

Abstract
Resistance selection by human immunodeficiency virus (HIV) towards known drug regimens necessitates the discovery of structurally novel antivirals with a distinct resistance profile. Based on our previously reported 3-hydroxypyrimidine-2,4-dione (HPD) core we have designed and synthesized a new integrase strand transfer (INST) inhibitor type featuring a 5-N-benzylcarboxamide moiety. Significantly, the 6-alkylamino variant of this new chemotype consistently conferred low nanomolar inhibitory activity against HIV-1. Extended antiviral testing against a few raltegravir-resistant HIV-1 clones revealed a resistance profile similar to that of the second generation INST inhibitor (INSTIs) dolutegravir. Although biochemical testing and molecular modeling also strongly corroborate the inhibition of INST as the antiviral mechanism of action, selected antiviral analogues also potently inhibited reverse transcriptase (RT) associated RNase H, implying potential dual target inhibition. In vitro ADME assays demonstrated that this novel chemotype possesses largely favorable physicochemical properties suitable for further development.
Introduction
HIV infects an estimated 34 million people worldwide. Clinical management of HIV / AIDS continues to rely on highly active antiretroviral therapy (HAART) that consists of five different classes of FDA-approved drugs1: nucleoside reverse transcriptase inhibitors (NRTIs); nonnucleoside reverse transcriptase inhibitors (NNRTIs); protease inhibitors (PIs); INSTIs; and entry inhibitors. However, HIV infection remains incurable2–3 due to the persistence of viral reservoirs. The resulting long duration of HAART typically amounts to two treatment challenges: toxicities and the selection of resistant viral strains. The emergence of multidrug resistance is particularly concerning as treatment options will be severely limited. Therefore, sustained success in HIV HAART requires novel antivirals with structurally unique pharmacophore and distinct resistance profile. HIV IN plays critical roles in viral infection and the establishment of proviral latency.4–6 As an antiviral target IN is particular attractive because 1) there is no host cellular counterpart, hence specific inhibitors should not interfere with cellular functions; and 2) IN uses the same active site (DD35E motif) for both the 3' processing and the ST steps, therefore, inhibitors could benefit from a potentially high genetic barrier to resistance selection. Specific INSTIs all feature a diketoacid (DKA) functionality or its heterocyclic bioisosteres7–11 along with a hydrophobic terminal benzyl moiety,12–17 as demonstrated by all three FDA-approved INSTIs (Figure 1): raltegravir (1)18–19, elvitegravir (2),20 and dolutegravir (3).21–22 Particularly significant is the second-generation INSTI 3 which retains potency against many raltegravir-resistant HIV strains.23 We have previously developed a few chemotypes featuring the HPD core24–27 that effectively inhibited HIV-1 in cell culture. The antiviral potency associated with these HPD subtypes is likely due to the dual inhibition of RT and IN as indicated by biochemical assays. However, the IN inhibition was typically much weaker than the inhibition of RT. Furthermore, cross resistance to 1 was also observed, suggesting that these early HPD subtypes may have the characteristics of first-generation INSTIs. Another variant of HPD was recently found to selectively inhibit the RT-associated RNase H without significantly inhibiting INST.28 We report herein a rationally designed new HPD variant (Figure 1, 4) featuring a unique C5 carboxamide moiety to specifically inhibit INST. Significantly, chemotype 4 has the two structural determinants essential for INST binding and inhibition (Figure 1). The overall shape and functionalities of 4 particularly resemble those of 3, suggesting that our novel inhibitors can be second generation INSTIs.
Figure 1.

Structures of FDA-approved INSTIs: raltegravir (1), elvitegravir (2), dolutegravir (3), and our newly designed HPD inhibitor subtype 4. Each approved drug features a chelating traid (red) and a terminal benzyl group (blue) that constitute the pharmacophore of HIV-1 INSTIs. Chemotype 4 fits the pharmacophore with the same two structural features.
Results and Discussion
Chemistry
Our target compounds 4 were prepared via a concise and diverse synthetic route shown in Scheme 1. The synthesis started from commercially available hydroxyurea 5 which was O-benzylated with BnBr in the presence of KOH under reflux to provide 1-(benzyloxy)urea 6 in high yield.29 Cyclocondensation of 1-(benzyloxy)urea 2 and diethyl malonate provided six-membered heterocycle compound 3-(benzyloxy)-6-hydroxypyrimidine-2,4(1H,3H)-dione 7 in moderate yield under microwave irradiation (150 °C, 20 min) or under conventional heating (reflux, overnight).30 Compound 7 was converted to key intermediate 3-(benzyloxy)-6-chloropyrimidine-2,4(1H,3H)-dione 8 by reacting with POCl3 in the presence of BnEt3NCl.31 Reaction of intermediate chloride 8 with amines in the presence of N,N-dimethylaniline under microwave condition provided 6-amination products 9 in moderate to good yield.32 The key C5 carboxamide was introduced by reacting 6-amino intermediates 9 with commercial or in situ generated isocyanates (Scheme 1),33 a highly efficient method for small scale synthesis of intermediate 10. The final debenzylation was achieved by treating compounds 10 with TFA under microwave condition34 or via catalytic hydrogenation.
Scheme 1a.

Synthesis of chemotype 4
Alternatively, the C5 carboxamidation can be achieved via a two-step reaction sequence (scheme 2) to avoid the use of the unpleasant nitrobenzene. In this case, the amino intermediate 9 was first treated with phenyl chloroformate and a base, such as pyridine, to give intermediate phenyl ester 11 which was converted to amide 10 upon reacting with a primary amine under conventional heating or microwave conditions. Debenzylation with the same protocol afforded the desired chemotype 4.
Scheme 2a.

Alternative synthesis of chemotype 4
Meanwhile our analogue synthesis also included a few variants of 4 which entailed slightly different synthetic routes or further functionalization (Scheme 3). In these events, direct debenzylation of intermediate 12 afforded compound 51 (Scheme 3, a), whereas methylation of intermediate 13 produced two regio-isomers 14 and 15, which upon debenzylation yielded compounds 52 and 53, respectively (Scheme 3, b). Interestingly, a 6-deamino analogue 54 was also synthesized from intermediate 7 via the carboxamidation and debenzylation sequence (Scheme 3, c).
Scheme 3a.

Synthesis of compounds 51–54
Finally, synthesis of analogues (55–57) with another six-membered ring fused to the HPD core via N1-C6 (55) or C6-C5 (56–57) was also attempted. Compound 55 was synthesized from 6-amino HPD intermediate 17 which was cyclized to 18 upon treating with 1,3-dibromopropane under basic condition (Scheme 4). The rest of the synthesis involved the same carboxamidation –debenzylation sequence as used for the synthesis of chemotype 4. The synthesis of C6-C5 fused analogues (56–57) was attempted based on Scheme 5. While the two intermediates 22–23 were obtained, the subsequent debenzylation was unsuccessful (Scheme 5).
Scheme 4a.


Synthesis of compound 55
Scheme 5a.

Attempted synthesis of compounds 56–57
Biology
All final compounds (24–55) were first screened in a viral cytopathic effect (CPE) based antiviral assay against HIV-1IIIB in CEM-SS cells.35–36 In this assay, the reduction of CPE was used to indicate the antiviral activity of a compound. The screening was conducted under single concentration (10 μM) and the percent inhibition and percent cell viability were calculated using a virus control and cell control, respectively. Best compounds from this assay were further evaluated in a single replication cycle MAGI assay. In addition, all compounds were tested in a biochemical assay with recombinant HIV-1 IN measuring INST activity. Selected ones were also tested against RT-associated RNase H activity. Both 3 and 1 were included in all assays as reference compounds. The results indicate a few prominent structure-activity-relationship (SAR) trends.
Alkylamino group is required at C6
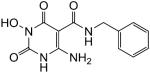
From the antiviral screening assay, the most striking SAR was that the two analogues with an aromatic amine at the C6 position (24–25) were completely inactive and largely cytotoxic in the cytoprotection antiviral assay, whereas a small aliphatic amine substitution at the same position (26–37, Table 1) conferred excellent antiviral activity without cytotoxicity, with the lone exception being the tbutylamino analogue (42) which was inactive and cytotoxic. The same dramatic SAR was observed from the INST biochemical assay in which neither of the arylamino analogues (24–25) showed appreciable inhibition at concentrations up to 100 μM whereas the alkylamino analogues (26–37) all demonstrated nanomolar inhibitory activity, including the tbutylamino analogue (35). Furthermore, although most alkyl analogues (26–31, 33–34) produced maximal cytoprotection at 10 μM without significant cytotoxicity, linear alkyl substituted analogues (26–31) are generally favored in the INST biochemical assay over the unsubstituted analogue (36) and the bulky alkyl substituted ones, such as the isopropyl (33) and cyclopropyl (34) groups. Biochemical analysis also showed that the ethyl substituent (30) confers the best inhibition against INST (IC50 = 33 nM).
Table 1.
SAR of the C6 amino group: antiviral screening and biochemical assay of 24–36
| Compd | Structure | Antiviral Screening (10 μM)a |
INST IC50 | |
|---|---|---|---|---|
| CPE reduction % | viability % | (μM)b | ||
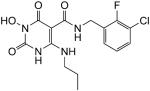
| 24 |
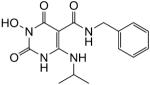
|
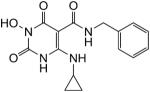
0 | 21 | >100 |
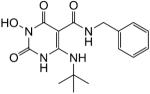
| 25 |
|
0 | 13 | >100 |
| 26 |
|
100 | 98 | 0.080 ± 0.007 |
| 27 |
|
100 | 95 | 0.25 ± 0.048 |
| 28 |
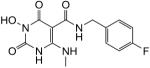
|
100 | 89 | 0.11 ± 0.021 |
| 29 |
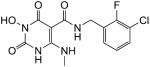
|
100 | 100 | 0.11 ± 0.033 |
| 30 |
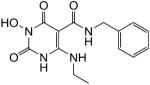
|
100 | 100 | 0.033 ± 0.004 |
| 31 |
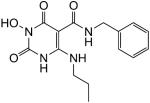
|
100 | 97 | 0.066 ± 0.016 |
| 32 |
|
33 | 48 | 0.058 ± 0.012 |
| 33 |
|
98 | 90 | 0.15 ± 0.034 |
| 34 |
|
98 | 91 | 0.21 ± 0.077 |
| 35 |
|
0 | 17 | 0.081 ± 0.015 |
| 36 |
|
99 | 100 | 0.90 ± 0.29 |
| 3 | -- | -- | -- | 0.068 ± 0.01 |
| 1 | -- | -- | -- | 0.65 ± 0.14 |
Cytoprotection assay using HIV-1 IIIB in CEM-SS cells with a viral control for CPE reduction and cell control for cell viability.
Concentration inhibiting INST by 50%, expressed as mean ± standard deviation from at least three independent experiments.
N-Benzyl substitution on the C5 carboxamide is highly desired
To explore the SAR concerning the C5 carboxamide moiety, analogues of compound 30 were synthesized and tested in the antiviral screening assay and the INST biochemical assay (Table 2). Remarkably, all analogues with a benzyl group on the C5 carboxamide (30, 37–46) demonstrated maximal cytoprotection (92–100%) without considerable cytotoxicity (cell viability 87–100%), with the only exception being compound 43 which provided 62% cytoprotection with 73% cell viability at 10 μM. Consistent with the maximal cytoprotection, exceptional biochemical inhibition (IC50 = 21–230 nM) was also observed for these analogues. Interestingly, compound with an extra methylene group (47) also produced maximal cytoprotection at 10 μM, though the biochemical inhibition was substantially reduced (47 vs 30). In stark contrast, replacing the benzyl group with a phenyl ring (48), alkyl group (50), or removing the N-substituent (49) completely abrogated both the antiviral activity and biochemical inhibition. Compound with a C5 phenyl ester (51) was also devoid of biochemical inhibition and yielded 50% cytoprotection at 10 μM. All these SAR observations are well aligned with the known pharmacophore model of INSTIs.
Table 2.
SAR around the C5 carboxamide: antiviral screening and biochemical assay of 30 analogues 47–51
| Compd | Structure | Antiviral Screening (10 μM)a |
INST IC50 | |
|---|---|---|---|---|
| CPE reduction % | viability % | (μM)b | ||
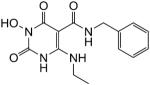
| 30 |
|
100 | 100 | 0.033 ± 0.004 |
| 37 |
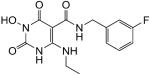
|
100 | 90 | 0.068 ± 0.007 |
| 38 |
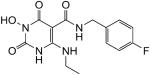
|
100 | 99 | 0.049 ± 0.004 |
| 39 |
|
92 | 87 | 0.068 ± 0.011 |
| 40 |
|
100 | 92 | 0.048 ± 0.007 |
| 41 |
|
100 | 100 | 0.23 ± 0.062 |
| 42 |
|
100 | 100 | 0.077 ± 0.011 |
| 43 |
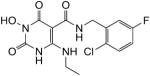
|
62 | 73 | 0.095 ± 0.019 |
| 44 |
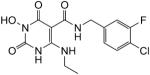
|
100 | 100 | 0.12 ± 0.021 |
| 45 |
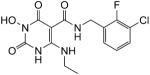
|
100 | 100 | 0.021 ± 0.002 |
| 46 |
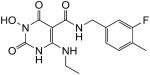
|
100 | 100 | 0.051 ± 0.013 |
| 47 |
|
99 | 87 | 0.10 ± 0.03 |
| 48 |
|
11 | 27 | 35 ± 17 |
| 49 |
|
-- | -- | >100 |
| 50 |
|
5.1 | 100 | >100 |
| 51 |
|
50 | 100 | >100 |
| 3 | -- | -- | -- | 0.068 ± 0.01 |
| 1 | -- | -- | -- | 0.65 ± 0.14 |
Cytoprotection assay using HIV-1 IIIB in CEM-SS cells with a viral control for CPE reduction and cell control for cell viability.
Concentration inhibiting INST by 50%, expressed as mean ± standard deviation from at least three independent experiments.
Scaffold analogues lack antiviral activity or biochemical INST inhibition
To further confirm the SAR trends shown in Tables 1–2, we also synthesized a few scaffold analogues of the new chemotype. Compounds 52–53 were synthesized to explore the impact of methylation on N1 or the C5 carboxamide. Remarkably, a simple methylation at N1 completely abrogated the antiviral activity, cell viability and the biochemical inhibition (52). Methylation of the C5 carboxamide also caused a complete loss of antiviral activity while substantially attenuating the biochemical inhibition (53 vs 38). Similarly, the 6-deamino analogue 61 demonstrated drastically reduced antiviral potency and biochemical inhibition (54 vs 30), confirming the importance of the amino group at C6 for biological activities. Finally, cyclized analogue 55 was inactive and cytotoxic in the antiviral screening assay, though the biochemical inhibition was largely preserved (IC50 = 430 nM).
Dose response antiviral testing
To better gauge the antiviral potency, selected inhibitors with excellent CPE reduction and cell viability from the antiviral screening assay were further tested in a dose response fashion, from which the EC50 and CC50 were determined (Table 4). All compounds tested showed nanomolar activity and five of the eight analogues were active in low nanomolar range (EC50 = 15–61 nM). Although these analogues showed mild cytotoxicity with CC50s in the range of 18–58 μM, the selectivity window (Table 4), particularly that for analogues 40 (TI =1500) and 45 (TI = 1200), is sufficiently large to warrant their further development. To confirm the observed antiviral potency, these same analogues were further evaluated in a distinct MAGI assay.36–37 This assay measures HIV infection in indicator cells (P4R5) through the expression of a Tat-dependent reporter (β-galactosidase) and requires only one replication cycle. While the EC50 values were generally higher in this assay, our compounds still demonstrated nanomolar antiviral potency with the exception of compounds 26 and 46. Interestingly, compound 45, while showing the best potency in both the INST assay and the CPE antiviral assay, exhibited a significantly higher EC50 (0.30 μM vs 0.015 μM) in the MAGI assay. By comparison, EC50 values for analogues 38 and 40 were comparable to 1 in the CPE assay and only 2–5 fold higher than 3 and 1 in the MAGI assay. Nevertheless, dose response antiviral testing in two distinct assays confirmed a highly potent antiviral profile of our newly designed compounds.
Table 4.
Dose-response antiviral results of selected analogues from two different assays
| Compound | Cytoprotection Assaya |
MAGI Assayb | ||
|---|---|---|---|---|
| EC50 (μM)c | CC50 (μM)d | TIe | EC50 (μM)f | |
| 26 | 0.83 | 58 | 70 | 3.7 ± 0.3 |
| 30 | 0.061 | 23 | 380 | 0.30 ± 0.01 |
| 38 | 0.033 | 19 | 580 | 0.090 ± 0.02 |
| 40 | 0.032 | 47 | 1500 | 0.070 ± 0.02 |
| 42 | 0.049 | 21 | 430 | 0.30 ± 0.06 |
| 44 | 0.19 | 19 | 100 | 0.80 ± 0.2 |
| 45 | 0.015 | 18 | 1200 | 0.30 ± 0.09 |
| 46 | 0.15 | 20 | 130 | 1.3 ± 0.2 |
| 3 | -- | -- | -- | 0.020 ± 0.002 |
| 1 | 0.023 | >10 | >430 | 0.030 ± 0.008 |
Concentration inhibiting INST by 50%,
Concentration of a compound inhibiting virus replication by 50%.
Concentration of a compound resulting in 50% cell death.
Therapeutic index, defined as CC50/EC50.
Expressed as mean ± standard deviation from two independent experiments.
Antiviral resistance profile
Mutations conferring resistance to INSTIs have been generated in cell culture and emerged in clinical studies.38–40 To establish the resistance profile of chemotype 4, a representative compound 45 was evaluated for antiviral potency against four raltegravir-resistant HIV-1 clones41 in the CPE assay. These clones contain one or more major mutations in HIV-1 IN that are associated with raltegravir resistance,23, 24 namely a Y143C or an N155H single mutation; a G140S / Q148H double mutation and a G140S / Y143H / Q148H triple mutation. AZT, 1 and 3 were included in the study for comparison purpose. The results are summarized in Table 5. As expected cross-resistance to 1 was not observed for AZT due to its orthogonal mechanism of action as an NRTI, whereas the resistance profile observed in our antiviral assay for 3 is largely consistent with the reported profile.23 Similar to 3, compound 45 retains its potency against clones with only one major IN mutation (clones 1556-1 and 4736-2) while HIV-1 clones with IN double mutations (clone 8070-1) and triple mutations (clone 8070-1) demonstrated moderate resistance to 45, with fold resistance of 13 and 6.3, respectively. The fold-resistance of 45 is strikingly similar to that observed with 3 for each of the four HIV-1 clones (Table 5). These observations strongly suggest that our chemotype 4 may confer antiviral activity via inhibiting INST in a manner similar to that of 3, a second generation INSTI.42
Table 5.
Resistance profile of 45 against raltegravir-resistant HIV-1 clones
| HIV-1 Clone | Major Mutations | Fold-resistancea |
|||
|---|---|---|---|---|---|
| 45 | AZT | 1 | 3 | ||
| 1556-1 | Y143C | 0.4 | 1.4 | 170 | 2.0 |
| 4736-2 | N155H | 2.4 | 0.7 | 14 | 1.8 |
| 8070-1 | G140S / Y143H / Q148H | 6.3 | 0.9 | 220 | 9.6 |
| 8070-2 | G140S / Q148H | 13 | 0.5 | 145 | 12 |
Fold-resistance is defined as EC50(mutant) / EC50 (WT).
Inhibition against RT-associated RNase H
Consistent with the aforementioned antiviral profile against raltegravir-resistant HIV-1 clones, which implies an antiviral mechanism of action (MOA) similar to second generation INSTI 3, biochemical data also closely correlate with the observed antiviral activity, as each of the selected analogues exhibited a biochemical IC50 against INST very similar to the antiviral EC50, particularly from the CPE assay (Table 6). In addition, none of the inactive compounds (24–25, 48 and 50–54) from the CPE antiviral assay inhibited INST (Tables 1–3), strongly suggesting that our new compounds target INST. However, HIV IN and RNase H share a similar active site fold,43 hence their inhibition entails similar pharmacophore elements, mainly a chelating triad and a hydrophobic aromatic moiety. Therefore, we also tested selected analogues in another biochemical assay measuring the RNase H function of RT. Remarkably, all these analogues demonstrated exceptional biochemical inhibition against RNase H with IC50s in the low nanomolar range (10—61 nM, Table 6), which correlates with the antiviral activity as well as the INST inhibition does in general. By contrast, the two reference compounds, 3 and 1, did not inhibit RNase H significantly at concentrations up to 10 μM. These observations indicate that the exceptional antiviral potency of our new chemotype could be due to dual target inhibition. Further virological characterizations of our compounds, including MOA studies via viral passage experiments for resistance selection, are currently underway and will be reported in due course.
Table 6.
Biochemical inhibition of RT-associated RNase H and the antiviral correlation
| Compound | RNase H | INST | CPE | MAGI |
|---|---|---|---|---|
| IC50 (μM)a | IC50 (μM)a | EC50 (μM)b | EC50 (μM)c | |
| 26 | 0.061 ± 0.004 | 0.080 ± 0.007 | 0.83 | 3.7 ± 0.3 |
| 30 | 0.042 ± 0.012 | 0.033 ± 0.004 | 0.061 | 0.30 ± 0.01 |
| 38 | 0.011 ± 0.003 | 0.049 ± 0.004 | 0.033 | 0.09 ± 0.02 |
| 40 | 0.010 ± 0.006 | 0.068 ± 0.011 | 0.032 | 0.07 ± 0.02 |
| 42 | 0.026 ± 0.009 | 0.077 ± 0.011 | 0.049 | 0.30 ± 0.06 |
| 44 | 0.022 ± 0.009 | 0.12 ± 0.021 | 0.19 | 0.80 ± 0.2 |
| 45 | 0.029 ± 0.015 | 0.021 ± 0.002 | 0.015 | 0.30 ± 0.09 |
| 46 | 0.020 ± 0.010 | 0.051 ± 0.013 | 0.15 | 1.3 ± 0.2 |
| 3 | >10 | 0.068 ± 0.01 | -- | 0.020 ± 0.002 |
| 1 | >10 | 0.65 ± 0.14 | 0.023 | 0.030 ± 0.008 |
Concentration with 50% enzyme inhibition, expressed as mean ± standard deviation from three independent experiments.
Concentration of a compound inhibiting virus replication by 50%.
Concentration of a compound inhibiting virus replication by 50%, expressed as mean ± standard deviation from three independent experiments.
Table 3.
Antiviral screening and biochemical assay of scaffold analogues 52–55
| Compd | Structure | Antiviral Screening (10 μM)a |
INST IC50 | |
|---|---|---|---|---|
| CPE reduction % | viability % | (μM)b | ||
| 52 |
|
6.5 | 10 | >100 |
| 53 |
|
0 | 89 | 7.0 ± 3.3 |
| 54 |
|
26 | 100 | 37 ± 21 |
| 55 |
|
0 | 5.5 | 0.43 ± 0.13 |
Cytoprotection assay using HIV-1 IIIB in CEM-SS cells with a viral control for CPE reduction and cell control for cell viability.
Concentration inhibiting INST by 50%, expressed as mean ± standard deviation from at least three independent experiments.
Mechanism of strand transfer inhibition
HIV IN is a 32-kDa protein encoded by viral pol gene consisting of three functional domains44: the N-terminal domain (NTD) with a conserved “HH-CC” zinc-binding motif; the catalytic core domain (CCD) containing the key D64-D116-E152 catalytic triad; and the C-terminal domain (CTD) important for DNA binding. Although crystal structures of each single domain as well as double domains had been reported;45–47 detailed understanding on HIV INST catalysis and inhibition remained elusive until the disclosure of crystal structures of full-length IN and viral DNA complex (intasome) for a homologous prototype foamy virus (PFV).48–49 Homologous models50–51 constructed based on these PFV intasome crystal structures have provided valuable details into HIV INST mechanism of action and formed the basis for structure-based INSTI design. Significantly these models corroborate the minimal pharmacophore embedded in major INSTIs as shown in Figure 1.
To illustrate the binding mode of our new chemotype at HIV IN active site, we have performed molecular docking on a representative analogue 45 using a PFV intasome model (Figure 2).50 Significantly, 45 fits perfectly into the IN binding site through two major binding domains: the 3-N hydroxyl group simultaneously chelates to both Mg2+ ions while allowing the placement of the benzyl group into the protein-DNA interfacial hydrophobic pocket involving π-stacking of the i) fluorobenzyl side chain with the deoxycytosine C16 of viral DNA, and ii) π -stacking of the central uracil ring with the terminal 3′-deoxyadenosine A17 (Figure 2a). In addition, a structure overlay of 45 with 3 reveals a high degree of similarity between their binding (Figure 2b), suggesting that our new molecular scaffold can effectively engage with HIV IN for ST inhibition.
Figure 2.
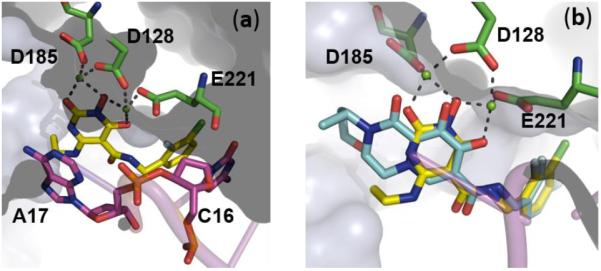
a) Predicted binding mode of 45 (yellow) as obtained by molecular docking in the PFV intasome catalytic site (PDB code: 3S3M).50 Magnesium cations depicted in green spheres, catalytic residues in green sticks and viral DNA in magenta cartoon. b) Overlay of 45 (yellow) and crystallographic 3 (cyan) in the catalytic active site of PFV intasome (PDB code: 3S3M) with Mg2+ ions in green spheres and viral DNA in magenta cartoon. Numbering in black is in accordance to the PFV intasome crystal structure (PDB code: 3S3M).
To understand the effect of mutations on antiviral profile of 1 and 45, molecular modeling was performed with these two compounds into the PFV IN Y212C mutant which corresponds to the Y143C of HIV IN (Figure 3). It was observed that the oxadiazole moiety of 1 makes a critical π-π stack interaction with Y143 (Figure 3a). This interaction is not available with the C143 residue and the binding of 1 to the Y143C mutant is significantly weakened (Figure 3b). On the other hand, 45 lacks the oxadiazole moiety to interact with Y143 (Figure 3a). Consequently, the Y143C mutation would not impact its binding to IN. These docking studies are consistent with the observations that Y143 mutation confers major resistance to 1 and no resistance to 45 (Table 5).
Figure 3.
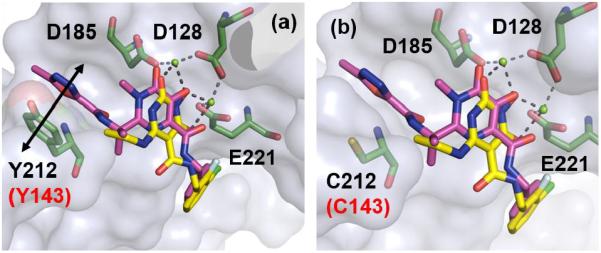
a) Overlay of 1 (magenta) and 45 (yellow) within the PFV IN active site in complex with magnesium (green spheres) (PDB code: 3OYA). π-π stack interaction between oxadiazole moiety within 1 and Y212 is shown by black double headed arrow. b) Overlay of 1 (magenta) and 45 (yellow) within the active site of PFV IN Y212C (corresponding to HIV IN Y143C) mutant. Key residues (D128, D185, E221 andY212) are represented as green sticks. Numbering in black is in accordance to the PFV intasome crystal structure (PDB code: 3S3M). The corresponding numbering for HIV IN is highlighted in red.
Physicochemical and in vitro ADME
To assess drug-like properties of chemotype 4, selected analogues were tested in various physicochemical and in vitro ADME studies (Table 7). First, aqueous stability and solubility were evaluated in Dulbecco's Phosphate-Buffered Saline (DPBS). The selected compounds showed excellent stability and good solubility in DPBS with the exception of 45 showed a lower but still acceptable solubility. Second, the selected compounds were found to be stable in both human and mouse plasma. Third, all compounds showed a higher protein binding in human plasma (98–99%) than that in mouse plasma (71–96%). Finally, while the selected compounds exhibited excellent phase I stability in both human and mouse liver microsomes, they were much less stable against phase II glucuronidation in microsomes from both species (Table 7). Taken together, although the selected compounds still need improvement in their phase II glucuronidation stability, they possess largely favorable physicochemical and in vitro ADME properties. It is also noteworthy that our chemotype has a significant smaller size (MW for 45 = 322; the MW for drugs 1–3 is in the range of 419–448) while supporting antiviral activities comparable to 1 (Table 4). The small size of our chemotype would allow significant room for further optimization.
Table 7.
Physicochemical and in vitro ADME profile of selected analogues
| Compd | Aqueous Solubility (μM) | Aqueous Stability t1/2 (h) | Plasma Stability t1/2 (h) | Plasma Protein Binding (%)a | Microsomal Stability (Phase I / Phase II, CLint)b | |||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Human | Mouse | Human | Mouse | Human | Mouse | |||
| 26 | 138 | > 24 | > 24 | > 24 | 99.2 | 70.9 | 0.8 / 51 | < 0.1 / 500 |
| 30 | 1091 | > 24 | > 24 | > 24 | 99.1 | 83.9 | < 0.1 / 355 | < 0.1 / 732 |
| 38 | 150 | > 24 | > 24 | > 24 | 99.4 | 86.5 | < 0.1 / 336 | < 0.1 / 1240 |
| 45 | 11 | > 24 | > 24 | > 24 | 98.4 | 94.0 | < 0.1 / 850 | < 0.1 / 1065 |
| 46 | 81 | > 24 | > 24 | > 24 | 98.9 | 95.9 | < 0.1 / 357 | 0.4 / 1342 |
Percent of fraction bound.
CLint: intrinsic clearance, μl/min/mg protein.
Conclusion
A new HPD subtype was designed and synthesized. Key to the design is the introduction of an N-benzyl-5-carboxamide moiety to mimic the second-generation INSTI 3. SAR revealed the requirement of an alkylamino group at C6 and an N-benzyl group on the C5 carboxamide for INST inhibition and antiviral activity. Numerous analogues demonstrated low nanomolar potencies against HIV-1 in cell culture and low nanomolar biochemical inhibition against INST. Molecular modeling showed that the representative inhibitor 45 binds to IN with a mode similar to 3. Antiviral testing of 45 against a panel of raltegravir-resistant HIV-1 clones showed a resistance profile similar to that of 3. However, analogues of our new chemotype also potently inhibited the RNase H function of RT, suggesting that the antiviral potency could be due to dual target inhibition. in vitro ADME assays also demonstrated largely favorable physicochemical properties for the new chemotype.
Experimental
Chemistry
General Procedures
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents were either purchased (dioxane and MeOH) or dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All moisture sensitive reactions were performed under an inert atmosphere of ultra-pure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Phenomenex Gemini, 5 micron C18 column (250mm × 4.6 mm). HPLC conditions: solvent A = H2O containing 0.1% TFA, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of 20% MeCN/H2O to 100% MeCN for 30 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥ 96%.
General method of 3-O-benzyl deprotection for the synthesis of final compounds 24–55
Method A: catalytic hydrogenation
To a solution of 100 mg of 3-(benzyloxy)-pyrimidine-2,4(1H,3H)-dione intermediate (10, 12, 14–16, 20) in 7.0 mL MeOH was added 20 mg Pearlman's catalyst (Pd-C, 20%). The reaction mixture was degassed using vacuum and refilling with H2 (40-50 psi) for three times. Then keep the reaction mixture being shaken under H2 (40-50 psi) atmosphere for appropriate time. The reaction was monitored by both TLC and LC-MS. The reaction mixture was filtered through a short celite column and then removed the solvent. Trituration with MeOH, ethyl acetate and DCM provided desired final compounds 24–55 in 82–97% yield.
Method B: acid hydrolysis
To a microwave reaction vessel were added 0.26 mmol of a 3-(benzyloxy)-pyrimidine-2,4(1H,3H)-dione intermediate (10, 12, 14–16, 20) and 6~10 mL of TFA. The reaction vessel was irradiated at 120 °C for the appropriate time. The reaction was monitored by both TLC and LC-MS. The reaction mixture was transferred to a round-bottom flask to remove the solvent under reduced pressure. Then the residue was purified by flash chromatography on C18 reverse phase column (H2O-MeOH) or trituration (MeOH, Ethylacetate and DCM) to provide the desired compounds 24–55 as solid in 50–77%.
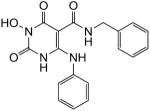
N-Benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxo-4-(phenylamino)pyrimidine-5-carboxamide (24)
White solid, 50% yield; 1H NMR (600 MHz, DMSO-d6): δ 12.72 (s, 1H), 11.26 (s, 1H), 10.32 (s, 1H), 10.03 (t, J = 2.4, 1H), 7.43–7.26 (m, 10H), 4.49 (s, 2H); 13C NMR (150 MHz, DMSO-d6): δ 167.7, 161.6, 154.0, 147.0, 139.3, 135.8, 129.5, 128.4, 127.2, 126.9, 126.5, 124.9, 81.7, 41.9; HRMS-ESI(−) m/z calcd for C18H16N4O4 351.1099 [M−H]−, found 351.1103.
N-Benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxo-4-(biphenylamino)pyrimidine-5-carboxamide (25)
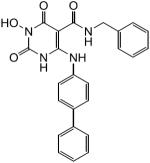
White solid, 77% yield; 1H NMR (600 MHz, DMSO-d6): δ 12.76 (s, 1H), 11.33 (s, 1H), 10.34 (s, 1H), 10.04 (dd, J = 6.0, 4.8 1H), 7.74–7.20 (m, 14H), 4.50 (d, J = 5.4, 2H); 13C NMR (150 MHz, DMSO-d6): δ 167.7, 161.6, 154.0, 147.0, 139.3, 138.1, 135.2, 129.0, 128.4, 127.7, 127.5, 127.2, 126.9, 126.5, 125.4, 80.9, 41.9; HRMS-ESI(−) m/z calcd for C24H20N4O4 427.1412 [M−H]−, found 427.1414.
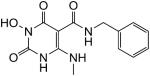
N-Benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-4-(methylamino)-2,6-dioxopyrimidine-5-carboxamide (26)
White solid, 39% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.05 (s, 1H), 10.88 (q, J = 4.8 Hz, 1H), 10.20 (s, 1H), 9.90 (t, J = 6.0 Hz, 1H), 7.33–7.22 (m, 5H), 4.43 (d, J = 6.0 Hz, 2H); 2.94 (d, J = 5.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.7, 161.3, 155.7, 147.3, 139.7, 128.3, 127.1, 126.7, 79.0, 41.7, 28.4; HRMS-ESI(−) m/z calcd for C13H14N4O4 289.0942 [M−H]−, found 289.0939.
N-(3-Fluorobenzyl)-1-(hydroxy)-1,2,3,6-tetrahydro-4-(methylamino)-2,6-dioxopyrimidine-5-carboxamide (27)
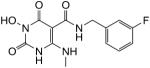
White solid, 20% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.05 (s, 1H), 10.80 (q, J = 4.8 Hz, 1H), 10.18 (s, 1H), 9.94 (t, J = 6.0 Hz, 1H), 7.35 (ddd, J = 7.8, 7.2, 6.0 Hz, 1H), 7.11 (d, J = 7.2 Hz, 1H), 7.06 (m, 2H), 4.44 (d, J = 6.0 Hz, 2H); 2.93 (d, J = 4.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.8, 162.2 (d, JCF = 241.5 Hz), 161.3, 155.9, 147.4, 143.0 (d, JCF = 6.9 Hz), 130.2 (d, JCF = 8.1 Hz), 123.1 (d, JCF = 2.4 Hz), 113.7 (d, JCF = 21.9 Hz), 113.4 (d, JCF = 20.0 Hz), 79.0, 41.2, 28.4; HRMS-ESI(−) m/z calcd for C13H13FN4O4 307.0848 [M−H]−, found 307.0852.
N-(4-Fluorobenzyl)-1-(hydroxy)-1,2,3,6-tetrahydro-4-(methylamino)-2,6-dioxopyrimidine-5-carboxamide (28)
White solid, 20% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.05 (s, 1H), 10.86 (q, J = 5.4 Hz, 1H), 10.19 (s, 1H), 9.87 (t, J = 6.0 Hz, 1H), 7.31 (dd, J = 8.4, 5.4 Hz, 2H), 7.14 (t, J = 9.0 Hz, 2H), 4.41 (d, J = 5.4 Hz, 2H); 2.94 (d, J = 4.8 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.7, 163.7, 161.1 (d, JCF = 241.5 Hz), 161.2, 155.7, 147.3, 136.0 (d, JCF = 2.4 Hz), 129.1 (d, JCF = 8.1 Hz), 115.0 (d, JCF = 20.7 Hz), 79.0, 40.9, 28.4; HRMS-ESI(−) m/z calcd for C13H13FN4O4 307.0848 [M−H]−, found 307.0850.
N-(3-Chloro-2-fluorobenzyl)-1-(hydroxy)-1,2,3,6-tetrahydro-4-(methylamino)-2,6-dioxopyrimidine-5-carboxamide (29)
White solid, 90 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.06 (s, 1H), 10.76 (q, J = 4.2 Hz, 1H), 10.22 (s, 1H), 9.95 (t, J = 6.0 Hz, 1H), 7.47 (t, J = 7.2 Hz, 1H), 7.27 (dd, J = 7.8, 6.0 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 4.50 (d, J = 4.8 Hz, 2H), 2.93 (d, J = 4.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.8, 161.3, 155.7, 155.2 (d, JCF = 246 Hz), 147.3, 129.0, 128.7 (d, JCF = 13.8 Hz), 128.2 (d, JCF = 4.5 Hz), 125.2 (d, JCF = 4.7 Hz), 119.4 (d, JCF = 17.3 Hz), 79.0, 35.9 (d, JCF = 3.5 Hz), 28.4; HRMS-ESI(−) m/z calcd for C13H12ClFN4O4 341.0458 [M−H]−, found 341.0462.
N-Benzyl-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (30)
White solid, 42% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 11.04 (t, J = 5.4 Hz, 1H), 10.20 (s, 1H), 9.93 (t, J = 6.0 Hz, 1H), 7.34–7.24 (m, 5H), 4.43 (d, J = 6.0 Hz, 2H); 3.36 (td, J = 6.6, 6.0 Hz, 2H), 1.14 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.8, 161.4, 154.7, 147.3, 139.6, 128.4, 127.1, 126.8, 78.8, 41.7, 36.0, 14.4; HRMS-ESI(−) m/z calcd for C14H16N4O4 303.1099 [M−H]−, found 303.1098.
N-Benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxo-4-(propylamino)pyrimidine-5-carboxamide (31)
White solid, 79% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.16 (s, 1H), 11.13 (s, 1H), 10.20 (s, 1H), 9.94 (s, 1H), 7.32-7.24 (m, 5H), 4.44 (d, J = 4.8 Hz, 2H); 3.30 (m, 2H), 1.54 (m, 2H), 0.91 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.4, 154.9, 147.3, 139.6, 128.4, 127.1, 126.8, 78.8, 42.6, 41.7, 22.1, 11.0; HRMS-ESI(−) m/z calcd for C15H18N4O4 317.1255 [M−H]−, found 317.1253.
N-(3-Chloro-2-fluorobenzyl)-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxo-4-(propylamino)pyrimidine-5-carboxamide (32)
White solid, 73 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.15 (s, 1H), 11.02 (s, 1H), 10.23 (s, 1H), 9.99 (s, 1H), 7.47–7.18 (m, 3H), 4.51 (d, J = 4.2 Hz, 2H), 3.30 (m, 2H), 1.52 (m, 2H), 0.89 (dd, J = 7.8, 6.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 168.0, 161.4, 155.2 (d, JCF = 246 Hz), 154.9, 147.3, 129.1, 128.6 (d, JCF = 14.9 Hz), 128.2 (d, JCF = 4.5 Hz), 125.3 (d, JCF = 3.5 Hz), 119.5 (d, JCF = 17.3 Hz), 78.8, 42.6, 35.9 (d, JCF = 3.5 Hz), 22.0, 11.0; HRMS-ESI(−) m/z calcd for C15H16ClFN4O4 369.0771 [M−H]−, found 369.0768.
N-Benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-4-(isopropylamino)-2,6-dioxopyrimidine-5-carboxamide (33)
White solid, 92% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.18 (d, J = 8.4 Hz 1H), 11.14 (s, 1H), 10.21 (s, 1H), 9.96 (d, J = 5.4 Hz 1H), 7.34–7.23 (m, 5H), 4.43 (d, J = 5.4 Hz, 2H); 4.08 (m, 1H), 1.16 (d, J = 6.0 Hz, 6H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.4, 153.8, 147.3, 139.6, 128.4, 127.1, 126.8, 78.7, 42.6, 41.7, 22.8; HRMS-ESI(−) m/z calcd for C15H18N4O4 317.1255 [M−H]−, found 317.1261.
N-Benzyl-1-(hydroxy)-4-(cyclopropylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (34)
White solid, 82% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.17 (s, 1 H), 11.01 (s, 1H), 10.24 (s, 1H), 9.87 (t, J = 6.0 Hz 1H), 7.31–7.21 (m, 5H), 4.39 (d, J = 5.4 Hz, 2H); 2.71 (m, 1H), 0.83 (m, 2H), 0.59 (m, 2 H); 13C NMR (150 MHz, DMSO-d6): δ 167.6, 161.2, 156.5, 147.1, 139.5, 128.4, 127.1, 126.8, 79.2, 41.7, 23.0, 7.7; HRMS-ESI(−) m/z calcd for C15H16N4O4 315.1099 [M−H]−, found 315.1101.
4-(Tert-butylamino)-N-benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (35)
White solid, 59% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.82 (s, 1H), 10.24 (s, 1H), 10.10 (t, J = 6.0 Hz, 1H), 10.03 (brs, 1H), 7.33–7.25 (m, 5H), 4.43 (d, J = 5.4 Hz, 2H), 1.41 (s, 9H); 13C NMR (150 MHz, DMSO-d6): δ 168.1, 161.4, 153.7, 146.8, 139.5, 128.4, 127.1, 126.8, 78.9, 52.0, 41.7, 29.0; HRMS-ESI(−) m/z calcd for C16H20N4O4 331.1412 [M−H]−, found 331.1416.
4-Amino-N-benzyl-1-(hydroxy)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (46)
White solid, 89% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.19 (brs, 1H), 10.13 (brs, 1H), 9.78 (s, 1H), 9.68 (s, 1H), 7.32-7.24 (m, 4H), 6.88 (brs, 1H), 4.43 (d, J = 4.8 Hz, 2H); 13C NMR (150 MHz, DMSO-d6): δ 167.1, 161.8, 156.4, 147.5, 139.7, 128.3, 127.1, 126.7, 79.8, 41.6; HRMS-ESI(−) m/z calcd for C12H12N4O4 275.0786 [M−H]−, found 275.0785.
N-(3-Fluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (47)
White solid, 76% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 11.98 (t, J = 4.8 Hz, 1H), 10.22 (s, 1H), 9.97 (t, J = 6.0 Hz, 1H), 7.36 (q, 7.2 Hz, 1H), 7.11 (d, J = 7.2 Hz, 1H), 7.06 (m, 2H), 4.45 (d, J = 6.0 Hz, 2H); 3.36 (td, J = 7.2, 6.6 Hz, 2H), 1.14 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.5 (d, JCF = 241.5 Hz), 161.4, 154.7, 147.3, 142.9 (d, JCF = 6.9 Hz), 130.3 (d, JCF = 8.0 Hz), 123.0, 113.7 (d, JCF = 21.8 Hz), 113.5 (d, JCF = 20.7 Hz), 78.8, 41.2, 36.1, 14.4; HRMS-ESI(−) m/z calcd for C14H15FN4O4 321.1005 [M−H]−, found 321.1005.
N-(4-Fluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (38)
White solid, 33% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.10 (s, 1H), 11.01 (t, J = 5.4 Hz, 1H), 10.20 (s, 1H), 9.93 (t, J = 6.0 Hz, 1H), 7.31 (dd, J = 8.4, 5.4 Hz, 2H), 7.14 (t, J = 9.0 Hz, 2H), 4.41 (d, J = 5.4 Hz, 2H), 3.36 (td, J = 7.2, 6.6 Hz, 2H), 1.14 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.8, 161.3, 161.1 (d, JCF = 240.3 Hz), 154.7, 147.3, 135.9 (d, JCF = 2.3 Hz), 129.1 (d, JCF = 8.0 Hz), 115.0 (d, JCF = 21.9 Hz), 78.8, 40.9, 36.0, 14.4; HRMS-ESI(−) m/z calcd for C14H15FN4O4 321.1005 [M−H]−, found 321.1003.
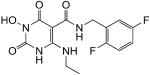
N-(2,5-difluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (39)
White solid, 32% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.12 (s, 1H), 11.90 (t, J = 5.4 Hz, 1H), 10.23 (s, 1H), 9.96 (t, J = 6.0 Hz, 1H), 7.25–7.07 (m, 3H), 4.45 (d, J = 5.4 Hz, 2H); 3.36 (td, J = 7.2, 6.6 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H); HRMS-ESI(−) m/z calcd for C14H14F2N4O4 339.091 [M−H]−, found 339.0911.
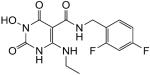
N-(2,4-difluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (40)
White solid, 26% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 10.91 (t, J = 5.4 Hz, 1H), 10.19 (s, 1H), 9.94 (t, J = 6.0 Hz, 1H), 7.36–7.04 (m, 3H), 4.43 (d, J = 6.0 Hz, 2H); 3.35 (td, J = 7.2, 6.6 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H); HRMS-ESI(−) m/z calcd for C14H14F2N4O4 339.091 [M−H]−, found 339.0915.
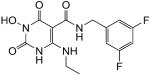
N-(3,5-difluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (41)
White solid, 61% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.12 (s, 1H), 10.92 (s, 1H), 10.23 (s, 1H), 9.99 (t, J = 5.4 Hz, 1H), 7.09–6.96 (m, 3H), 4.45 (d, J = 5.4 Hz, 2H); 3.36 (m, 2H), 1.14 (dd, J = 7.2, 6.6 Hz, 3H); HRMS-ESI(−) m/z calcd for C14H14F2N4O4 339.091 [M−H]−, found 339.0914.
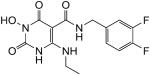
N-(3,4-difluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (42)
White solid, 56% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 10.96 (s, 1H), 10.21 (s, 1H), 9.96 (s, 1H), 7.38–7.16 (m, 3H), 4.41 (d, J = 3.6 Hz, 2H); 3.36 (m, 2H), 1.14 (dd, J = 7.2. 4.8 Hz, 3H); HRMS-ESI(−) m/z calcd for C14H14F2N4O4 339.091 [M−H]−, found 339.0915.
N-(2-Chloro-5-fluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (43)
White solid, 20 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.13 (s, 1H), 10.88 (dd, J = 5.4, 4.2 Hz, 1H), 10.24 (s, 1H), 10.03 (t, J = 6.0 Hz, 1H), 7.50 (dd, J = 9.0, 5.4 Hz, 1H), 7.16 (td, J = 8.4, 3.0 Hz, 1H), 7.09 (dd, J = 9.6, 2.4 Hz, 1H), 4.47 (d, J = 6.0 Hz, 2H), 3.35 (td, J = 7.2, 6.6 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 168.0, 161.4, 160.8 (d, JCF = 242.7 Hz), 154.7, 147.3, 139.5 (d, JCF = 6.9 Hz), 130.9 (d, JCF = 8.1 Hz), 127.2 (d, JCF = 2.3 Hz), 116.2 (d, JCF = 24.2 Hz), 115.4 (d, JCF = 23.1 Hz), 78.8, 39.8, 36.1, 14.3; HRMS-ESI(−) m/z calcd for C14H14ClFN4O4 355.0615 [M−H]−, found 355.0618.
N-(4-Chloro-3-fluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (44)
White solid, 51 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.12 (s, 1H), 10.94 (t, J = 5.4 Hz, 1H), 10.22 (s, 1H), 9.98 (t, J = 6.0 Hz, 1H), 7.53 (dd, J = 8.4, 7.2 Hz, 1H), 7.28 (d, J = 10.8 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H), 3.36 (td, J = 7.2, 6.6 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 168.0, 161.3, 157.0 (d, JCF = 244.8 Hz), 154.7, 147.3, 142.0 (d, JCF = 5.7 Hz), 130.4, 124.2 (d, JCF = 3.4 Hz), 117.4 (d, JCF = 17.3 Hz), 115.4 (d, JCF = 20.7 Hz), 78.8, 40.8, 36.1, 14.3; HRMS-ESI(−) m/z calcd for C14H14ClFN4O4 355.0615 [M−H]−, found 355.0621.
N-(3-Chloro-2-fluorobenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (45)
White solid, 90 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.12 (s, 1H), 10.90 (t, J = 5.4 Hz, 1H), 10.23 (s, 1H), 9.99 (t, J = 6.0 Hz, 1H), 7.48 (t, J = 7.2 Hz, 1H), 7.27 (dd, J = 7.2 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 4.50 (d, J = 4.2 Hz, 2H), 3.35 (td, J = 7.2, 6.6 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.4, 155.2 (d, JCF = 244.8 Hz), 154.7, 147.3, 129.1, 128.6 (d, JCF = 13.8 Hz), 128.2 (d, JCF = 3.5 Hz), 125.2 (d, JCF = 4.5 Hz), 119.4 (d, JCF = 17.3 Hz), 78.8, 36.0, 35.9 (d, JCF = 3.5 Hz), 14.4; HRMS-ESI(−) m/z calcd for C14H14ClFN4O4 355.0615 [M−H]−, found 355.0620.
N-(3-Fluoro-4-methylbenzyl)-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (46)
White solid, 37 % yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 11.00 (t, J = 5.4 Hz, 1H), 10.21 (s, 1H), 9.93 (t, J = 5.4 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.01 (m, 2H), 4.39 (d, J = 6.0 Hz, 2H), 3.36 (td, J = 6.6, 6.0 Hz, 2H), 2.19 (s, 3H), 1.13 (dd, J = 7.2, 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.4, 160.5 (d, JCF = 239.3 Hz), 154.7, 147.3, 139.9 (d, JCF = 6.9 Hz), 131.5 (d, JCF = 5.7 Hz), 122.8 (d, JCF = 3.5 Hz), 122.3 (d, JCF = 17.3 Hz), 113.5 (d, JCF = 21.8 Hz), 78.8, 41.0, 36.0, 14.4, 13.8 (d, JCF = 3.5 Hz); HRMS-ESI(−) m/z calcd for C15H17FN4O4 335.1161 [M−H]−, found 335.1164.
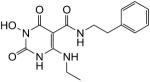
1-(Hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxo-N-phenethylpyrimidine-5-carboxamide (47)
White solid, 79%yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (t, J = 5.4 Hz, 1H), 11.06 (s, 1H), 10.17 (s, 1H), 9.56 (t, J = 5.4 Hz, 1H), 7.30–7.19 (m, 5H), 3.45 (ddd, J = 7.2, 6.6, 6.0 Hz, 2H); 3.35 (qd, J = 7.2, 6.6 Hz, 2H), 2.77 (t, J = 7.2 Hz, 3H), 1.14 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.8, 161.2, 154.6, 147.3, 139.4, 128.6, 128.4, 126.1, 78.9, 39.8, 36.0, 35.3, 14.4; HRMS-ESI(−) m/z calcd for C15H18N4O4 317.1255 [M−H]−, found 317.1257.
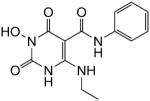
1-(Hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxo-N-phenylpyrimidine-5-carboxamide (48)
White solid, 83% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.89 (s, 1H), 11.26 (s, 1H), 10.89 (t, J = 5.4 Hz, 1H), 10.38 (s, 1H), 7.55 (d, J = 7.8 Hz, 2H), 7.31 (dd, J = 7.8, 7.2 Hz, 2H), 7.31 (dd, J = 7.8, 7.2 Hz, 1H), 3.42 (qd, J = 6.6, 6.0 Hz, 2H), 1.18 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 166.3, 161.8, 154.9, 147.1, 138.5, 128.9, 123.1, 119.8, 79.2, 36.3, 14.4; HRMS-ESI(−) m/z calcd for C13H14N4O4 289.0942 [M−H]−, found 289.0942.
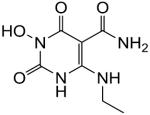
1-(Hydroxy)-1,2,3,6-tetrahydro-2,6-dioxo-4-(phenylamino)pyrimidine-5-carboxamide (49)
White solid; 1H NMR (600 MHz, DMSO-d6): δ 12.90 (s, 1H), 11.20 (s, 1H), 10.27 (s, 1H), 8.96 (s, 1H), 7.44–7.27 (m, 5H); 13C NMR (150 MHz, DMSO-d6): δ 169.9, 161.4, 154.2, 147.0, 135.8, 129.6, 126.4, 124.9, 80.7; HRMS-ESI(−) m/z calcd for C11H10N4O4 261.0629 [M−H]−, found 261.0632.
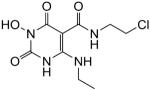
1-(Hydroxy)-N-(2-chloroethyl)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxamide (50)
White solid, 83% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.11 (s, 1H), 11.98 (s, 1H), 10.22 (s, 1H), 9.78 (s, 1 H), 3.68 (m, 2H), 3.55 (m, 2H), 3.37 (m, 2H), 1.15 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 168.0, 161.2, 154.7, 147.3, 78.7, 43.97, 40.1, 36.1, 14.4; HRMS-ESI(−) m/z calcd for C9H13ClN4O4 275.0553 [M−H]−, found 275.0552.
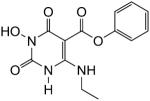
Phenyl-1-(hydroxy)-4-(ethylamino)-1,2,3,6-tetrahydro-2,6-dioxopyrimidine-5-carboxylate (51)
White solid, 92% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.16 (s, 1H), 10.12 (s, 1H), 9.49 (s, 1H), 7.41–7.09 (m, 5H), 3.42 (m, 2H), 1.14 (m, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.2, 157.2, 155.6, 150.7, 147.6, 129.3, 125.3, 122.1, 78.2, 36.5, 14.2; HRMS-ESI(−) m/z calcd for C13H13N3O5 290.0782 [M−H]−, found 290.0784.
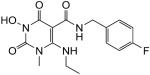
N-(4-Fluorobenzyl)-3-(hydroxy)-6-(ethylamino)-1,2,3,4-tetrahydro-1-methyl-2,4-dioxopyrimidine-5-carboxamide (52)
White solid, 38% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.89 (s, 1H), 10.29 (s, 1H), 9.93 (s, 1H), 7.32 (m, 2H), 7.14 (m, 2H), 4.43 (d, J = 4.2 Hz, 2H), 4.00 (s, 3H), 3.47 (m, 2H), 1.16 (dd, J = 6.6, 5.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.6, 161.1 (d, JCF = 240.3 Hz), 160.5, 159.8, 154.2, 135.9, 129.1 (d, JCF = 8.1 Hz), 115.0 (d, JCF = 20.7 Hz), 84.4, 55.6, 41.0, 35.2, 14.8; HRMS-ESI(−) m/z calcd for C15H17FN4O4 335.1161 [M−H]−, found 335.1158.
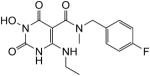
3-(Benzyloxy)-6-(ethylamino)-N-(4-fluorobenzyl)-N-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (53)
White solid, 28% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.25 (s, 1H), 9.99 (s, 1H), 7.52–7.13 (m, 9H), 4.98 (s, 2H), 4.43 (s, 2H), 3.43 (m, 2H), 3.38 (s, 3H), 1.19 (dd, J = 6.0, 5.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.5, 161.2 (d, JCF = 240.3 Hz), 161.0, 159.9, 148.5, 135.7, 134.4, 129.34, 129.31 (d, JCF = 8.1 Hz), 128.8, 128.3, 115.1 (d, JCF = 20.7 Hz), 83.4, 77.3, 41.6, 41.2, 35.9, 15.5; HRMS-ESI(−) m/z calcd for C22H23FN4O4 425.1631 [M−H]−, found 425.1631.
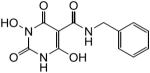
N-Benzyl-1,6-dihydroxy-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (54)
Pale white solid, 53% yield; 1H NMR (600 MHz, DMSO-d6): δ 11.91 (s, 1H), 10.40 (s, 1H), 9.97 (s, 1H), 7.36–7.29 (m, 5H), 4.57 (brs, 2H); 13C NMR (150 MHz, DMSO-d6): δ 170.0, 147.6, 137.6, 7, 128.5, 127.41, 127.36, 79.0, 42.8; HRMS-ESI(−) m/z calcd for C12H11N3O5 276.0626 [M−H]−, found 276.0628.
N-(4-Fluorobenzyl)-7-(hydroxy)-2,3,4,6,7,8-hexahydro-6,8-dioxo-1H-pyrimido[1,6-a]pyrimidine-9-carboxamide (55)
Pale white solid, 96 yield; 1H NMR (600 MHz, DMSO-d6): δ 11.52 (s, 1H), 10.29 (s, 1H), 10.02 (br s, 1H), 7.31–7.13 (m, 4H), 4.42 (d, J = 4.8, 2H), 3.82 (m, 2H), 3.38 (m, 2H), 1.94 (m, 2H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 161.1 (d, JCF = 241.5 Hz), 160.4, 154.1, 147.4, 136.0 (d, JCF = 2.3 Hz), 129.1 (d, JCF = 8.1 Hz), 115.0 (d, JCF = 20.1 Hz), 78.6, 41.0, 40.8, 38.1, 18.8; HRMS-ESI(−) m/z calcd for C15H15FN4O4 333.1005 [M−H]−, found 333.1001.
Biology
INST assay
HIV integrase was expressed and purified as previously reported.52 Inhibition assays were performed using a modified protocol of our reported method.52 Briefly, 2.1 μL of compound suspended in DMSO was placed in duplicate into a Black 96 well non-binding plate (corning 3991). Compounds were plated in duplicate to a final concentration of 0.13 — 100 μM. To each well of the plate 186.9 μL of reaction mixture without DNA substrate was added (10 mM HEPES pH 7.5, 10 % glycerol w/v, 10 mM MnCl2, 1 mM DTT, 1 μM integrase). The enzyme was incubated with inhibitor for 10 min at 25 °C after which the reaction was initiated by the addition of 21 μL of 500 nM oligo (5' biotin ATGTGGAAAATCTCTAGCA annealed with ACTGCTAGAGATTTTCCACAT 3' Cy5). Reactions were incubated at 37 °C for 30 min and then quenched by the addition of 5.2 μL 500 mM EDTA. Each reaction was moved (200 μL) to a MultiScreen HTS PCR plate (Millipore MSSLBPC10) containing 20 μL streptavidin agarose beads (Life Technologies S951) and incubated with shaking for 30 min. A vacuum manifold was used to remove the reaction mixture and the beads were similarly washed 3 times with wash buffer (.05% SDS, 1 mM EDTA in PBS). The plates were further washed 3 times with 200 μL 50 mM NaOH to denature DNA not covalently linked to the biotin modification. For each denaturation step the plate was incubated with shaking at 25 °C for 5 min and the NaOH was removed by centrifugation at 1000 g for 1 min. The reaction products were eluted from the beads by the addition of 150 μL formamide. The plate was incubated at 25 °C for 10 min and read directly at 635/675 in a SpectraMax i3 plate reader (Molecular Devices).
Expression and purification of the recombinant IN in Escherichia coli were performed as previously reported39, 53 with addition of 10% glycerol to all buffers. Preparation of oligonucleotide substrates has been described.54 Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5'-end [32P]-labeled oligonucleotide substrate and inhibitors at various concentrations. Solutions of 10% DMSO without inhibitors were used as controls. Reactions were incubated at 37 °C (60 minutes) in buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Reactions were stopped by addition of 10 μL of loading dye (10 mM EDTA, 98% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were then subjected to electrophoresis in 20% polyacrylamide–7 M urea gels. Gels were dried and reaction products were visualized and quantitated with a Typhoon 8600 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analyses were performed using ImageQuant from Molecular Dynamics Inc. The concentrations at which enzyme activity was reduced by 50% (IC50) were determined using “Prism” software (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
RNase H assay
RNase H activity was measured essentially as previously described.55 Full-length HIV RT was incubated with the RNA/DNA duplex substrateHTS-1 (RNA 5'-gaucugagccugggagcu -3'-fluorescein annealed to DNA 3'-CTAGACTCGGACCCTCGA -5'-Dabcyl), a high sensitivity duplex that assesses non-specific internal cleavage of the RNA strand.
HIV-1 cytoprotection assay35
The HIV Cytoprotection assay used CEM-SS cells and the IIIB strain of HIV-1. Briefly virus and cells were mixed in the presence of test compound and incubated for 6 days. The virus was pre-titered such that control wells exhibit 70 to 95% loss of cell viability due to virus replication. Therefore, antiviral effect or cytoprotection was observed when compounds prevent virus replication. Each assay plate contained cell control wells (cells only), virus control wells (cells plus virus), compound toxicity control wells (cells plus compound only), compound colorimetric control wells (compound only) as well as experimental wells (compound plus cells plus virus). Cytoprotection and compound cytotoxicity were assessed by MTS (CellTiter® 96 Reagent, Promega, Madison WI) and the EC50 (concentration inhibiting virus replication by 50%), CC50 (concentration resulting in 50% cell death) and a calculated TI (therapeutic index CC50/EC50) were provided. Each assay included AZT as a positive control.
Antiviral MAGI assays
MAGI assays were carried out using P4R5 indicator cells essentially as previously described.36 P4R5 cells were cultured in 96-well microplates with 4×103 cells per well and maintained in DMEM/10% FBS supplemented with puromycin (1 μg/ml). Cells were incubated with either 1% DMSO or varying concentrations of the drugs for 24h and then exposed to HIV (MOI of 1.25) followed by an additional incubation period of 48h. The extent of infection was assessed using a fluorescence-based β-galactosidase detection assay, as previously described with minor modifications.56 After the 48h incubation period, cells were lysed and substrate 4-methylumbelliferyl-galactoside (MUG) was added. The β-galactosidase produced during infection acts on the substrate MUG and yields a fluorescent product 4-methylumbelliferone (4-MU) that could be detected fluorimetrically with excitation wavelength 365 nm and emission wavelength 446 nm.
Modeling and docking
Molecular modeling was performed using the Schrodinger small molecule drug discovery suite 2014-3. The crystal structure of PFV intasome in complex with magnesium and 3 was used as a starting point (PDB code: 3S3M).50 This model was subjected to Protein Preparation Wizard57–58 (Schrödinger Inc.) in which missing hydrogens atoms were added, zero-order bonds to metals were created followed by the generation of metal binding states. The structure of protein was minimized using OPLS 2005 force field59 to optimize hydrogen bonding network and converge heavy atoms to an rmsd of 0.3 Å. The receptor grid generation tool in Maestro (Schrodinger Inc.) was used to define an active site around the native ligand (3) to cover all the residues within 12 Å from it with both the metal cofactors (Mg2+) as a constraint to identify the chelating triad during docking.
All the compounds synthesized were drawn using Maestro and subjected to Lig Prep58 to generate conformers, possible protonation at pH of 7 ± 3, and metal binding states that serve as an input for docking process. All the dockings were performed using Glide XP60 (Glide, version 6.4) mode with both the Mg2+ metal cofactors as a constraint. The van der Waals radii of nonpolar atoms for each of the ligands were scaled by a factor of 0.8.
To understand the effect of observed mutations on resistance profile for 1, 3 and the representative compound 45, a crystal structure of PFV intasome in complex with magnesium and 3 was used as a starting point (PDB code: 3S3M). The observed mutants were changed in the crystal structure manually using Maestro (PFV intasome numbering of G209S, Y212C, Q217H and N224H corresponds to HIV intasome numbering of G140S, Y143C, Q148H and N155H) followed by a protein preparation.
Three compounds (1, 3 and 45) were drawn using Maestro and subjected to Lig Prep to generate conformers, possible protonation at pH of 7 ± 3, and metal binding states that serve as an input for docking process. These compounds were docked using a similar protocol as above.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (AI100890) and partially by the Center for Drug Design, University of Minnesota.
ABBREVIATIONS USED
- HIV
human immunodeficiency virus
- INST
integrase strand transfer
- HPD
3-hydroxypyrimidine-2,4-dione
- INST
integrase strand transfer
- INSTIs
integrase strand transfer inhibitors
- RT
reverse transcriptase
- HAART
highly active antiretroviral therapy
- NRTIs
nucleoside reverse transcriptase inhibitors
- NNRTIs
nonnucleoside reverse transcriptase inhibitors
- PIs
protease inhibitors
- DKA
diketoacid
- CPE
cytopathic effect
- SAR
structure-activity-relationship
- MOA
mechanism of action
- NTD
N-terminal domain
- CCD
catalytic core domain
- CTD
C-terminal domain
- PFV
prototype foamy virus.
Footnotes
Supporting Information Available. Synthesis and characterization data, including 1H NMR, 13C NMR and HRMS data, of intermediates 7–23. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kinch MS, Patridge E. An analysis of FDA-approved drugs for infectious disease: HIV/AIDS drugs. Drug Discov. Today. 2014;19:1510–1513. doi: 10.1016/j.drudis.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science. 2014;345:169–174. doi: 10.1126/science.1255512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun TW, Fauci AS. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS. 2012;26:1261–1268. doi: 10.1097/QAD.0b013e328353f3f1. [DOI] [PubMed] [Google Scholar]
- 4.Bagasra O. A unified concept of HIV latency. Expert Opin. Biol. Ther. 2006;6:1135–1149. doi: 10.1517/14712598.6.11.1135. [DOI] [PubMed] [Google Scholar]
- 5.Chun T-W, Stuyver L, Mizell SB, Ehler LA, Mican JAM, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 7.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discovery. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 8.Cotelle P. Patented HIV-1 integrase inhibitors (1998–2005) Recent Pat. Anti-Infect. Drug Discovery. 2006;1:1–15. doi: 10.2174/157489106775244082. [DOI] [PubMed] [Google Scholar]
- 9.Henao-Mejia J, Goez Y, Patino P, Rugeles MT. Diketo acids derivatives as integrase inhibitors: the war against the acquired immunodeficiency syndrome. Recent Pat.Anti-Infect. Drug Discovery. 2006;1:255–265. doi: 10.2174/157489106777452700. [DOI] [PubMed] [Google Scholar]
- 10.Dubey S, Satyanarayana YD, Lavania H. Development of integrase inhibitors for treatment of AIDS: An overview. Eur. J. Med. Chem. 2007;42:1159–1168. doi: 10.1016/j.ejmech.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 11.Marchand C, Maddali K, Metifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr. Top. Med. Chem. 2009;9:1016–1037. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dayam R, Sanchez T, Neamati N. Diketo acid pharmacophore. 2. discovery of structurally diverse inhibitors of HIV-1 integrase. J. Med. Chem. 2005;48:8009–8015. doi: 10.1021/jm050837a. [DOI] [PubMed] [Google Scholar]
- 13.Dayam R, Sanchez T, Clement O, Shoemaker R, Sei S, Neamati N. b-Diketo acid pharmacophore hypothesis. 1. discovery of a novel class of HIV-1 integrase inhibitors. J. Med. Chem. 2005;48:111–120. doi: 10.1021/jm0496077. [DOI] [PubMed] [Google Scholar]
- 14.Deng J, Sanchez T, Neamati N, Briggs JM. Dynamic pharmacophore model optimization: identification of novel HIV-1 integrase inhibitors. J. Med. Chem. 2006;49:1684–1692. doi: 10.1021/jm0510629. [DOI] [PubMed] [Google Scholar]
- 15.Barreca ML, Ferro S, Rao A, De Luca L, Zappala M, Monforte A-M, Debyser Z, Witvrouw M, Chimirri A. Pharmacophore-based design of HIV-1 integrase strand-transfer inhibitors. J. Med. Chem. 2005;48:7084–7088. doi: 10.1021/jm050549e. [DOI] [PubMed] [Google Scholar]
- 16.Mustata GI, Brigo A, Briggs JM. HIV-1 integrase pharmacophore model derived from diverse classes of inhibitors. Bioorg. Med. Chem. Lett. 2004;14:1447–1454. doi: 10.1016/j.bmcl.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 17.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 18.Cocohoba J, Dong BJ. Raltegravir: the first HIV integrase inhibitor. Clin. Ther. 2008;30:1747–1765. doi: 10.1016/j.clinthera.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-Integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 20.Shimura K, Kodama EN. Elvitegravir: a new HIV integrase inhibitor. Antiviral Chem. Chemother. 2009;20:79–85. doi: 10.3851/IMP1397. [DOI] [PubMed] [Google Scholar]
- 21.Vandekerckhove L. GSK-1349572, a novel integrase inhibitor for the treatment of HIV infection. Curr. Opin. Invest. Drugs. 2010;11:203–212. [PubMed] [Google Scholar]
- 22.Min S, Song I, Borland J, Chen S, Lou Y, Fujiwara T, Piscitelli SC. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2009;54:254–258. doi: 10.1128/AAC.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shafer RW. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006;194(Suppl 1):S51–8. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang Z. N-3 hydroxylation of pyrimidine-2,4-diones yields dual inhibitors of HIV reverse transcriptase and integrase. ACS Med. Chem. Lett. 2011;2:63–67. doi: 10.1021/ml1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang ZQ. 6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2011;21:2400–2402. doi: 10.1016/j.bmcl.2011.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang J, Maddali K, Metifiot M, Sham YY, Vince R, Pommier Y, Wang ZQ. 3-Hydroxypyrimidine-2,4-diones as an inhibitor scaffold of HIV integrase. J. Med. Chem. 2011;54:2282–2292. doi: 10.1021/jm1014378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang ZQ. 6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2011;21:2400–2402. doi: 10.1016/j.bmcl.2011.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vernekar SK, Liu Z, Nagy E, Miller L, Kirby KA, Wilson DJ, Kankanala J, Sarafianos SG, Parniak MA, Wang Z. Design, synthesis, biochemical, and antiviral evaluations of C6 benzyl and C6 biarylmethyl substituted 2-hydroxylisoquinoline-1,3-diones: dual inhibition against HIV reverse transcriptase-associated RNase H and polymerase with antiviral activities. J. Med. Chem. 2015;58:651–664. doi: 10.1021/jm501132s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mai X, Lu XS, Xia HY, Cao YS, Liao YJ, Lv XL. Synthesis, antitumor evaluation and crystal structure of hydroxyurea derivatives. Chem. Pharm. Bull. 2010;58:94–97. doi: 10.1248/cpb.58.94. [DOI] [PubMed] [Google Scholar]
- 30.Stadlbauer W, Badawey ES, Hojas G, Roschger P, Kappe T. Malonates in cyclocondensation reactions. Molecules. 2001;6:338–352. [Google Scholar]
- 31.Arnott EA, Chan LC, Cox BG, Meyrick B, Phillips A. POCl3 chlorination of 4-quinazolones. J. Org. Chem. 2011;76:1653–1661. doi: 10.1021/jo102262k. [DOI] [PubMed] [Google Scholar]
- 32.Kotera M, Ishii K, Tamura O, Sakamoto M. 1,3-dipolar cycloadditions of photoinduced carbonyl ylides. Part 2. Photoreactions of alpha,beta-unsaturated gamma,delta-epoxy dinitriles and ethyl vinyl ether. J. Chem. Soc. Perk T. 1. 1998:313–318. [Google Scholar]
- 33.Lee IY, Lee JY, Gong YD. Microwave-assisted facile one-step carbamoylation of 6-aminouracils. Synthesis. 2005:2713–2717. [Google Scholar]
- 34.Fletcher S, Gunning PT. Mild, efficient and rapid O-debenzylation of ortho-substituted phenols with trifluoroacetic acid. Tetrahedron Lett. 2008;49:4817–4819. [Google Scholar]
- 35.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Sirivolu VR, Vernekar SKV, Ilina T, Myshakina NS, Parniak MA, Wang ZQ. Clicking 3 '-azidothymidine into novel potent inhibitors of human immunodeficiency virus. J. Med. Chem. 2013;56:8765–8780. doi: 10.1021/jm401232v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metifiot M, Marchand C, Maddali K, Pommier Y. Resistance to integrase inhibitors. Viruses. 2010;2:1347–1366. doi: 10.3390/v2071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry. 2010;49:3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.da Silva D, Van Wesenbeeck L, Breilh D, Reigadas S, Anies G, Van Baelen K, Morlat P, Neau D, Dupon M, Wittkop L, Fleury H, Masquelier B. HIV-1 resistance patterns to integrase inhibitors in antiretroviral-experienced patients with virological failure on raltegravir-containing regimens. J. Antimicrob. Chemother. 2010;65:1262–1269. doi: 10.1093/jac/dkq099. [DOI] [PubMed] [Google Scholar]
- 41.Reuman EC, Bachmann MH, Varghese V, Fessel WJ, Shafer RW. Panel of prototypical raltegravir-resistant infectious molecular clones in a novel integrase-deleted cloning vector. Antimicrob. Agents Chemother. 2010;54:934–936. doi: 10.1128/AAC.01345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fantauzzi A, Mezzaroma I. Dolutegravir: clinical efficacy and role in HIV therapy. Ther. Adv. Chronic Dis. 2014;5:164–177. doi: 10.1177/2040622314530461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009;10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esposito D, Craigie R. HIV integrase structure and function. Adv. Virus Res. 1999;52:319–333. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 45.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. USA. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 47.O'Brien C. HIV integrase structure catalyzes drug search. Science. 1994;266:1946. doi: 10.1126/science.7801119. [DOI] [PubMed] [Google Scholar]
- 48.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hare S, Vosb AM, Claytonb RF, Thuringb JW, Cummingsb MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA. 2010;157:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hare S, Smith SJ, Metifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol. Pharmacol. 2011;80:565–572. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15910–15915. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J. Med. Chem. 2007;50:3416–3419. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]
- 53.Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, Mouscadet JF. Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry. 2000;39:9285–9294. doi: 10.1021/bi000398b. [DOI] [PubMed] [Google Scholar]
- 54.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by α-hydroxytropolones. Mol. Pharmacol. 2006;69:1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 55.Parniak MA, Min KL, Budihas SR, Le Grice SF, Beutler JA. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal. Biochem. 2003;322:33–39. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Abram ME, Parniak MA. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J. Virol. 2005;79:11952–11961. doi: 10.1128/JVI.79.18.11952-11961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 58.Schrödinger Release 2014-3: Schrödinger Suite 2014-3 Protein Preparation Wizard; Epik version 2.9, Schrödinger. LLC; New York, NY: 2014. Impact version 6.4, Schrödinger, LLC, New York, NY, 2014; Prime version 3.7, Schrödinger, LLC, New York, NY, 2014. In. [Google Scholar]
- 59.Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 60.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1139–1149. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.