

Abstract
Esophageal adenocarcinoma and its precursor Barrett's esophagus have been rapidly increasing in incidence for half a century, for reasons not adequately explained by currently identified risk factors such as gastroesophageal reflux disease and obesity. The upper gastrointestinal microbiome may represent another potential cofactor. The distal esophagus has a distinct microbiome of predominantly oral-derived flora, which is altered in Barrett's esophagus and reflux esophagitis. Chronic low grade inflammation or direct carcinogenesis from this altered microbiome may combine with known risk factors to promote Barrett's metaplasia and progression to adenocarcinoma.
Keywords: Esophageal adenocarcinoma, Barrett's esophagus, Gastroesophageal reflux disease, Human microbiome
Introduction
Esophageal adenocarcinoma (EAC) is becoming an increasingly common cause of morbidity and mortality among Western populations. Overall five-year survival of patients with EAC is approximately 17%, as most tumors are discovered in later stages, after local invasion and/or metastasis has occurred (1). Esophageal adenocarcinoma develops from the precursor lesion Barrett's esophagus (BE), a characteristic intestinal metaplasia of the distal esophagus (1). The presence of BE is associated with a 10-40 fold increased risk for the development of esophageal cancer; however, the rate of progression from BE to EAC is very low at only 0.1-0.3% per year (2-4). As such, there is considerable interest in understanding how BE develops and who progresses to EAC, as to identify biomarkers of EAC risk or therapeutic targets to prevent the development of EAC. The major identified modifiable risk factors for BE and EAC include gastroesophageal reflux disease, obesity, high dietary fat, and smoking (1). However, there are significant gaps in our understanding of the origins of BE and its progression to EAC. Here we review the upper gastrointestinal microbiome, a new potential co-factor in Barrett's neoplasia (Figure 1). We summarize recent developments in characterizing the microbiome in esophageal adenocarcinoma and its precursors, and postulate potential future avenues of investigation and therapeutic targets.
Figure 1.
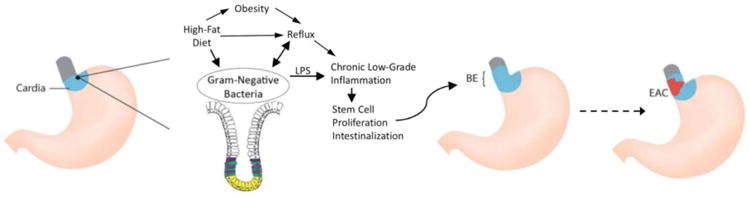
Factors promoting Barrett's esophagus and esophageal adenocarcinoma. Chronic low grade inflammation of the gastric cardia, from a combination of factors including reflux, diet and an altered microbiome, promotes stem cell proliferation and intestinal metaplasia, leading to Barrett's esophagus, which in some cases can progress to esophageal adenocarcinoma
Historical Trends in BE and EAC
Although previously uncommon, EAC has the most rapidly increasing incidence of any tumor type in Western populations (5). There has been a slight decrease in the rise in incidence in more recent years, particularly with early stage tumors (5). However, the incidence continues to rise in younger birth cohorts, suggesting that the overall incidence will continue to rise in the near future (6). Data from the Surveillance, Epidemiology and End Results (SEER) database demonstrates steadily increasing incidence of EAC since data collection began in 1975 (7). However, there was no baseline plateau in this data set, which suggested that the increasing incidence began before this time frame. Our group analyzed data from the Connecticut Tumor Registry, which dates to the 1930s, and demonstrated that the age-adjusted incidence of esophageal adenocarcinoma was very low (∼0.4 cases per 100,000 person-years) and relatively stable until the late 1960s, and has had an overall approximately ten-fold increase since that time (8) (Figure 2). Similar findings have also been reported in Scandinavian cohorts (9). Incidence rates of BE are more difficult to estimate, as the rates of diagnosis are strongly influenced by endoscopy utilization. Like EAC, until recently BE incidence trends also seemed to show a sharp increase (10-12). Interestingly, however, a recent population-based study using diagnosis codes found a decrease in BE diagnoses from 2001-2010, suggesting that the incidence of BE may no longer be on the rise (13).
Figure 2.
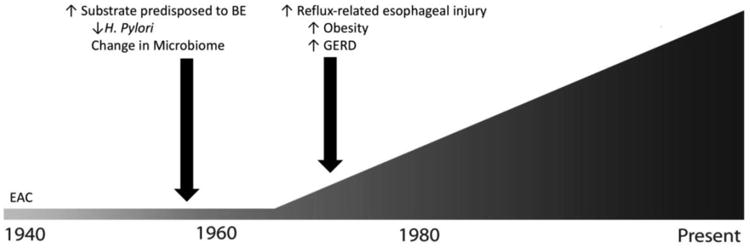
Historical trend of esophageal adenocarcinoma incidence. Esophageal adenocarcinoma began dramatically increasing in incidence in the 1960s, with a rise in Barrett's esophagus presumably preceding this by at least 5-10 years. This predates the increasing incidence of known risk factors for esophageal adenocarcinoma, including gastroesophageal reflux disease and obesity, but coincides with major changes in the upper gastrointestinal microbiome.
Although the reasons underlying the initial rise in EAC incidence are unknown, two hypothetical scenarios, either alone or in combination, could explain this historical pattern: the risk of progression of BE to EAC may have dramatically increased, and/or the incidence of BE sharply increased at some point predating the rise of EAC. While neither can definitively be proven, we believe that the latter scenario is more likely. BE and EAC share virtually identical risk factors, so changes in exposure leading to increased EAC likely also increased the risk of BE. It is difficult to extrapolate exactly when a rise in BE incidence may have occurred, as definitive data is lacking in the time of progression from BE to EAC. Long-term surveillance studies of patients with BE have demonstrated ∼5 years time-to-progression in patients that develop cancer (14). However, the date of BE onset is unknown, and some estimates suggest that progression time may be as long as 20-30 years (15). Regardless, changes in exposures in the mid-20th century are likely responsible for the initial rise in incidence of EAC.
Currently identified modifiable risk factors, including gastroesophageal reflux disease, obesity, smoking and high-fat diet cannot fully explain the chronology and magnitude of the rise of incidence in EAC (16) (Figure 2). Gastro-esophageal reflux disease (GERD) is the strongest known risk factor for the development of BE and EAC (1). However, analyses using claims data suggest that GERD prevalence began to rise in the late 1970s, after the initial 3-4 fold rise in EAC incidence (17, 18). Likewise, obesity, another strong risk factor for EAC, did not begin markedly increasing in the United States until the mid-1980s, and increasing obesity is estimated to account for only 6.5% of the observed rise of EAC (19, 20). Smoking is another known risk factor for EAC, and smoking prevalence rose dramatically in the first half of the 20th century. While this trend fits chronologically with the initial increase in EAC, it does not explain the continued rise, as smoking rates have dropped while EAC becomes increasingly common (21). Additionally, smoking is only a moderate risk factor for EAC, and other GI tumors with similar strengths of association did not have the same dramatic increase seen with EAC (22, 23). Changes in dietary habits, and specifically a diet high in fat, have been epidemiologically linked with the development of EAC (24). While accurate national survey-based dietary data do not exist prior to the 1970s, significant changes occurred in the normal western diet in the early 20th century, following the development of industrial methods for productions of oils and sweeteners (25). However, the proportion of daily calories from fat has decreased since the late 1970s, while EAC continues to rise (26-28).
Development and widespread use of antibiotics began in the 1940s and likely caused major shifts in the upper gastrointestinal microflora (29). Helicobacter pylori dominates the gastric microbiome and causes gastric cancer, but is inversely associated with risk of EAC (30). H. pylori infection rates began to decline in the middle of the 20th century, prior to the observed rise of EAC (31, 32). The temporal associations between increased antibiotic use, decreased H. pylori prevalence, and subsequent rise in EAC incidence suggest that the upper gastrointestinal microbiome may play a key causative role in the development of BE and EAC.
Microbiome and Cancer
Much recent work has focused on the gastrointestinal microbiome, the complex symbiotic community of bacteria colonizing the human GI tract, made up of some 1014 individual bacteria and 3 million bacterial genes (33). Whereas former bacteriological work was founded on the Koch hypotheses of a single pathogen leading to disease, modern research has focused on characterizing these bacterial communities, and identifying global differences in healthy and disease states (33, 34). The rise of molecular techniques and sequencing in the early 21st century has revealed the diversity of the gastrointestinal microbiome (35). Alterations of the gut microbiome have been implicated in many gastrointestinal diseases, including cancers along the GI tract, in particular gastric and colorectal cancer(33).
Several mechanisms by which specific bacteria contribute to the development of GI cancers have been elucidated. For example, in the stomach the H. pylori CagA protein activates the oncogenic tyrosine phosphatase SHP2, leading to cell morphology changes and loss of cell polarity (33). Certain bacterial toxins, such as cytolethal distending toxin (CDT), produced by several Gram-negative species, and colibactin, produced by pks+ Escherichia coli strains, cause direct DNA damage and promote tumorigenesis through activation of DNA repair pathways in colorectal cancer (36, 37). Bacterial virulence factors can also activate host-derived tumor-promoting pathways. In colorectal cancer, FadA-producing Fusobacterium nucleatum interacts with E-cadherin to activate the beta-catenin pathway (38). The esophageal microbiome is less well-studied than other parts of the human gastrointestinal system, but work in characterizing the human esophageal microbiome in normal and pathologic states has revealed significant alterations in resident flora that may contribute to the pathogenesis of metaplasia and dysplasia.
Native Esophageal Microbiome
The esophagus, like all other parts of the human gastrointestinal system, harbors a distinct, diverse microbiome, which is broadly similar to the oropharyngeal microbiome in terms of phyla distribution, although with key taxa-level differences. Like the stomach, the esophagus was long thought to be a relatively sterile environment. Early studies using cultivation of esophageal washes isolated few organisms, which were typically attributed to transient passage of oropharyngeal contents (39-41). However, Pei et al, using culture-independent techniques, characterized a diverse esophageal microbiome using mucosal biopsies of four patients undergoing upper endoscopy and without esophageal disease (42). Using sequencing of the unique bacterial 16S ribosomal RNA gene, they isolated ∼100 unique taxa, made up of 6 major phyla: Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Fusobacteria and TM7. The majority of clones isolated belonged to the Streptococcus genus, making up 39% of the total isolates. Prevotella (17%) and Veilonella (14%) were also common. While the esophageal flora was made up of oropharyngeal species, the overall makeup of the characterized esophageal microbiome was not identical to that of a typical oropharynx (43). Certain common oropharyngeal genera such as Spirochaetes were not found in the esophagus, indicating that the esophagus has its own, distinct microbiome, and is not merely a reflection of oropharyngeal colonization.
BE and Reflux-associated Microbiome
A history of gastroesophageal reflux is the strongest modifiable risk factor for EAC. Published data suggest that patients with BE and reflux esophagitis harbor a distinct esophageal microbiome, characterized by an increased relative abundance of Gram-negative bacteria, including Fusobacterium, Neisseria, Campylobacter, Bacteroides, Proteobacteria and Veilonella taxa, while the Gram-positive Streptococcus is reduced in reflux-related conditions. These microbiome alterations are potentially due to reflux of gastric and bile acids, intestinal and gastric species, or alteration of the distal esophagus microenvironment.
The initial understanding of the BE-associated microbiome was derived from culture-based studies. Osias et al. demonstrated that resident bacteria colonized the Barrett's mucosa, and were not simply transiently deposited, based on close mucosal association of cultivable bacteria on microscopic examination of biopsy specimens (44). Macfarlane et al cultured mucosal biopsy and gastric aspirate samples collected at upper endoscopy from patients with and without BE, and used 16S rRNA gene sequencing to identify isolated organisms. A broader range of bacteria was isolated in BE patients as compared to non-BE patients (23 species from 11 genera vs. 12 species of 7 genera), indicating a potentially increased microbiological diversity in patients with reflux-related BE (45). Notably, several Gram-negative genera, including Fusobacterium, Neisseria and Campylobacter were identified only in Barrett's patients, with none isolated in controls.
Yang et al more comprehensively defined the reflux-related microbiome using culture-independent techniques in a cross-sectional study of 34 patients with BE, reflux esophagitis, and controls (46). Biopsy samples of patients undergoing endoscopy for upper gastrointestinal symptoms (heartburn, fecal occult blood, nausea, etc.) were taken 2 cm above the gastroesophageal junction, and were then classified as normal, esophagitis or BE based on histology. They performed unsupervised clustering on 16S rRNA gene sequencing data from biopsies and identified microbiomes clustered into one of two broad categories, deemed Type I and Type II. The Type I microbiome was characterized by a predominance of Gram-positive taxa, especially Streptococcus, with a relative abundance of 79%. The Type II microbiome had an overall greater diversity, relatively less abundant Streptococcus (30%), and a predominance of Gram-negative taxa (53%), including notable increases in Bacteroides, Proteobacteria and Fusobacterium. Eleven of twelve (92%) of the type I microbiome subjects were controls, while the Type II microbiome was strongly linked with reflux esophagitis (odds ratio 15.4) and Barrett's esophagus (odds ratio 16.5).
A similar study in Japanese patients again characterized a rich reflux-associated esophageal microbiome (47). Streptococcus was the most abundant taxon in controls, while a diverse array of Gram-negative taxa, including Veilonella, Neisseria, and Fusobacterium predominated in BE. Normal, reflux esophagitis and BE patients all harbored similar total bacterial DNA by qPCR (106-107 CFU equivalent), indicating that relative species abundance may be more significantly associated with inflammation and metaplasia of the distal esophagus than total bacterial load. Amir et al compared the esophageal squamous microbiome in patients with symptomatic non-erosive reflux, reflux esophagitis, and BE. Notably, patients taking acid-suppressive therapy prior to the study were excluded. Using 16S pyrosequencing, they found a diverse microbiome composed of mostly oral flora. This study did not note statistically significant differences in microbiome composition between these groups with GERD, indicating that acidic gastric reflux may be a key determinant of microbiome diversity and composition in the distal esophagus.
A recently published study by Gall et al used 16S pyrosequencing to characterize the microbiome at several upper gastrointestinal sites in patients with known BE (48). Samples were taken from esophageal squamous mucosa, Barrett's mucosa, stomach corpus and stomach antrum. Like previous studies, they described an esophageal microbiome consisting of the phyla Firmicutes, Actinobacteria, Bacteroidetes, Proteobacteria and Fusobacteria. No substantial difference in phylogenetic diversity was noted between squamous and Barrett's mucosa microbiome within subjects, and the stomach antrum microbiome more closely resembled the BE microbiome than the contiguous corpus. Relative abundance of species varied greatly between patients, and intra-subject variability across biopsy sites was less than inter-subject comparison at each site. This study used mucosal brushings as well as biopsies, and showed that brushings give a superior yield of bacterial DNA and significantly improve the ratio of bacterial to host DNA, allowing detection of a greater number of taxa (48). We thus favor the use of brushings to sample the esophageal microbiome.
Microbiome in Esophageal Adenocarcinoma
The microbiome in esophageal adenocarcinoma is less well characterized than that of Barrett's esophagus. Several early culture-based studies of esophagectomy specimens from both EAC and squamous cell cancer carried out in the 1980s did not note any difference in organisms isolated in benign versus malignant tissue, however these studies were more concerned with identifying pathogens associated with post-surgical infections than characterizing differences in non-pathogenic bacteria between patients with EAC and control patients (39-41). A more recent study using 16S sequencing of normal versus tumor tissue in esophageal cancer patients found increased Treponema denticola in tumor tissue, but the histologic subtypes of the tumors analyzed was not specified (49). Blackett et al. performed quantitative 16S rRNA sequencing of pre-specified species chosen based on culture isolation, comparing controls without symptomatic reflux and patients with GERD, BE and EAC (50). A “U-shaped” trend was noted of decreased relative abundance of several genera, including Bifodobacteria, Bacteroides, Fusobacteria, Veillonella, Staphylococcus and Lactobacilli in GERD and BE, with a rise toward the control level in EAC. Campylobacter species had a reversed trend, with low abundance in control and cancer patients, but higher prevalence in GERD and BE. The analyses did not include several potentially key genera, such as Streptococcus and Neisseria species. The microbiome in esophageal adenocarcinoma therefore remains a significant gap in knowledge, and there is insufficient data to date on which to base any firm conclusions.
The Gastric Cardia, BE, and the Microbiome
Studies to date on microbiome risk factors for EAC have focused on characterizing the resident esophageal microbiome in both esophageal squamous and columnar tissue in reflux esophagitis and BE. The origin of Barrett's is controversial, and there is increasing evidence that BE may in fact originate from the gastric cardia, a short region of tissue just distal to the esophagus that is chronically inflamed and exposed to a more acidic environment than the rest of the stomach (51, 52). Quante et al. developed a transgenic mouse model of Barrett's metaplasia and EAC, where IL-1β is overexpressed in esophageal tissue, leading to rapidly progressive BE and EAC upon exposure to bile acids and nitrosamine. Using this model, they were able to demonstrate that markers of intestinal progenitor cells, absent in normal squamous esophageal epithelium, were expressed first in the gastric cardia and later in metaplastic esophageal tissue (53).
In humans, Barrett's glands are typically a mix of gastric and intestinal epithelium (type II and type III intestinal metaplasia), so the presence of goblet cells cannot discount a gastric origin. A recent excellent review on the subject argued that, compared to a squamous origin for BE, a cardia origin is better able to account for the presence of the multiple gland phenotypes seen in BE and their distribution in Barrett's glands (52). There is no published data comprehensively describing the microbiome of the cardia, and it is plausible that the cardia microbiome may directly influence BE risk. Future understanding of microbiologic risk factors of BE and EAC should include characterization of the gastric cardia microbiome in addition to that of the distal esophagus.
The Role of H. pylori and the Gastric Microbiome
As previously noted, H. pylori infection, especially with the cagA+ strain, is associated with a decreased risk of development of both BE and EAC (30, 54). Conversely, H. pylori infection strongly increases the risk of gastric cancer, and this paradox is not well understood. The inverse association with BE and EAC is frequently ascribed to decreased acid production in the presence of H. pylori infection, especially in patients with resulting atrophic gastritis. However, while H. pylori infection is associated with reduced risk of BE, no significant association has been noted with GERD or reflux esophagitis; thus, the protective effect is likely not entirely due to reduced gastric acid production (55). Alternatively, the observed associations may be due to more direct microbiome-mediated effects. The gastric microbiome differs greatly in H. pylori infected and uninfected patients. However, H. pylori does not typically colonize the esophagus in high abundance in infected individuals (56). In a mouse model, gastric infection with H. pylori led to an altered esophageal microbiome with increased taxonomic diversity, and H. pylori eradication resulted in a decrease in lower esophageal diversity (57). Thus, H. pylori infection of the stomach in humans may have a direct impact on the lower esophageal microbiome and disease risk.
When present, H. pylori dominates the gastric microbiome, making up 75-99% of total clones in samples from stomach corpus samples (56, 58). In individuals not infected with H. pylori, far fewer total bacteria inhibit the stomach, and the microbiome, like that of the esophagus, is comprised of many oropharyngeal taxa. Actinobacteria, Firmicutes, Bacteroidetes, Proteobacteria and Fusobacteria are the most abundant phyla, with either Proteobacteria or Actinobacteria tending to be most common (56, 58-61). In the absence of H. pylori, the gastric microbiome more closely resembles the Barrett's microbiome than the normal squamous esophageal microbiome, with decreased Firmicutes, and increased Gram-negative phyla such as Fusobacteria, Bacteroidetes and Proteobacteria (42, 46, 58, 59).
Proton Pump Inhibitors and the Upper GI Microbiome
Proton pump inhibitors (PPIs) are a mainstay of treatment of GERD and BE. However, alteration of the ecologic niche through reduced acidity affects the composition of the microbiome. Additionally, PPIs may have direct antibiotic activity against bacteria such as H. pylori and S. pneumoniae that contain P-type ATPase enzymes. In studies of the fecal microbiome, PPIs have been shown to increase the relative abundance of pharyngeal flora, including Streptococcus species, relative to resident colonic bacteria, likely through removing the barrier of a highly acidic environment (62, 63). PPI treatment increases total bacterial concentrations in the stomach, and significantly alters the gastric microbiome, with statistically significant increases in several genera, including Streptococcus, Staphylococcus, Veilonella (64). Amir et al. compared esophageal biopsies and refluxate aspirate of 34 patients before and after PPI treatment. After PPI administration, significant increases in relative abundance were seen in members of the Firmicutes phylum, with decreases in Proteobacteria and Bacteroidetes phyla in both aspirate and biopsy samples. These microbiome alterations are similar to the differences seen between the BE and non-reflux related microbiomes, indicating the key role an acidic environment may play in the microbiota of this region. Given the abundance of patients with upper GI symptoms regularly taking these medications, PPI use must be accounted for in studies of the upper gastrointestinal microbiome.
Diet and the Upper GI microbiome
As previously discussed, dietary factors have been identified as risk factors for BE and EAC (24). Although not studied in the esophageal microbiome, dietary choices cause rapid microbiome alterations and likely potentiate cancer risk in other parts of the human gastrointestinal system (65, 66). A diet high in fat has been shown to cause microbiome-mediated carcinogenesis in the small intestine in mice (67). In humans, reducing dietary fat and increasing dietary fiber leads to decreased colonic inflammation and a less pro-carcinogenic microbiome (68). Potentially similar effects of diet on the upper GI microbiome deserve further investigation.
Potential Mechanisms for Microbiome-Mediated EAC Risk
Several theories have been put forward to explain how microbiome alterations could predispose to Barrett's esophagus and EAC. These tend to revolve around the concept that certain bacteria can promote chronic low grade tissue inflammation, which in turn contributes to the development of metaplasia and progression to cancer. There appears to be a high relative abundance of Gram-negative bacteria in the reflux-associated esophageal microbiome. Although causation is difficult to determine from observational studies, there are plausible mechanisms through which Gram-negatives could lead to metaplasia. Bacterial antigens specific to Gram-negative organisms, such as lipopolysaccharide (LPS), promote tissue inflammation. LPS binding induces increased expression of NF-κβ, which plays a central role in many tissue inflammatory responses, and has increased expression along the spectrum from BE to EAC (69, 70). Interestingly, LPS may also promote BE via increased reflux. In animal studies, LPS has been shown to relax the lower esophageal sphincter and delay gastric emptying, both important mechanisms contributing to gastro-esophageal reflux in humans (71, 72). Gram-negative bacteria can also reduce dietary nitrates to nitrites, which in turn can become carcinogenic N-nitroso compounds in the acidic environment of the cardia and distal esophagus in reflux disease (73).
Studies of colon carcinogenesis have focused on specific bacteria such as F. nucleatum, and genotoxins such as CDT and colibactin that induce DNA damage or promote oncogenic signaling pathways as discussed above. The roles that these bacteria may play in progression from BE to EAC are unknown.
Key Questions
Although progress has been made toward an understanding of the native esophageal microbiome in healthy and pathological states, several key questions remain unanswered.
Are esophageal microbiome alterations causative of BE or just associated with disease?
As mentioned previously, studies to date on the esophageal microbiome have been predominantly cross-sectional in nature, and increased taxa abundance in disease does not necessarily infer a causal role. Experimental alteration of the microbiome, in germ-free or gnotobiotic animals, will be necessary to clarify a correlative or causatve role for the microbiome in BE.
Are there specific pathogenic bacteria that drive the development of BE and/or progression to EAC?
Thus far little is known with regard to specific taxa that are associated with Barrett's dysplasia and EAC. However, lessons can be drawn from studies in colon cancer. Fusobacterium nucleatum has been shown to directly promote colorectal carcinogenesis through the E-cadherin/β-catenin pathway (38). Increased abundance of Fusobacterium spp. has been noted in BE (46, 47). Further investigations are warranted to assess the role of LPS-induced inflammation, F. nucleatum, and other potential pro-neoplastic bacterial species and genotoxins such as colibactin and CDT in esophageal carcinogenesis.
Is the microbiome of this region stable over time?
Existing studies of the esophageal microbiome have been predominantly cross-sectional, so little is known about the stability of the esophageal microbiome over time. Only a small subset of BE patients progress to EAC, and alterations in the microbiome over time may be important drivers of neoplastic progression.
What exposures influence the esophageal and cardia microbiome?
A full understanding of the role of the microbiome in the development of EAC requires that it be viewed within the context of the esophageal environment (Figure 1). The major modifiable risk factors for BE and EAC (gastro-esophageal reflux, obesity, high-fat diet, and smoking) all have the potential to alter significantly the esophageal microbiome. GERD, in particular, may alter the esophageal microbiome either through reflux of gastric acid, bile acids, and intestinal flora, or alteration in the distal esophageal microenvironment. Thus, effects of these exposures on the esophageal microbiome need to be described, and these effects need to be accounted for in future studies aimed at elucidating the microbiome-EAC relationship.
How can the microbiome be modified to mediate BE and EAC risk?
If the esophageal microbiome directly contributes to the development of EAC, then therapeutic interventions to alter the microbiome should be investigated as a means of reducing EAC risk. The use of systemic antibiotics is not a reasonable option for chemoprevention, as long-term use will likely have detrimental effects elsewhere in the body. Probiotic treatment with benign flora is an alternative that may not carry such risk of systemic alterations. Gastric acid suppression may have chemopreventive effects via alterations in the microbiome, and the effects of other medications used in chemoprevention, such as aspirin or statins, on the esophageal microbiome have not been described. Direct treatment of the esophageal lining with topical prebiotics or antibiotics and targeting the oral microbiome may also represent approaches to reduce the risk of EAC.
Conclusion
In summary, increasing rates of esophageal adenocarcinoma are not adequately explained by currently identified risk factors such as GERD, obesity, smoking and diet, and the upper GI microbiome may represent another potential cofactor. The normal distal esophagus has a distinct microbiome of predominantly oral flora. This microbiome is altered in Barrett's esophagus and reflux esophagitis, with relatively increased Gram-negative bacteria and reduced Streptococcus, similar to the H. pylori-negative gastric microbiome. The presence or absence of H. pylori may serve as a biomarker of the microbiome of the gastric cardia, a crucial region from which Barrett's esophagus potentially arises. Future multi-omic analyses should aim to define microbiome alterations in Barrett's esophagus and its progression to esophageal adenocarcinoma, combined with assessment of alterations in key tissue-level pathways, in order to more fully understand the role of the microbiome in esophageal carcinogenesis and to identify potential risk biomarkers and therapeutic targets.
Table 1.
Most common bacterial phyla along the upper gastrointestinal tract. Percentages represent the approximate relative abundances of each phylum.
| Oral Cavity38 | Firmicutes (67%) | Proteobacteria (11%) | Actinobacteria (8%) | Bacteroidetes (7%) | Fusobacteria (4%) | Spirochaetes (2%) |
| Normal esophagus37,41 | Firmicutes (70-87%) | Bacteroidetes (5-20%) | Proteobacteria (2-5%) | Actinobacteria (2-4%) | Fusobacteria (1-2%) | TM7 (0-1%) |
| BE41,42 | Firmicutes (50-55%) | Proteobacteria (20-22%) | Bacteroidetes (14-19%) | Fusobacteria (2-9%) | Actinobacteria (2-7%) | TM7 (0-1%) |
| Stomach (H. pylori negative)53,54 | Firmicutes (22-30%) | Actinobacteria (8-47%) | Proteobacteria (11-37%) | Bacteroidetes (11-28%) | Fusobacteria (1-4%) | TM7 (<1%) |
Acknowledgments
The authors are supported in part by a Columbia Physicians and Surgeon's Dean's Research Fellowship (EJS), a mentored career development award through the National Center for Advancing Translational Sciences' Clinical and Translational Science awards program (NIH KL2 TR000081; DEF), a U54 award from the National Cancer Institute (U54 CA163004; JAA), and an Irving Scholars Award (JAA).
References
- 1.Rustgi AK, El-Serag HB. Esophageal Carcinoma. New England Journal of Medicine. 2014;371(26):2499–509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 2.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett's esophagus. N Engl J Med. 2011;365(15):1375–83. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 3.Bhat S, Coleman HG, Yousef F, et al. Risk of Malignant Progression in Barrett's Esophagus Patients: Results from a Large Population-Based Study. J Natl Cancer Inst. 2011;103(13):1049–57. doi: 10.1093/jnci/djr203. doi:djr203[pii]10.1093/jnci/djr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wani S, Falk G, Hall M, et al. Patients with nondysplastic Barrett's esophagus have low risks for developing dysplasia or esophageal adenocarcinoma. Clin Gastroenterol Hepatol. 2011;9(3):220–7. doi: 10.1016/j.cgh.2010.11.008. quiz e26. doi:S1542-3565(10)01188-2[pii]10.1016/j.cgh.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Pohl H, Sirovich B, Welch HG. Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol Biomarkers Prev. 2010;19(6):1468–70. doi: 10.1158/1055-9965.EPI-10-0012. [DOI] [PubMed] [Google Scholar]
- 6.Thrift AP, Whiteman DC. The incidence of esophageal adenocarcinoma continues to rise: analysis of period and birth cohort effects on recent trends. Ann Oncol. 2012;23(12):3155–62. doi: 10.1093/annonc/mds181. [DOI] [PubMed] [Google Scholar]
- 7.Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100(16):1184–7. doi: 10.1093/jnci/djn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abrams JA, Sharaiha RZ, Gonsalves L, Lightdale CJ, Neugut AI. Dating the rise of esophageal adenocarcinoma: analysis of Connecticut Tumor Registry data, 1940-2007. Cancer Epidemiol Biomarkers Prev. 2011;20(1):183–6. doi: 10.1158/1055-9965.EPI-10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansson LE, Sparen P, Nyren O. Increasing incidence of both major histological types of esophageal carcinomas among men in Sweden. Int J Cancer. 1993;54(3):402–7. doi: 10.1002/ijc.2910540309. [DOI] [PubMed] [Google Scholar]
- 10.Post PN, Siersema PD, Van Dekken H. Rising incidence of clinically evident Barrett's oesophagus in The Netherlands: a nation-wide registry of pathology reports. Scand J Gastroenterol. 2007;42(1):17–22. doi: 10.1080/00365520600815654. [DOI] [PubMed] [Google Scholar]
- 11.Coleman HG, Bhat S, Murray LJ, McManus D, Gavin AT, Johnston BT. Increasing incidence of Barrett's oesophagus: a population-based study. Eur J Epidemiol. 2011;26(9):739–45. doi: 10.1007/s10654-011-9596-z. [DOI] [PubMed] [Google Scholar]
- 12.van Soest EM, Dieleman JP, Siersema PD, Sturkenboom MC, Kuipers EJ. Increasing incidence of Barrett's oesophagus in the general population. Gut. 2005;54(8):1062–6. doi: 10.1136/gut.2004.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petrick JL, Nguyen T, Cook MB. Temporal trends of esophageal disorders by age in the Cerner Health Facts database. Ann Epidemiol. 2016;26(2):151–4 e4. doi: 10.1016/j.annepidem.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma P, Falk GW, Weston AP, Reker D, Johnston M, Sampliner RE. Dysplasia and cancer in a large multicenter cohort of patients with Barrett's esophagus. Clin Gastroenterol Hepatol. 2006;4(5):566–72. doi: 10.1016/j.cgh.2006.03.001. doi:S1542-3565(06)00227-8[pii]10.1016/j.cgh.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 15.den Hoed CM, van Blankenstein M, Dees J, Kuipers EJ. The minimal incubation period from the onset of Barrett's oesophagus to symptomatic adenocarcinoma. Br J Cancer. 2011;105(2):200–5. doi: 10.1038/bjc.2011.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engel LS, Chow WH, Vaughan TL, et al. Population attributable risks of esophageal and gastric cancers. J Natl Cancer Inst. 2003;95(18):1404–13. doi: 10.1093/jnci/djg047. [DOI] [PubMed] [Google Scholar]
- 17.El-Serag HB. Time trends of gastroesophageal reflux disease: a systematic review. Clin Gastroenterol Hepatol. 2007;5(1):17–26. doi: 10.1016/j.cgh.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 18.el-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer and reflux disease. Gut. 1998;43(3):327–33. doi: 10.1136/gut.43.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960-1994. Int J Obes Relat Metab Disord. 1998;22(1):39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 20.Kong CY, Nattinger KJ, Hayeck TJ, et al. The impact of obesity on the rise in esophageal adenocarcinoma incidence: estimates from a disease simulation model. Cancer Epidemiol Biomarkers Prev. 2011;20(11):2450–6. doi: 10.1158/1055-9965.EPI-11-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giovino GA. Epidemiology of tobacco use in the United States. Oncogene. 2002;21(48):7326–40. doi: 10.1038/sj.onc.1205808. [DOI] [PubMed] [Google Scholar]
- 22.Cook MB, Kamangar F, Whiteman DC, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst. 2010;102(17):1344–53. doi: 10.1093/jnci/djq289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 24.Lagergren K, Lindam A, Lagergren J. Dietary proportions of carbohydrates, fat, and protein and risk of oesophageal cancer by histological type. PLoS One. 2013;8(1):e54913. doi: 10.1371/journal.pone.0054913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70(1):3–21. doi: 10.1111/j.1753-4887.2011.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Austin GL, Ogden LG, Hill JO. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971-2006. Am J Clin Nutr. 2011;93(4):836–43. doi: 10.3945/ajcn.110.000141. [DOI] [PubMed] [Google Scholar]
- 27.Wright JD, Wang CY. NCHS Data Brief. 49. 2010. Trends in intake of energy and macronutrients in adults from 1999-2000 through 2007-2008; pp. 1–8. [PubMed] [Google Scholar]
- 28.Centers for Disease C, Prevention. Trends in intake of energy and macronutrients--United States, 1971-2000. MMWR Morb Mortal Wkly Rep. 2004;53(4):80–2. [PubMed] [Google Scholar]
- 29.Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12(5):371–87. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 30.Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res (Phila) 2008;1(5):329–38. doi: 10.1158/1940-6207.CAPR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis. 1993;168(1):219–21. doi: 10.1093/infdis/168.1.219. [DOI] [PubMed] [Google Scholar]
- 32.Rehnberg-Laiho L, Rautelin H, Koskela P, et al. Decreasing prevalence of helicobacter antibodies in Finland, with reference to the decreasing incidence of gastric cancer. Epidemiol Infect. 2001;126(1):37–42. [PMC free article] [PubMed] [Google Scholar]
- 33.Abreu MT, Peek RM., Jr Gastrointestinal Malignancy and the Microbiome. Gastroenterology. 146(6):1534–46.e3. doi: 10.1053/j.gastro.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13(11):800–12. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraher MH, O'Toole PW, Quigley EM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9(6):312–22. doi: 10.1038/nrgastro.2012.44. [DOI] [PubMed] [Google Scholar]
- 36.Smith JL, Bayles DO. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit Rev Microbiol. 2006;32(4):227–48. doi: 10.1080/10408410601023557. [DOI] [PubMed] [Google Scholar]
- 37.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lau WF, Wong J, Lam KH, Ong GB. Oesophageal microbial flora in carcinoma of the oesophagus. Aust N Z J Surg. 1981;51(1):52–5. doi: 10.1111/j.1445-2197.1981.tb05905.x. [DOI] [PubMed] [Google Scholar]
- 40.Finlay IG, Wright PA, Menzies T, McArdle CS. Microbial flora in carcinoma of oesophagus. Thorax. 1982;37(3):181–4. doi: 10.1136/thx.37.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mannell A, Plant M, Frolich J. The microflora of the oesophagus. Ann R Coll Surg Engl. 1983;65(3):152–4. [PMC free article] [PubMed] [Google Scholar]
- 42.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004;101(12):4250–5. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192(19):5002–17. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osias GL, Bromer MQ, Thomas RM, et al. Esophageal bacteria and Barrett's esophagus: a preliminary report. Dig Dis Sci. 2004;49(2):228–36. doi: 10.1023/b:ddas.0000017443.44802.4b. [DOI] [PubMed] [Google Scholar]
- 45.Macfarlane S, Furrie E, Macfarlane GT, Dillon JF. Microbial colonization of the upper gastrointestinal tract in patients with Barrett's esophagus. Clin Infect Dis. 2007;45(1):29–38. doi: 10.1086/518578. [DOI] [PubMed] [Google Scholar]
- 46.Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137(2):588–97. doi: 10.1053/j.gastro.2009.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu N, Ando T, Ishiguro K, et al. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett's esophagus. BMC Infect Dis. 2013;13:130. doi: 10.1186/1471-2334-13-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gall A, Fero J, McCoy C, et al. Bacterial Composition of the Human Upper Gastrointestinal Tract Microbiome Is Dynamic and Associated with Genomic Instability in a Barrett's Esophagus Cohort. PLoS One. 2015;10(6):e0129055. doi: 10.1371/journal.pone.0129055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narikiyo M, Tanabe C, Yamada Y, et al. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci. 2004;95(7):569–74. doi: 10.1111/j.1349-7006.2004.tb02488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blackett KL, Siddhi SS, Cleary S, et al. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett's and oesophageal carcinoma: association or causality? Aliment Pharmacol Ther. 2013;37(11):1084–92. doi: 10.1111/apt.12317. [DOI] [PubMed] [Google Scholar]
- 51.Clarke AT, Wirz AA, Seenan JP, Manning JJ, Gillen D, McColl KE. Paradox of gastric cardia: it becomes more acidic following meals while the rest of stomach becomes less acidic. Gut. 2009;58(7):904–9. doi: 10.1136/gut.2008.161927. [DOI] [PubMed] [Google Scholar]
- 52.McDonald SA, Lavery D, Wright NA, Jansen M. Barrett oesophagus: lessons on its origins from the lesion itself. Nat Rev Gastroenterol Hepatol. 2015;12(1):50–60. doi: 10.1038/nrgastro.2014.181. [DOI] [PubMed] [Google Scholar]
- 53.Quante M, Bhagat G, Abrams JA, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21(1):36–51. doi: 10.1016/j.ccr.2011.12.004S1535-6108(11)00474-0[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10(6):403–14. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rubenstein JH, Inadomi JM, Scheiman J, et al. Association between Helicobacter pylori and Barrett's esophagus, erosive esophagitis, and gastroesophageal reflux symptoms. Clin Gastroenterol Hepatol. 2014;12(2):239–45. doi: 10.1016/j.cgh.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103(3):732–7. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian Z, Yang Z, Gao J, Zhu L, Jiang R, Jiang Y. Lower esophageal microbiota species are affected by the eradication of infection using antibiotics. Exp Ther Med. 2015;9(3):685–92. doi: 10.3892/etm.2015.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3(7):e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li XX, Wong GL, To KF, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One. 2009;4(11):e7985. doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stearns JC, Lynch MD, Senadheera DB, et al. Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170. doi: 10.1038/srep00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, et al. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5(4):574–9. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2015 doi: 10.1136/gutjnl-2015-310861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freedberg DE, Toussaint NC, Chen SP, et al. Proton Pump Inhibitors Alter Specific Taxa in the Human Gastrointestinal Microbiome: A Crossover Trial. Gastroenterology. 2015 doi: 10.1053/j.gastro.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosen R, Amirault J, Liu H, et al. Changes in gastric and lung microflora with acid suppression: acid suppression and bacterial growth. JAMA Pediatr. 2014;168(10):932–7. doi: 10.1001/jamapediatrics.2014.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Albenberg LG, Wu GD. Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology. 2014;146(6):1564–72. doi: 10.1053/j.gastro.2014.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schulz MD, Atay C, Heringer J, et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514(7523):508–12. doi: 10.1038/nature13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Keefe SJ, Li JV, Lahti L, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun. 2015;6:6342. doi: 10.1038/ncomms7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysaccharide induces physical proximity between CD14 and toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-kappa B. J Immunol. 2000;165(7):3541–4. doi: 10.4049/jimmunol.165.7.3541. [DOI] [PubMed] [Google Scholar]
- 70.O'Riordan JM, Abdel-latif MM, Ravi N, et al. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol. 2005;100(6):1257–64. doi: 10.1111/j.1572-0241.2005.41338.x. [DOI] [PubMed] [Google Scholar]
- 71.Calatayud S, Garcia-Zaragoza E, Hernandez C, et al. Downregulation of nNOS and synthesis of PGs associated with endotoxin-induced delay in gastric emptying. Am J Physiol Gastrointest Liver Physiol. 2002;283(6):G1360–7. doi: 10.1152/ajpgi.00168.2002. [DOI] [PubMed] [Google Scholar]
- 72.Fan YP, Chakder S, Gao F, Rattan S. Inducible and neuronal nitric oxide synthase involvement in lipopolysaccharide-induced sphincteric dysfunction. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G32–42. doi: 10.1152/ajpgi.2001.280.1.G32. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki H, Iijima K, Scobie G, Fyfe V, McColl KE. Nitrate and nitrosative chemistry within Barrett's oesophagus during acid reflux. Gut. 2005;54(11):1527–35. doi: 10.1136/gut.2005.066043. [DOI] [PMC free article] [PubMed] [Google Scholar]