent with these earlier findings, the behavioral phenotypes of tim mutant transgenes were not dose-dependent, and were indistinguishable in animals heterozygous or homozygous for these transgenes (Figure 5B). Similarly, we could not detect phenotypic differences among transgenes integrated at different chromosomal sites (data not shown). Finally, blocking phosphorylation on TIM does not change protein levels with respect to wild-type (Figure S5). Therefore, the phenotypes caused by the mutant TIM proteins reflect their intrinsic activity rather than gene dosage or positional effects caused by transgene integration.
We next sought to test whether CK2 phosphorylation of TIM is SGG-dependent in vivo, as suggested by our in vitro analysis (Figure 3). To this end, we examined the ability of tim mutant flies to block or permit the behavioral effects of CK2 overexpression either in all circadian neurons (Figure 5C) or just LNvs (Figure 5D). Although CK2 overexpression shortens behavioral rhythmicity in wild-type flies, it does not alter it in flies carrying mutations on tim CK2 sites (tim3A or tim3D). This indicates that the CK2 sites within the TIM ST are essential and rate-limiting steps, necessary to observe the effect on rhythmic behavior caused by CK2 overexpression. This correlates well with the TIM ST-dependent phosphorylation of the TIM ST-distal region by CK2, in vitro (Figure 2, S2). On the other hand, the rhythm-shortening effect of CK2 overexpression is blocked in tim2A flies, but permitted in tim2D flies. Since tim2A and tim2D mutants encode forms of TIM that respectively block and mimic SGG phosphorylation, these results support a mechanism in which CK2 activity is subsequent to, and dependent on, SGG activity in the TIM ST. Therefore this dual kinase, phosphorylation cascade is active in vivo, mirroring our results in vitro (Figure 2).
SGG and CK2 phosphorylation sites on TIM regulate PER/TIM nuclear accumulation in pacemaker neurons in vivo
To test whether altered behavioral rhythmicity of adult flies is associated with changes in nuclear accumulation in circadian regulatory neurons, we analyzed fly brains using immunofluorescence microscopy. We entrained flies in a 12h:12h light-dark cycle (zeitgeber time; ZT), shifted them to constant darkness (circadian time; CT), and isolated brains from wild type and tim2A mutant flies at 2-h intervals between ZT14 and CT08. Brains were stained for TIM, PER and PDF, and the localizations of these proteins were assessed in the primary pacemaker neurons, the s-LNvs. In wild-type transgenic flies, we first observed TIM in the nucleus of s-LNvs at ZT20 (Figures 5E–F). By contrast, tim2A mutant flies revealed a >4 h delay in the nuclear localization of TIM, between ZT24-CT02 (Figure 5E–F). This correlates well with the increased period (~30.5 h) of behavioral rhythmicity of this mutant (Figure 5B). In the tim3A and tim3D mutant flies, TIM proteins accumulated in s-LNv nuclei with an ~2-h delay or ~2-h advance, respectively, whereas the tim2D mutant exhibited near-wild-type nuclear accumulation (Figure 5F). Once again, the delays and advances in the timing of nuclear accumulation correlate well with changes in the behavioral rhythmicity period observed in these mutants (Figure 5B), though they do not account for the entire behavioral deficit. The slight delay or advance in degradation of the mutant proteins accounts for the remaining deficit, as shown in Figure 5F. These data suggest that SGG- and CK2-dependent phosphorylations of TIM ST determine circadian periodicity principally by affecting the timing of PER/TIM nuclear accumulation.
The SGG/CK2-regulated molecular clock is restricted to the s-LNvs
We have shown that the tim mutants that block SGG and CK2 phosphorylation (tim2A and tim3A) have long behavioral rhythms, and exhibit delayed PER/TIM nuclear accumulation in the s-LNvs (Figures 5B, 5F). Strikingly, we found that the pattern of PER/TIM nuclear accumulation in the large ventral lateral neurons (l-LNvs) of the tim2A and tim3A mutants was the same as wild type (Figures 5G, S6A). These data correlate with the shorter behavioral rhythmicity caused by SGG and CK2 overexpression in s-LNVs, but not l-LNvs (Figure 1). Consistent with previous findings that the s-LNvs affect morning anticipation behavior (Stoleru et al., 2005), the tim2A and tim3A mutants exhibit a shift in the peak of morning anticipation, and a change in the range of evening anticipation with little effect on its peak (Figure S6B). This supports our hypothesis that these phosphorylation sites are relevant only in the s-LNvs. To explore any further differences between neuron clusters, we also measured the nuclear localization of TIM in the dorsal neurons (DN1s, DN2s, DN3s), and in dorsal lateral neurons (LNds) (Figure 5G). We observed a varying effect of tim2A as compared to wild type tim on nuclear entry. While the l-LNvs showed the same nuclear accumulation of TIM2A as wild-type, DN3s showed no nuclear accumulation at all. Within the remaining clusters, the LNds exhibited a delay of ~2 h, while the DN1s and DN2s exhibited a 4–6 h delay. These data suggest that a mutation affecting the nuclear translocation of the PER/TIM repressor complex is differently effective in different neuronal contexts. This supports the hypothesis that different neurons harbor differently regulated molecular clocks that converge to generate rhythmic behavior. Taken together, our data present a model in which SGG and CK2 participate in a sequential, two-step phosphorylation cascade that is required for nuclear translocation and accumulation of the TIM/PER repressor complex. This two-step regulatory mechanism acts specifically in the master regulatory circadian neurons (s-LNVs) and appears to underlie the dominance of these neurons in regulating the overall rhythmic locomotor activity of the whole adult animal.
Discussion
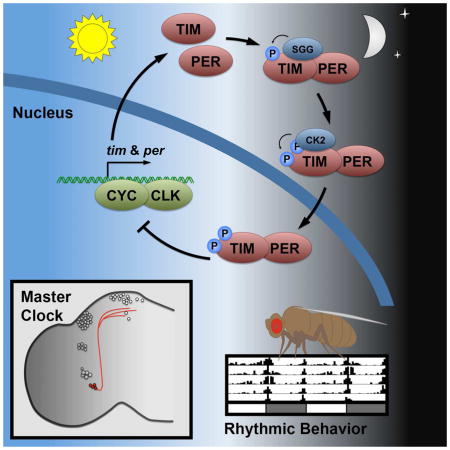
Although genetic and molecular studies have shown that SGG and CK2 play critical roles in the regulation of circadian rhythmicity in animal cells, the mechanism through which these two kinases converge to exert their control has not been previously explored (Lin et al., 2002; Martinek et al., 2001; Meissner et al., 2008; Smith et al., 2008). In this study, we present a model that establishes central and cooperative roles for SGG- and CK2-dependent phosphorylation of TIM in the regulation of subcellular localization of the PER/TIM transcriptional repressor complex (Figure 6). (1) After TIM-PER association, (2) TIM-bound SGG acts as the priming kinase for (3) CK2 in a phosphorylation cascade of specific TIM sites to determine (4) the timing of PER/TIM nuclear accumulation, thereby setting the fly behavioral period. This mechanism, which is only found in the master pacemaker neurons, is required to maintain coherent 24-h behavioral rhythmicity.
Figure 6. Model for regulating TIM-mediated nuclear entry.

Model describing SGG and CK2 cooperation in regulating the pace of the s-LNv clock. (1) TIM and PER interaction facilitates (2) SGG-mediated phosphorylation of the 297/301 sites within TIM, which (3) triggers CK2-mediated phosphorylation of the 305/309/313 sites. (4) This cascade regulates the accumulation of the PER/TIM transcriptional repressor complex in the nucleus of the small ventral lateral neurons (shown in red), which governs behavioral rhythmicity. The other circadian neurons are shown in grey.
Recently, the phospho-dependent regulation of FRQ stability was distinguished from circadian activity in Neurospora crassa (Larrondo et al., 2015). Similarly, the PER/TIM nuclear entry mechanism we describe is distinct from the mechanism that regulates protein stability. These two mechanisms have previously been difficult to separate, since post-translational modifications of PER that affect protein abundance, such as phosphorylation and O-GlcNAcylation, also affect nuclear entry (Kaasik et al., 2013; Kim et al., 2012; Lin et al., 2005; Meissner et al., 2008; Smith et al., 2008). Therefore it is unsurprising that changes in PER expression levels alter behavioral rhythmicity (Baylies et al., 1987). In contrast, changes in TIM expression levels do not change behavioral rhythmicity (Ashmore et al., 2003). Molecularly, TIM nuclear entry is PER dependent (Saez and Young, 1996), however we demonstrate that phosphomimetic mutant forms of TIM can accumulate in the nucleus without PER (Figure 4C–D). This suggests that a PER-TIM interaction is required for the TIM phosphorylation events that regulate nuclear entry.
We have shown that CK2 is a part of the mechanism that regulates nuclear entry. While we and others show that changes in CK2 activity influence both nuclear translocation and protein stability (Meissner et al., 2008; Smith et al., 2008), we also show that mutation of specific CK2 target sites in TIM3A can separate the effect of CK2 on nuclear entry from its effect on protein stability (Figures 4, 5, S5). By contrast, the timUL mutation, which falls within the TIM ST-proximal 260–292 region that is predicted to be a target for CK2 phosphorylation (Meissner et al., 2008), stabilizes TIM but has no effect on nuclear entry (Rothenfluh et al., 2000). The involvement of CK2 in both TIM nuclear translocation and TIM stability positions this kinase to couple nuclear entry of the PER/TIM repressor complex to its degradation, in that order. Thus, CK2 ensures that nuclear translocation precedes regulated degradation of the nuclear complex. It will be interesting to use phospho-specific antibodies in future immunofluorescence studies to determine in which cellular compartment phosphorylation of the TIM ST region occurs.
Our work also resolves whether SGG regulates TIM indirectly through CRY, the light-sensing protein that destabilizes TIM during the day to reset the clock (Emery et al., 1998). A lack of evidence for a TIM-SGG interaction, and the stabilization of TIM in constant light by SGG overexpression, suggest that SGG acts on TIM through CRY (Stoleru et al., 2007). However, several lines of evidence argue against a role for CRY in SGG-mediated regulation of TIM. First, we demonstrate that TIM and SGG interact independently of CRY (Figure 2, S2); second, cry01 flies exhibit wild-type rhythmicity (Dolezelova et al., 2007); third, SGG overexpression in cry01 and cry+ genetic backgrounds show the same 3-h reduction in behavioral rhythmicity (Table S1). Finally, since SGG stabilizes TIM (Figure 2B) and TIM protein oscillations are not necessary for rhythmic behavior (Ashmore et al., 2003), it is likely that SGG overexpression stabilizes TIM sufficiently to restore rhythmicity to flies in constant light. Taken together, these data demonstrate that SGG regulates TIM and the behavioral rhythm independently of CRY.
The fact that SGG initiates a dual-kinase phosphorylation cascade of TIM may provide clues to the coordination of non-light environmental inputs, such as feeding cues, to the master regulatory neuronal clock. SGG activity is regulated by a wide range of signaling pathways, including Wnt, receptor tyrosine kinases, and G-protein-coupled receptors, through phosphorylation of its Ser9/Ser21 residues (Doble, 2003; Harwood, 2001). In Drosophila, as in mammals, two essential pathways mediating endocrine signaling and nutrient/energy homeostasis, AKT and TOR-S6K, converge by virtue of their common, post-translational regulation of SGG activity. Overexpression of either AKT or TOR-S6K alter the period length of Drosophila circadian rhythmicity through their regulation of SGG (Zheng and Sehgal, 2010). Thus, the specific regulatory interactions between SGG and TIM identified in this study provide a possible mechanism for directly coupling physiological signals to phase and period responses of the circadian clock. Indeed, the other regulated steps of the circadian clock involve protein degradation or production, which do not offer the short time-scale control over rhythmicity that is necessary for adjusting behavior in response to physiological cues.
Surprisingly, the SGG/CK2-regulated mechanism we have described, which establishes the behavioral period of the fly, appears to reside in the small ventral lateral neurons (s-LNvs), two four-neuron clusters that express the neurotransmitter pigment-dispersing factor (PDF). Flies that express the mutant gene tim2A exhibit delayed nuclear entry in the s-LNvs, but not the large ventral lateral neurons (l-LNvs), relative to wild type flies (Figure 5). Additionally, overexpression of either SGG or CK2 in s-LNvs produces short-period behavioral rhythms, but no behavioral change occurs when overexpression is restricted to the l-LNvs (Figure 1). First, this suggests that the l-LNvs harbor an alternate mechanism of nuclear accumulation of the PER/TIM complex. Second, this alternate mechanism is not affected by the slowed oscillator in the s-LNvs. Since PDF is a critical neuropeptide in synchronizing and resetting circadian neurons (Renn et al., 1999) and l-LNvs do not express the PDF receptor (PDFR), it is not unexpected that the l-LNvs are not influenced by rhythmic PDF release from the s-LNvs (Park et al., 2000; Shafer et al., 2008). There is evidence however that the PDF+ s-LNvs project to the DNs and LNds, some of which express the PDFR, suggesting that there is a signaling mechanism through which the delayed s-LNv clock slows the molecular clock in these clusters (Figure 5G) (Guo et al., 2014; Im and Taghert, 2010; Seluzicki et al., 2014; Stoleru et al., 2005; Yao and Shafer, 2014; Yasuyama and Meinertzhagen, 2010). Indeed, a change in transcription activity in these downstream PDF− clusters has been observed with the overexpression of SGG kinase expression in the PDF+ s-LNvs (Stoleru et al., 2005). Although overexpression of SGG in the PDF− clusters does not alter behavioral rhythmicity (Figure 1B), it is possible that endogenous SGG is sufficient to mediate local regulatory mechanisms, as the extent of SGG involvement, if any, in PDF− clusters is currently unknown. Immunofluorescence of CK2α indicates that CK2 is expressed in the PDF+ LNvs (Lin et al., 2002), but not in the PDF− DNs or LNds (Figure S1). While it is possible that undetected CK2 expression affects the local molecular clock in the PDF− neurons, our ability to fully block the behavioral effect of CK2 overexpression in the PDF+ neurons through mutation of phosphorylation sites on TIM suggests that CK2 is unlikely to play a major role in behavioral rhythmicity in the PDF− neurons. Further, TIM2A and TIM3A variants are not delayed in nuclear entry in the l-LNvs suggesting that the dual kinase regulatory mechanism for nuclear entry is not present in the l-LNvs, despite high CK2 expression levels. Therefore, given the differences in CK2 expression in different neural clusters and CK2’s integral role in the molecular mechanism of PER/TIM nuclear accumulation, the DNs and LNds, like the l-LNvs, may control rhythmicity through a different mechanism that is dependent on signals from the s-LNvs. The mechanism of regulated nuclear entry that we describe likely influences the DNs and LNds downstream of the s-LNvs via interneuron communication (Yao and Shafer, 2014). However, these PDF− neurons may employ some of the same components as the sLNvs in a local context. Further work is needed to describe local regulatory mechanisms in the different neural clusters.
In summary, circadian rhythmic behavior is the sum product of multiple oscillating clocks within an increasingly complex network of pacemaker neurons. While it is difficult to assign a specific circadian behavior, such as morning activity, to specific neural clusters (Stoleru et al., 2005) given the fluid nature of these cell identities (Im and Taghert, 2010; Rieger et al., 2009), the hypothesis that rhythmicity is an emergent property of differently regulated clocks within the circadian network appears more likely (Yao and Shafer, 2014). These earlier studies were primarily conducted by manipulating the expression level of various kinases, without identifying the mechanistic consequences. Our study suggests that a major difference between these neural clusters is their unique biochemical environments, which give rise to the different mechanisms that regulate the local molecular clock. We describe a phospho-cluster within TIM, regulated by a SGG-triggered CK2 phosphorylation cascade, which in turn regulates nuclear translocation of the PER/TIM transcriptional repressor complex. Although TIM and PER are expressed throughout the ~150 circadian neurons, this regulatory mechanism appears to reside in the s-LNvs. The distinct biochemistry of the s-LNvs sets the s-LNv clock apart from other clocks as that of the master clock that ultimately sets the pace of fly behavioral rhythmicity.
Experimental Procedures
Additional details concerning all experimental procedures are found in the Supplemental Experimental Procedures.
Plasmids, S2 cell culture, transfection, RNAi treatment, fluorescence microscopy
Timeless and its variants, Period, Shaggy, CK2α and CFP cDNA was subcloned into the HS-Casper (Saez and Young, 1996) and pAc5.1/V5-HisA vector plasmids (Invitrogen) with the indicated tags fused to their C-termini, using standard cloning techniques. S2 cells were maintained in Schneider’s Medium supplemented with 10% FBS and transfected with effectene (Qiagen). RNAi experiments were conducted as per manufacturer’s protocol (Ambion). dsRNA was generated from the first 400 base pairs or third 400 base pairs of each target gene. S2 cells were imaged and analyzed as previously described (Meyer et al., 2006; Syed et al., 2011). Briefly, S2 cells in chamber slides were heat shock induced to exogenously express TIM-YFP, PER-mCherry and free CFP. Cells were imaged using a DeltaVision system (Applied Precision) equipped with an inverted Olympus IX70 microscope. Imaged cells were analyzed using a locally written algorithm in Matlab (Mathworks Inc) to measure fluorescence intensity in the nuclei and cytoplasm.
Immunoblotting, immunoprecipitation and immunocytochemistry
Fly heads and S2 cells were lysed in lysis buffer, diluted, either immunoprecipitated or frozen, and analyzed by immunoblot as detailed in supplemental experimental procedures. Fly brains were collected, fixed, mounted, and imaged using Leica confocal microscopy as previously described (Saez et al., 2011).
Protein purification and in vitro kinase assay
Wild type and TIM variant fragments were purified from bacteria using an N-terminal GST tag. Purified protein bound to glutathione beads was used in an in vitro kinase assay and analyzed by radiography as previously described (Martinek et al., 2001). Briefly, protein-bound beads were washed with the appropriate buffer, and incubated with 1 μl 32PγATP and 0.2 μl of kinase. A fraction of the protein was subsequently analyzed by SDS-PAGE and radiography.
Transgenic flies and behavioural analysis
TIM transgenic variants were generated as previously described (Ousley et al., 1998; Rutila et al., 1998). Briefly, the tim minigene was cloned into Casper4 vector, injection into embryos, and selected for P-element based insertion. R6-Gal4 and C929-Gal4 flies were gifts from Paul Taghert. cry01 flies were a gift from Jeff Hall. Individual flies were analyzed for locomotor activity after light dark entrainment using the Drosophila Activity Monitor System IV (Trikinetics). Period was determined using ClockLab Software (Actimetrics).
Supplementary Material
Highlights.
SGG and CK2 interact with TIM
SGG phosphorylation of TIM triggers a CK2 phosphorylation cascade
Phosphorylation site mutation alters TIM/PER nuclear entry and behavioral period
Mechanism of TIM/PER nuclear accumulation is restricted to master pacemaker neurons
Acknowledgments
We would like to thank Jenna L. O’Neil for expert technical assistance, and Alina Patke, Wanhe Li, and Philip B Kidd for critical review of the manuscript. This work was supported by a grant from the National Institutes of Health (GM054339) to Michael W Young. DT would like to thank MWY for his full support in honoring our friend and colleague LS.
Footnotes
Author Contributions
D.T. conceived and performed experiments, analyzed data, wrote the manuscript. E.H. and E.L.A. performed experiments. S.S. developed software. L.S. conceived and performed experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abruzzi KC, Rodriguez J, Menet JS, Desrochers J, Zadina A, Luo W, Tkachev S, Rosbash M. Drosophila CLOCK target gene characterization: implications for circadian tissue-specific gene expression. Genes & Development. 2011;25:2374–2386. doi: 10.1101/gad.178079.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akten B, Jauch E, Genova GK, Kim EY, Edery I, Raabe T, Jackson FR. A role for CK2 in the Drosophila circadian oscillator. Nat Neurosci. 2003;6:251–257. doi: 10.1038/nn1007. [DOI] [PubMed] [Google Scholar]
- Allada R, Chung BY. Circadian organization of behavior and physiology in Drosophila. Annu Rev Physiol. 2010;72:605–624. doi: 10.1146/annurev-physiol-021909-135815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen VW, O’Connor RM, Ulgherait M, Zhou CG, Stone EF, Hill VM, Murphy KR, Canman JC, Ja WW, Shirasu-Hiza MM. period-Regulated Feeding Behavior and TOR Signaling Modulate Survival of Infection. Current Biology. 2015 doi: 10.1016/j.cub.2015.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore LJ, Sathyanarayanan S, Silvestre DW, Emerson MM, Schotland P, Sehgal A. Novel insights into the regulation of the timeless protein. Journal of Neuroscience. 2003;23:7810–7819. doi: 10.1523/JNEUROSCI.23-21-07810.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylies MK, Bargiello TA, Jackson FR, Young MW. Changes in abundance or structure of the per gene product can alter periodicity of the Drosophila clock. Nature. 1987;326:390–392. doi: 10.1038/326390a0. [DOI] [PubMed] [Google Scholar]
- Boothroyd CE, Wijnen H, Naef F, Saez L, Young MW. Integration of Light and Temperature in the Regulation of Circadian Gene Expression in Drosophila. PLoS Genet. 2007;3:e54. doi: 10.1371/journal.pgen.0030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane BR, Young MW. Interactive Features of Proteins Composing Eukaryotic Circadian Clocks. Annu Rev Biochem. 2014;83:191–219. doi: 10.1146/annurev-biochem-060713-035644. [DOI] [PubMed] [Google Scholar]
- Doble BW. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezelova E, Dolezel D, Hall JC. Rhythm defects caused by newly engineered null mutations in Drosophila’s cryptochrome gene. Genetics. 2007;177:329–345. doi: 10.1534/genetics.107.076513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery P, So WV, Kaneko M, Hall JC, Rosbash M. CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell. 1998;95:669–679. doi: 10.1016/s0092-8674(00)81637-2. [DOI] [PubMed] [Google Scholar]
- Grima B, Chélot E, Xia R, Rouyer F. Morning and evening peaks of activity rely on different clock neurons of the Drosophila brain. Nature. 2004;431:869–873. doi: 10.1038/nature02935. [DOI] [PubMed] [Google Scholar]
- Guo F, Cerullo I, Chen X, Rosbash M. PDF neuron firing phase-shifts key circadian activity neurons in Drosophila. eLife. 2014;3 doi: 10.7554/eLife.02780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin PE. Molecular Genetic Analysis of Circadian Timekeeping in Drosophila. The Genetics of Circadian Rhythms (Elsevier) 2011:141–173. doi: 10.1016/B978-0-12-387690-4.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood AJ. Regulation of GSK-3: a cellular multiprocessor. Cell. 2001;105:821–824. doi: 10.1016/s0092-8674(01)00412-3. [DOI] [PubMed] [Google Scholar]
- Helfrich-Förster C. Robust circadian rhythmicity of Drosophila melanogaster requires the presence of lateral neurons: a brain-behavioral study of disconnected mutants. J Comp Physiol A. 1998;182:435–453. doi: 10.1007/s003590050192. [DOI] [PubMed] [Google Scholar]
- Iitaka C, Miyazaki K, Akaike T, Ishida N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. Journal of Biological Chemistry. 2005;280:29397–29402. doi: 10.1074/jbc.M503526200. [DOI] [PubMed] [Google Scholar]
- Im SH, Taghert PH. PDF receptor expression reveals direct interactions between circadian oscillators in Drosophila. Journal of Comparative Neurology. 2010;518:1925–1945. doi: 10.1002/cne.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang AR, Moravcevic K, Saez L, Young MW, Sehgal A. Drosophila TIM Binds Importin α1, and Acts as an Adapter to Transport PER to the Nucleus. PLoS Genet. 2015;11:e1004974. doi: 10.1371/journal.pgen.1004974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, Baer K, Kissel H, Burlingame AL, Shokat KM, Ptácek LJ, et al. Glucose Sensor O-GlcNAcylation Coordinates with Phosphorylation to Regulate Circadian Clock. Cell Metabolism. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Jeong EH, Park S, Jeong HJ, Edery I, Cho JW. A role for O-GlcNAcylation in setting circadian clock speed. Genes & Development. 2012;26:490–502. doi: 10.1101/gad.182378.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Kim EY, Chiu J, Vanselow JT, Kramer A, Edery I. A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3beta/SGG in circadian clocks. Journal of Neuroscience. 2010;30:12664–12675. doi: 10.1523/JNEUROSCI.1586-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntamalla PP, Kunttas-Tatli E, Karandikar U, Bishop CP, Bidwai AP. Drosophila protein kinase CK2 is rendered temperature-sensitive by mutations of highly conserved residues flanking the activation segment. Mol Cell Biochem. 2008;323:49–60. doi: 10.1007/s11010-008-9963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrondo LF, Olivares-Yanez C, Baker CL, Loros JJ, Dunlap JC. Decoupling circadian clock protein turnover from circadian period determination. Science. 2015;347:1257277–1257277. doi: 10.1126/science.1257277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JM, Kilman VL, Keegan K, Paddock B, Emery-Le M, Rosbash M, Allada R. A role for casein kinase 2alpha in the Drosophila circadian clock. Nature. 2002;420:812–816. doi: 10.1038/nature01235. [DOI] [PubMed] [Google Scholar]
- Lin JM, Schroeder A, Allada R. In Vivo Circadian Function of Casein Kinase 2 Phosphorylation Sites in Drosophila PERIOD. Journal of Neuroscience. 2005;25:11175–11183. doi: 10.1523/JNEUROSCI.2159-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Wendt S, Vanselow JT, Wallach T, Reischl S, Oehmke S, Schlosser A, Kramer A. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes & Development. 2009;23:708–718. doi: 10.1101/gad.512209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinek S, Inonog S, Manoukian AS, Young MW. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell. 2001;105:769–779. doi: 10.1016/s0092-8674(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Meissner RA, Kilman VL, Lin JM, Allada R. TIMELESS is an important mediator of CK2 effects on circadian clock function in vivo. Journal of Neuroscience. 2008;28:9732–9740. doi: 10.1523/JNEUROSCI.0840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P, Saez L, Young MW. PER-TIM interactions in living Drosophila cells: an interval timer for the circadian clock. Science. 2006;311:226–229. doi: 10.1126/science.1118126. [DOI] [PubMed] [Google Scholar]
- Myers MP, Wager-Smith K, Rothenfluh-Hilfiker A, Young MW. Light-induced degradation of TIMELESS and entrainment of the Drosophila circadian clock. Science. 1996;271:1736–1740. doi: 10.1126/science.271.5256.1736. [DOI] [PubMed] [Google Scholar]
- Ousley A, Zafarullah K, Chen Y, Emerson M, Hickman L, Sehgal A. Conserved regions of the timeless (tim) clock gene in Drosophila analyzed through phylogenetic and functional studies. Genetics. 1998;148:815–825. doi: 10.1093/genetics/148.2.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Helfrich-Förster C, Lee G, Liu L, Rosbash M, Hall JC. Differential regulation of circadian pacemaker output by separate clock genes in Drosophila. Proc Natl Acad Sci USA. 2000;97:3608–3613. doi: 10.1073/pnas.070036197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn SC, Park JH, Rosbash M, Hall JC, Taghert PH. A pdf neuropeptide gene mutation and ablation of PDF neurons each cause severe abnormalities of behavioral circadian rhythms in Drosophila. Cell. 1999;99:791–802. doi: 10.1016/s0092-8674(00)81676-1. [DOI] [PubMed] [Google Scholar]
- Rieger D, Wülbeck C, Rouyer F, Helfrich-Förster C. Period gene expression in four neurons is sufficient for rhythmic activity of Drosophila melanogaster under dim light conditions. J Biol Rhythms. 2009;24:271–282. doi: 10.1177/0748730409338508. [DOI] [PubMed] [Google Scholar]
- Rothenfluh A, Young MW, Saez L. A TIMELESS-independent function for PERIOD proteins in the Drosophila clock. Neuron. 2000;26:505–514. doi: 10.1016/s0896-6273(00)81182-4. [DOI] [PubMed] [Google Scholar]
- Ruel L, Pantesco V, Lutz Y, Simpson P, Bourouis M. Functional significance of a family of protein kinases encoded at the shaggy locus in Drosophila. Embo J. 1993;12:1657–1669. doi: 10.1002/j.1460-2075.1993.tb05811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutila JE, Maltseva O, Rosbash M. The timSL Mutant Affects a Restricted Portion of the Drosophila melanogaster Circadian Cycle. J Biol Rhythms. 1998;13:380–392. doi: 10.1177/074873098129000200. [DOI] [PubMed] [Google Scholar]
- Saez L, Young MW. Regulation of nuclear entry of the Drosophila clock proteins period and timeless. Neuron. 1996;17:911–920. doi: 10.1016/s0896-6273(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Saez L, Derasmo M, Meyer P, Stieglitz J, Young MW. A key temporal delay in the circadian cycle of Drosophila is mediated by a nuclear localization signal in the timeless protein. Genetics. 2011;188:591–600. doi: 10.1534/genetics.111.127225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluzicki A, Flourakis M, Kula-Eversole E, Zhang L, Kilman V, Allada R. Dual PDF Signaling Pathways Reset Clocks Via TIMELESS and Acutely Excite Target Neurons to Control Circadian Behavior. PLoS Biol. 2014;12:e1001810. doi: 10.1371/journal.pbio.1001810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer OT, Kim DJ, Dunbar-Yaffe R, Nikolaev VO, Lohse MJ, Taghert PH. Widespread receptivity to neuropeptide PDF throughout the neuronal circadian clock network of Drosophila revealed by real-time cyclic AMP imaging. Neuron. 2008;58:223–237. doi: 10.1016/j.neuron.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Lin JM, Meissner RA, Allada R. Dominant-negative CK2alpha induces potent effects on circadian rhythmicity. PLoS Genet. 2008;4:1–11. doi: 10.1371/journal.pgen.0040012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoleru D, Nawathean P, de Fernández MLP, Menet JS, Ceriani MF, Rosbash M. The Drosophila Circadian Network Is a Seasonal Timer. Cell. 2007;129:207–219. doi: 10.1016/j.cell.2007.02.038. [DOI] [PubMed] [Google Scholar]
- Stoleru D, Peng Y, Agosto J, Rosbash M. Coupled oscillators control morning and evening locomotor behaviour of Drosophila. Nature. 2004;431:862–868. doi: 10.1038/nature02926. [DOI] [PubMed] [Google Scholar]
- Stoleru D, Peng Y, Nawathean P, Rosbash M. A resetting signal between Drosophila pacemakers synchronizes morning and evening activity. Nature. 2005;438:238–242. doi: 10.1038/nature04192. [DOI] [PubMed] [Google Scholar]
- Syed S, Saez L, Young MW. Kinetics of Doubletime Kinase-dependent Degradation of the Drosophila Period Protein. Journal of Biological Chemistry. 2011;286:27654–27662. doi: 10.1074/jbc.M111.243618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosshall LB, Price JL, Sehgal A, Saez L, Young MW. Block in nuclear localization of period protein by a second clock mutation, timeless. Science. 1994;263:1606–1609. doi: 10.1126/science.8128247. [DOI] [PubMed] [Google Scholar]
- Wijnen H, Naef F, Boothroyd C, Claridge-Chang A, Young MW. Control of Daily Transcript Oscillations in Drosophila by Light and the Circadian Clock. PLoS Genet. 2005:e39. doi: 10.1371/journal.pgen.0020039. preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Shafer OT. The Drosophila Circadian Clock Is a Variably Coupled Network of Multiple Peptidergic Units. Science. 2014;343:1516–1520. doi: 10.1126/science.1251285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuyama K, Meinertzhagen IA. Synaptic connections of PDF-immunoreactive lateral neurons projecting to the dorsal protocerebrum of Drosophila melanogaster. J Comp Neurol. 2010;518:292–304. doi: 10.1002/cne.22210. [DOI] [PubMed] [Google Scholar]
- Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001;2:702–715. doi: 10.1038/35088576. [DOI] [PubMed] [Google Scholar]
- Zheng X, Sehgal A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr Biol. 2010;20:1203–1208. doi: 10.1016/j.cub.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Sehgal A. Speed control: cogs and gears that drive the circadian clock. Trends in Neurosciences. 2012;35:574–585. doi: 10.1016/j.tins.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.