

Abstract
For decades, protein engineers have endeavored to reengineer existing proteins for novel applications. Overall, protein folds and gross functions can be readily transferred from one protein to another by transplanting large blocks of sequence (i.e., domain recombination). However, predictably fine-tuning function (e.g., by adjusting ligand affinity, specificity, catalysis, and/or allosteric regulation) remains a challenge. One approach has been to use the sequences of protein families to identify amino acid positions that change during the evolution of functional variation. The rationale is that these nonconserved positions could be mutated to predictably fine-tune function. Evolutionary approaches to protein design have had some success, but the engineered proteins seldom replicate the functional performances of natural proteins. This Biophysical Perspective reviews several complexities that have been revealed by evolutionary and experimental studies of protein function. These include 1) challenges in defining computational and biological thresholds that define important amino acids; 2) the co-occurrence of many different patterns of amino acid changes in evolutionary data; 3) difficulties in mapping the patterns of amino acid changes to discrete functional parameters; 4) the nonconventional mutational outcomes that occur for a particular group of functionally important, nonconserved positions; 5) epistasis (nonadditivity) among multiple mutations; and 6) the fact that a large fraction of a protein’s amino acids contribute to its overall function. To overcome these challenges, new goals are identified for future studies.
Main Text
Since the dawn of recombinant DNA technology, a significant effort has been made to engineer new protein functions. The holy grail of protein engineering is de novo rational design of novel sequences and functions. However, success in this area has been limited to small proteins (1, 2, 3). Thus, to engineer proteins with more complex functions, researchers have developed strategies to modify naturally evolved proteins. One strategy directly incorporates evolutionary information. Since sequence alignments can be used to reverse engineer functional changes that occurred during protein evolution (e.g., (4, 5)), the same information should be useful for forward engineering novel protein functions.
Several groups have made efforts to bridge the fields of evolutionary biology and protein chemistry. Harms and Thornton (5) described historical disparities that hobbled this effort, along with efforts to reconcile them. The relationships among evolution, biophysics, and structure were recently reviewed (6). In parallel, dozens of computer algorithms have been developed to analyze amino acid (a.a.) changes in protein sequence alignments, with the rationale that nonrandom patterns of change reflect evolutionary constraints at important positions (7, 8, 9, 10, 11, 12, 13, 14, 15, 16). One class of algorithms identifies positions for which a.a. changes correlate with phylogeny. A second class identifies positions that change together (pairwise or higher-order coevolution). A third class predicts the outcomes of individual a.a. substitutions, and often includes sequence alignments as input.
Despite their varied mathematical bases and desired outputs, many of these algorithms share common assumptions about a.a. substitutions. This Biophysical Perspective reviews studies that were designed to explicitly test these assumptions. The results illuminate several factors that currently confound predictive protein engineering.
Definitions and context
Despite the efforts that have been made to bridge disciplines, many communication gaps persist. Thus, the text below defines our use of the terms “conservation” and “neutrality” to provide a context for the subject of this Biophysical Perspective: a.a. positions that are not conserved and for which mutations are not neutral. Likewise, Fig. 1 provides a context for the type of mutational outcomes that are covered here.
Figure 1.

Simple hierarchy of the protein structure-function relationship. When a protein is mutated, bonds and/or motions change. Motion changes can alter many factors in this hierarchy (gray arrows). Bond changes (black arrows) can change the conformational ensemble, which includes subensembles of folded (i.e., functional), intermediate, and unfolded structures; redistribution among these species is detected as altered stability. Folded structures carry out function (dashed box). Functional parameters are only detected upon ligand binding, which also alters bonds and motions within the protein molecule. In some proteins, particularly intrinsically disordered proteins, stability is tightly coupled to functional parameters. In many of the studies reviewed here, this does not appear to be so. To see this figure in color, go online.
Amino acid conservation is identified by two orthogonal methods. First, for each column in a sequence alignment, the degree of conservation can be quantified by its sequence entropy, which accounts for the number and frequency of distinct a.a. side chains at each position (17). Some studies use all 20 a.a. to calculate sequence entropy, whereas others group a.a. by chemically similar side chains. Our studies used all 20 a.a. and a stringent value for sequence entropy to define conserved positions (7, 18, 19); this context is used in this Biophysical Perspective. Second, conservation can be identified via the use of a.a. substitution matrices (e.g., BLOSUM-62 (20)). These matrices are empirically determined from groups of related sequences, and different input sequences produce different matrix values.
Neutrality has alternative meanings in different fields. The term can refer to mutations that do not change the overall protein structure (21). Alternatively, neutrality can refer to a.a. variants that function like wild-type protein (as in this Biophysical Perspective). Kimura (22) discussed functional neutrality in 1968, when he hypothesized that most individual a.a. substitutions are neutral, else catastrophe would preclude protein evolution. More recently, some analyses implicitly expanded this idea to include neutral positions in sequence alignments, i.e., positions at which any a.a. variant functions like a wild-type protein.
Nonneutral mutations can have a wide range of outcomes (Fig. 1). Simplistically, they are divided into two levels: conserved and nonconserved. Conserved positions convey overall protein folds and gross functions, which can be fairly easily transferred by transplanting large blocks of sequence (i.e., domain recombination); within these blocks, mutagenesis of individual conserved positions is often catastrophic. Nonconserved positions can be mutated to fine-tune functional parameters without disrupting the overall structure or function. Detailed parameters include the binding affinity, ligand specificity, magnitude of the allosteric response, catalytic rate, catalytic mechanism, and stability. Setting aside stability as distinct, this Biophysical Perspective focuses primarily on our ability to predictably modify functional parameters of the folded ensemble (Fig. 1, dashed box) by changing a.a. at nonconserved positions.
Justification for this focus comes from comparing paralogs, which exhibit functional variation without a significant change in the overall protein fold. For example, the LacI and PurR transcription regulators have the same function of binding DNA under allosteric regulation by a small molecule. Superimposition of LacI (1efa (23)) and PurR (1wet (24)) monomer structures yields a Cα root mean-square deviation of 1.7 Å. However, these two proteins have evolved sequence differences that fine-tune this function for different biological purposes: they bind different DNA ligands and thereby regulate different operons, they bind different small molecules, and they have inverted allosteric responses (25).
Finally, explicit definitions are needed for the functional parameters in Fig. 1. For most protein functions, the primordial step is a binding event, quantified as the binding affinity. Two or more binding affinities are needed to define the allosteric response (the change in binding for one ligand in the presence of a second one; cooperativity is a subtype of allostery) and ligand specificity (the rank order of binding affinities for all ligands). For enzymes, the chemical rates for catalytic steps can be quantified, whereas the order and types of steps that comprise the catalytic mechanism are descriptive.
A brief history of evolution-guided protein redesign
Evolution-based protein redesign has been approached from two directions. The first uses evolutionary information to rationally identify positions that can be mutated to achieve a desired functional change. The second—directed evolution—relies on selection or screening of random changes to identify a desired function. These approaches have also been combined with structural and energetic calculations to yield the desired functional variation. (The extensive field of structure-based redesign is beyond the scope of this Biophysical Perspective.) The successes and shortcomings of the evolutionary approaches are briefly reviewed here.
Rationalization
As soon as the first homologs were identified, their sequences were compared to predict which a.a. changes convey functional variation. The thought was that a few key a.a. could then be exchanged between homologs to exchange their functions. However, exchange experiments have had modest success: designed proteins showed some transfer of the desired function, but performance seldom (if ever) reached that of natural proteins.
Exchange experiments produced complicated results even between closely related proteins. For example, in an early study by Park and Plapp (26), various combinations of a.a were exchanged between two isozymes of alcohol dehydrogenase that differed by 10 a.a. widely distributed over the protein structure. The results provided an early example that changes that do not occur directly in the active site can alter function. Further, many of the intermediate alcohol dehydrogenase variants (those with a subset of the 10 possible changes) had worse function than either of the natural proteins. In the years since that early work, similar studies using a wide range of proteins have suggested that these two results are rules rather than exceptions. Indeed, the fact that long-range effects are difficult to identify from structural comparisons provides a strong motivation for using evolutionary information to guide protein redesign.
For proteins with a large number of a.a. differences, more complicated sequence comparisons have been devised to identify key a.a. for exchange. Yin and Kirsch (27) used Venn diagrams to predict five positions that differentiate malate dehydrogenase and lactate dehydrogenase (LDH). When the five LDH positions were exchanged into malate dehydrogenase, the desired substrate specificity improved by nearly nine orders of magnitude. However, the catalytic efficiency was still three orders of magnitude below that of natural LDH proteins (27). In another example, Rodriguez et al. (28) used evolutionary trace analysis to identify key positions that differ among G-protein-coupled receptors with alternative specificities for dopamine or serotonin. Individually swapping high-scoring positions conveyed enhanced serotonin binding to the dopamine receptor, whereas swapping low-scoring positions did not. However, when the set of substitutions were combined into one construct, the resulting protein could not be expressed. Thus, the high-scoring positions did not capture all of the key differences between the two proteins, and a.a. contributions to function were not cleanly parsed from those to stability.
Directed evolution
Given the partial success of rationalization, the strategy of directed evolution was developed. The rationale is that when an organism is subjected to the appropriate environmental stress, it will generate mutations that convey the desired functional change (e.g., (29)). A variation of this approach is to build a library of protein mutants and screen or select for the desired function (e.g., (30, 31)). Both approaches generate proteins with functional efficiencies similar to those of natural proteins, but they are limited by the number of mutants that can be sampled: a library of 1010 variants only samples complete a.a. diversity at a maximum of seven to eight positions. Further, some protein functions are not easily adapted for high-throughput screening or the biological fitness assays that are needed for directed evolution.
Several strategies have been developed to enhance the success of directed evolution. For example, structural comparisons were used to exclude mutations that were predicted to disrupt structure (32). Other studies iterated between rationalization (sequence or structure based) and selection/screening (e.g., (33)). For example, a heroic effort was recently made to redesign LacI so that DNA binding could be regulated by four nonnatural inducers (34). However, this required the combined efforts of several large labs and the generation and characterization of several thousand variants. Many of the best-performing variants required 100 mM ligand to elicit a strong allosteric response, whereas wild-type LacI had a strong response at 0.1 mM isopropyl β-D-1-thiogalactopyranoside and the strongest response at 10 mM. Thus, even a combination of many strategies requires expensive resources and person hours.
Factors that confound the rational design of functional variation
The limitations of directed evolution and library screening justify continued efforts to improve rational protein design. One avenue is to examine the common assumptions that underlie computational analyses of protein families. The results described below come from many types of experiments and computations, but together they identify current limitations and specify new directions that must be explored before we can fully understand the protein sequence-structure-function relationship.
Data thresholding greatly influences results
One of the biggest hurdles in our studies, both computational and experimental, has been to identify thresholds that discriminate between significant and insignificant for each type of results.
First, to use evolutionary information to guide engineering, one must choose a sequence identity threshold to define the protein family. A common threshold has been ∼40%, although dozens of studies have used the LacI/GalR paralogs with a threshold of ∼15% (19). However, depending on the threshold, various sequence analyses generate entirely different results. For example, some positions that are nonconserved in the full LacI/GalR family (15% threshold) are conserved in the subfamilies (40% threshold). The recognition of different thresholds allows nested analyses that identify family- and subfamily-specific positions, thereby identifying a greater number of important positions (18, 28).
Second, thresholding is necessary to interpret output scores from computer algorithms. For example, algorithms that detect nonrandom patterns of a.a. changes calculate a score for every position (or pair of positions), and top scores presumably indicate the most important a.a. positions. However, we have never seen a natural break in the scores that delineates the difference between important and nonimportant (Fig. 2). A conservative solution to this problem is to take the top percent of scores or to set a Z-score threshold, but this carries the risk of truncating important results. More information can be gleaned by assessing the results as a function of the threshold cutoff. For example, in a comparison of two sets of scores, Jaccard analyses can quantify the similarities across all possible thresholds (18). A second approach is to defer thresholding until the final step in the analysis, which prevents the loss of top positions that only emerge from downstream calculations (7).
Figure 2.

Computational scores do not contain obvious thresholds. The histogram shows the distribution of pairwise coevolution scores determined for the aldolase family (7) using the McBASC algorithm (11). A few scores are excerpted for the bar centered at 0.8 and the score at Z = 4 is highlighted. No natural break occurs around this or any other potential threshold. To see this figure in color, go online.
Ideally, the choice of a computational threshold should be guided by biology: a significant sequence change should cause a functional change large enough to alter the host organism. However, and contrary to many expectations, this can be surprisingly small. For example, <2-fold differences in the Km for the tetracycline resistance protein proved adaptive at clinically relevant drug concentrations (35). In humans, various normal phenotypes have been predicted to arise through combinations of weakly nonneutral protein variants (36).
Further, biological thresholds can be difficult to approximate in the lab because in real life, various conditions impose different thresholds (37). For example, de Vos et al. (38) showed that the same mutation can have different outcomes in alternative environments, subsequently leading to different trajectories of evolutionary change. Steinberg and Ostermeier (39) showed that transient environmental changes early in protein evolution can allow a temporary tolerance to deleterious mutations, which in turn allows subsequent mutations that become beneficial in the final environment. Rockah-Shmuel et al. (40) showed that neutral substitutions in DNA methyl-transferase are actually deleterious under some conditions.
Indeed, the influence of experimental thresholds and assay resolution has perhaps been underappreciated in computational studies. For example, many researchers do not realize that in the large LacI mutational data set (41), wild-type variants have a repression range of ∼40-fold. The influence of biological thresholds is especially important to consider with regard to the newly popular approach of deep mutational scanning, which uses biological competition and next-generation sequencing to infer functional changes from massively mutated libraries (42). The influences of threshold on such experiments were recently summarized (43).
Based on these observations, the ideal experiments for benchmarking algorithms would directly report protein function, with minimal contributions from other cellular or environmental processes. One could then use the resulting data in a variety of settings after applying the relevant biological threshold(s). Computer algorithms should strive to predict mutation outcomes at this same level of resolution. If large sets of mutational variants are assessed with in vivo functional assays, they should be benchmarked to in vitro studies. For example, LacI/GalR studies monitored in vivo repression under conditions in which repression was highly correlated with the Kd for DNA binding (44). In another example, Firnberg et al. (43) performed deep mutational scanning in 13 different environments to avoid thresholds that would have truncated the functional results.
The sequences of protein families show multiple patterns of a.a. change
When using evolutionary information to understand protein function, one assumes that a.a. changes at important positions will be constrained (nonrandom) during evolution. Many algorithms have been developed to identify nonrandom patterns, including (with representative citations) overall conservation/sequence entropy (8), a.a. changes that mirror the branching of phylogenetic trees (9, 45), combinations of conservation and phylogeny (10), pairwise and higher-order coevolving positions (11, 12, 13, 46), and coevolutionary network centrality (7).
These patterns are often treated as alternative and competing options, with the supposition that one pattern predicts important positions better than the others do. Alternatively, each pattern may contain distinct information, with the combination of multiple patterns giving rise to the emergent protein function. This idea is supported by experiment: when calculations were compared with experimental mutation outcomes in LacI, different algorithms identified different top-scoring positions, but all of the top-scoring positions were mutationally sensitive (7, 18, 19).
Furthermore, the distinct patterns may or may not comprise overlapping positions. For example, high-scoring positions for the patterns of network centrality and phylogeny were shown to have significant overlap in the LacI/GalR family (7). In contrast, for the aldolase family, positions in the two patterns were largely separate (7). To our knowledge, the ramifications of pattern overlap have only been considered for coevolution and phylogeny, with phylogeny treated as a contaminant in coevolutionary signals (47, 48). However, another possibility is that some coevolving positions track with phylogeny due to biologically meaningful constraints. Since both coevolution and phylogenetic patterns identify mutationally sensitive positions, discerning between these alternatives will be difficult.
Another complication is that a common structural scaffold can be plastic with respect to the locations of constrained positions. For example, conserved and coevolving positions in the full LacI/GalR family were compared with those in six LacI/GalR subfamilies (18). As expected, all subfamilies had a common set of constrained positions. In addition, each subfamily had uniquely constrained positions that were unconstrained in other subfamilies. Other protein families show similar plasticity in the locations of constrained positions (10), and thus plasticity must be widespread.
Individual evolutionary patterns may not correlate with specific functional parameters
Some studies have attempted to match specific patterns of a.a. change with specific functional parameters. For example, coevolutionary patterns were proposed to identify allosteric positions, but this approach is unlikely to generalize to a wide range of allosteric proteins (49). In another example, paralog sequences were analyzed for specificity determinants, and nonconserved positions that correlate with phylogeny were proposed to alter ligand specificity (45). However, ligand specificity is not the sole parameter that changes during paralog evolution. Enzyme evolution can change the catalytic mechanism and/or the final chemistry (50), the evolution of allosterically regulated proteins can alter the degree of the allosteric response (51, 52), and if host organisms thrive at different temperatures, mutations can be required to alter protein stability (53). Changes in affinity may also be present in the evolutionary record. For example, one paralog may have 10-fold stronger binding affinities for its ligands compared with another paralog. Indeed, for several LacI/GalR homologs, mutations at specificity-determinant positions most often altered the overall affinity, and changes in ligand specificity occurred for <25% of the variants (52, 54).
Nor should changes in binding affinities (or other parameters) be treated as contamination in the evolutionary record. Affinity changes in both paralog and ortholog evolution can allow organisms to adapt to new environments. Affinity changes are also needed in protein engineering: even in domain recombination, affinity modulation is often needed to compensate for the far- and mid-range influences of nonconserved positions on domain-domain or subunit-subunit interfaces (55, 56, 57).
Mutations at some nonconserved positions do not follow conventional substitution rules
In many computational studies, the results were validated according to the criterion that important nonconserved positions were mutationally sensitive. Further, mutations at these positions were expected to follow the conventional rules (Box 1) that arose from our collective laboratory experiences. However, most laboratory experiments have been biased to mutations at conserved positions (58), which are expected to abolish function (toggle off) unless the substitution is chemically similar (e.g., Ser for Thr; toggle on). Notably, nonconserved positions have seldom been the subject of experiment, and therefore we must consider whether the rules apply to these positions.
Box 1. Conventional Rules for Mutating Important Positions.
Most substitutions damage function or structure
- A few amino acids allow normal function
- Physicochemically similar
- Presence allowed during evolution
Substitution = same outcome in any homolog
In experiments to directly test these rules, multiple a.a. were substituted at nonconserved positions in LacI/GalR proteins (59). Instead of partitioning variants into the expected on/off pattern, the mutational outcomes ranged progressively over orders of magnitude (Fig. 3), which was described as a rheostatic pattern of change (59). This rheostat mutational behavior was also observed for variants of Bcl-2 family proteins (60), a PDZ domain (61), an E3 ubiquitin ligase (62), and pyruvate kinase (51). Thus, rheostat positions are probably widespread in the protein universe, and a variety of protein functions appear to have the potential for rheostatic changes.
Figure 3.
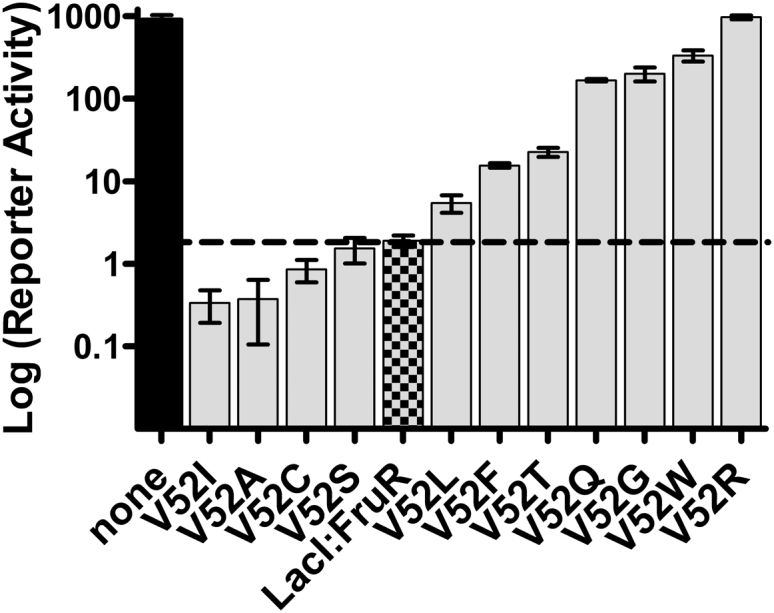
Example of an evolutionary rheostat. Data are shown for 12 variants of a synthetic LacI/GalR transcription repressor (LacI:FruR) (59). The checkered bar and dashed line represent the activity of the parent protein. The gray bars represent the activities for variants at position 52. Mutations with increased repression fall below the dashed line; those with diminished repression fall above the line. The black bar depicts reporter gene activity in the absence of repressor protein.
Notably, rheostat mutations in the LacI/GalR family do not follow any of the conventional rules for mutational outcomes (59). Nor is the rule-breaking limited to this family: in one study, evolutionary frequency did not correlate with mutation outcomes in Hsp90 (63), and in another, mutational outcomes in human growth hormone did not correlate with a.a. chemistry (64). The idea of conservative substitutions based on chemical similarities was challenged in a study that compared a.a. distributions on the structures of natural proteins (65).
Thus, the existence of rheostat positions has important ramifications for computational predictions of a.a. changes. As far as we can tell, most evolutionary algorithms and/or their interpretation explicitly or implicitly assume the rules in Box 1. As such, they will incorrectly handle mutations at rheostat positions. To improve predictions, new rules must be devised to explain mutational outcomes at rheostat positions. One challenge will be to distinguish nonconserved positions at which similar a.a. are interchangeable from rheostat positions that do not follow this rule. It also remains to be seen whether rheostat behaviors correlate with evolutionary patterns or with specific functional parameters. For the LacI/GalR proteins, rheostat positions might correlate with phylogenetic patterns (59). For these and other proteins, we expect that changes in affinity, catalytic rates, and stability can be incrementally modified by mutating various rheostat positions. The behavior would then propagate to any derived parameters, such as the magnitude of the allosteric response.
Epistasis among multiple a.a. positions
In every analysis that uses a protein sequence alignment, an explicit assumption is that the a.a. present in a column provide information about the importance of that position in individual homologs. The interpretative corollary is that when a mutational outcome is known for one homolog, similar outcomes are expected for all other homologs. As with the other rules listed in Box 1, this assumption was largely derived from mutations at conserved positions. However, for nonconserved positions, this need not be true.
To test this possibility, mutations must be created in multiple proteins at analogous nonconserved positions. Such a study was carried out using 10 members of the LacI/GalR family (55, 66, 67). The results showed that changes at nonconserved positions had protein-specific outcomes: commonly, a given a.a. substitution was detrimental in one protein, neutral in another, and enhancing in yet another. Disparate outcomes were common even among closely related homologs.
This behavior (i.e., the same variant causing different mutational outcomes in different sequence contexts) is the hallmark of epistasis. Epistasis has confounded many attempts to design proteins. For example, epistasis was observed during efforts to transplant protein-protein interfaces among PDZ domains (68). Epistasis is now recognized to be a dominant feature in the evolution of natural proteins such the influenza A surface proteins (69) and TEM-1 β-lactamase (70). Since a huge number of mutated proteins are needed to explore epistasis experimentally, this will be one of the most significant challenges to the field of rational protein design.
A high percentage of amino acids contribute to a protein’s function
As far as we are aware, attempts to identify a discrete subset of amino acids for functional exchange have never yielded a protein that functions as well as natural proteins. One reason may be that numerous a.a. positions contribute to overall function. This notion is based on the aggregate results of many different mutational experiments. One example comes from a screen for novel aminotransferase activity, which yielded 12 variants with desirable function (71). Together, these proteins had more than 40 substitutions, seven of which were common to all. When only these seven a.a. were mutated on the parent protein, the resulting protein had only 60% of the functionality of the selected proteins (71). Thus, the other 30-plus positions must contribute to the final function.
Another example comes from a study involving whole-protein mutagenesis of wild-type LacI, in which 12–13 a.a. were substituted at nearly every position (41). The resulting data set has been used in dozens of studies to verify that high scores from computational algorithms identify important positions. However, an examination of the full data set shows that at least 50% of all LacI positions contribute to function. Indeed, when the full data set was compared with three algorithms, mutationally sensitive positions were found to occur over the full range of scores (see Fig. 5 in (19)). Thus, no single evolutionary pattern has captured all of the relevant functional information for this family.
Future directions
The results described above have led us to consider what kinds of studies are needed in the future to advance protein engineering. An immediate and economical approach would be to glean more information from existing directed-evolution experiments by retrospectively comparing the mutations with analyses of sequence alignments. However, one must remember that such experiments were designed to generate “winners” and do not yield much information about “losers.”
Another approach would be to expand the number of experimental variants available for comparison with sequence analyses by characterizing multiple a.a. substitutions at each position studied. For example, as noted above, no correlation has yet been documented between specific patterns and specific functional parameters. A more fruitful approach could be to correlate an orthogonal classification of mutation outcomes (e.g., toggle, rheostat, and neutral behaviors) with patterns of evolutionary change. Toggle behavior strongly correlates with mutations at conserved positions. Rheostat behavior may correlate with positions that show phylogenetic patterns of change; this should be tested in other protein families. And although it is commonly assumed that positions that show unconstrained, random changes during evolution are functionally neutral, to my knowledge, this has never been tested.
Rheostat behavior, epistasis, and the fact that many a.a. positions contribute to protein function are likely to affect structure-based, de novo, and evolution-based engineering. This issue could be addressed by tying functional changes back to other areas shown in Fig. 1. To that end, the biophysical community has begun useful studies. Several have explored the apparent trade-off between function and protein stability (e.g., (72, 73)), and a compelling link was noted between changes in protein dynamics during the evolution of functional variation (74, 75). It will be particularly interesting to understand the biophysics that underlies mutational outcomes at evolutionary rheostat positions, and this simply requires that common tools be systematically applied to a series of a.a. at each rheostat position. To illuminate epistatic relationships, researchers will need to generate all possible (pairwise through n-wise) mutational combinations, which would be orders of magnitude larger than the number generated in studies completed to date. Deep mutational scanning (42) has been used to compare functions for large sets of protein variants, but the characterization strategies must be scaled up to yield more detailed functional and biophysical information.
Another area to consider is the different classes of protein structures (globular soluble, integral membrane, and intrinsically disordered). To date, most attempts to integrate protein design with evolutionary information have been carried out on soluble proteins, and only a few studies have focused on membrane proteins. However, these three classes have been subjected to fundamentally different structural constraints during evolution, which may give rise to different correlations between evolutionary patterns of change and mutational outcomes (76).
Finally, some protein structures may have evolved to evolve, that is, the underlying structural scaffold of a protein family can support a wide range of functional variation. For example, the >45 paralog groups of the LacI/GalR family (19) have a common structure that tolerates a large number of a.a. changes to effect wide functional variation. As evolvable scaffolds may be more useful for redesign, it is imperative to understand what features make one structure more amenable to functional variation than another one. Bloom et al. (21, 77) found that proteins with enhanced stability can accept a wider range of a.a. mutations. Tóth-Petróczy and Tawfik (78) proposed that highly evolvable proteins have a high fraction of functional positions in flexible regions. It will be interesting to compare efforts to redesign proteins that do and do not have these features. (This will almost certainly pose another gradient/threshold problem, with different protein families tolerating varied mutation loads.) At the same time, it may be harder to predict mutational outcomes in highly evolvable proteins than in proteins with stronger structural, functional, and evolutionary constraints.
Conclusions
The applications for which proteins are engineered often require complex, nuanced functions. Directed evolution and library screening have limitations that could be circumvented by rational protein design. Likewise, the widely used structure/energy calculations used to guide protein redesign (too extensive to review here) are limited by the need for 1) a protein structure and 2) user input about the targeted region, which is difficult to identify outside of binding sites. Evolution-guided redesign has the potential to circumvent these problems and to complement structural/energetic approaches.
The confounding factors described above suggest several reasons why attempts to guide rational protein engineering with evolutionary information have not been fully successful. Further, these factors impact more than protein engineering: they hamper the ability to predict mutation outcomes for genomic diagnoses, and they hamper the ability to assign functions to uncharacterized proteins.
Although these confounding factors are challenging, I remain hopeful that they can be overcome. Since directed evolution and selection from massive libraries are successful strategies, the correct mutational answers exist in the protein universe. Rational protein design is not limited by nature, only by our own understanding.
Acknowledgments
I thank Drs. Sarah Bondos (Texas A&M Health Science Center), Joff Silberg (Rice University), Aron Fenton and Dan Parente (University of Kansas Medical Center), James Horn (Northern Illinois University), and an anonymous reviewer for helpful discussions and critical reviews of the manuscript.
This study was supported by an internal Lied Basic Science Grant from the KUMC Research Institute, with support from a National Institutes of Health Clinical and Translational Science Award (UL1TR000001, formerly UL1RR033179), and private funds.
Editor: Brian Salzberg.
References
- 1.Joh N.H., Wang T., DeGrado W.F. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science. 2014;346:1520–1524. doi: 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith B.A., Hecht M.H. Novel proteins: from fold to function. Curr. Opin. Chem. Biol. 2011;15:421–426. doi: 10.1016/j.cbpa.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlman B., Dantas G., Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 4.Harms M.J., Eick G.N., Thornton J.W. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc. Natl. Acad. Sci. USA. 2013;110:11475–11480. doi: 10.1073/pnas.1303930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms M.J., Thornton J.W. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev. Genet. 2013;14:559–571. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liberles D.A., Teichmann S.A., Whelan S. The interface of protein structure, protein biophysics, and molecular evolution. Protein Sci. 2012;21:769–785. doi: 10.1002/pro.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parente D.J., Ray J.C.J., Swint-Kruse L. Amino acid positions subject to multiple coevolutionary constraints can be robustly identified by their eigenvector network centrality scores. Proteins. 2015;83:2293–2306. doi: 10.1002/prot.24948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau M., Mayrose I., Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mihalek I., Res I., Lichtarge O. Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics. 2006;22:1656–1657. doi: 10.1093/bioinformatics/btl157. [DOI] [PubMed] [Google Scholar]
- 10.Gu X., Vander Velden K. DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics. 2002;18:500–501. doi: 10.1093/bioinformatics/18.3.500. [DOI] [PubMed] [Google Scholar]
- 11.Dekker J.P., Fodor A., Yellen G. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics. 2004;20:1565–1572. doi: 10.1093/bioinformatics/bth128. [DOI] [PubMed] [Google Scholar]
- 12.Fodor A.A., Aldrich R.W. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins. 2004;56:211–221. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 13.Brown C.A., Brown K.S. Validation of coevolving residue algorithms via pipeline sensitivity analysis: ELSC and OMES and ZNMI, oh my! PLoS One. 2010;5:e10779. doi: 10.1371/journal.pone.0010779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty A., Chakrabarti S. A survey on prediction of specificity-determining sites in proteins. Brief. Bioinform. 2015;16:71–88. doi: 10.1093/bib/bbt092. [DOI] [PubMed] [Google Scholar]
- 15.Grimm D.G., Azencott C.A., Borgwardt K.M. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015;36:513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Juan D., Pazos F., Valencia A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 17.Valdar W.S. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 18.Parente D.J., Swint-Kruse L. Multiple co-evolutionary networks are supported by the common tertiary scaffold of the LacI/GalR proteins. PLoS One. 2013;8:e84398. doi: 10.1371/journal.pone.0084398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tungtur S., Parente D.J., Swint-Kruse L. Functionally important positions can comprise the majority of a protein’s architecture. Proteins. 2011;79:1589–1608. doi: 10.1002/prot.22985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bloom J.D., Silberg J.J., Arnold F.H. Thermodynamic prediction of protein neutrality. Proc. Natl. Acad. Sci. USA. 2005;102:606–611. doi: 10.1073/pnas.0406744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 23.Bell C.E., Lewis M. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- 24.Schumacher M.A., Glasfeld A., Brennan R.G. The X-ray structure of the PurR-guanine-purF operator complex reveals the contributions of complementary electrostatic surfaces and a water-mediated hydrogen bond to corepressor specificity and binding affinity. J. Biol. Chem. 1997;272:22648–22653. doi: 10.1074/jbc.272.36.22648. [DOI] [PubMed] [Google Scholar]
- 25.Swint-Kruse L., Matthews K.S. Allostery in the LacI/GalR family: variations on a theme. Curr. Opin. Microbiol. 2009;12:129–137. doi: 10.1016/j.mib.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park D.H., Plapp B.V. Interconversion of E and S isoenzymes of horse liver alcohol dehydrogenase. Several residues contribute indirectly to catalysis. J. Biol. Chem. 1992;267:5527–5533. [PubMed] [Google Scholar]
- 27.Yin Y., Kirsch J.F. Identification of functional paralog shift mutations: conversion of Escherichia coli malate dehydrogenase to a lactate dehydrogenase. Proc. Natl. Acad. Sci. USA. 2007;104:17353–17357. doi: 10.1073/pnas.0708265104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez G.J., Yao R., Wensel T.G. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc. Natl. Acad. Sci. USA. 2010;107:7787–7792. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloom J.D., Arnold F.H. In the light of directed evolution: pathways of adaptive protein evolution. Proc. Natl. Acad. Sci. USA. 2009;106(Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miersch S., Li Z., Sidhu S.S. Scalable high throughput selection from phage-displayed synthetic antibody libraries. J. Vis. Exp. 2015;17:51492. doi: 10.3791/51492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murtaugh M.L., Fanning S.W., Horn J.R. A combinatorial histidine scanning library approach to engineer highly pH-dependent protein switches. Protein Sci. 2011;20:1619–1631. doi: 10.1002/pro.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silberg J.J., Nguyen P.Q., Stevenson T. Computational design of chimeric protein libraries for directed evolution. Methods Mol. Biol. 2010;673:175–188. doi: 10.1007/978-1-60761-842-3_10. [DOI] [PubMed] [Google Scholar]
- 33.Goldsmith M., Tawfik D.S. Directed enzyme evolution: beyond the low-hanging fruit. Curr. Opin. Struct. Biol. 2012;22:406–412. doi: 10.1016/j.sbi.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Taylor N.D., Garruss A.S., Raman S. Engineering an allosteric transcription factor to respond to new ligands. Nat. Methods. 2016;13:177–183. doi: 10.1038/nmeth.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walkiewicz K., Benitez Cardenas A.S., Shamoo Y. Small changes in enzyme function can lead to surprisingly large fitness effects during adaptive evolution of antibiotic resistance. Proc. Natl. Acad. Sci. USA. 2012;109:21408–21413. doi: 10.1073/pnas.1209335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bromberg Y., Kahn P.C., Rost B. Neutral and weakly nonneutral sequence variants may define individuality. Proc. Natl. Acad. Sci. USA. 2013;110:14255–14260. doi: 10.1073/pnas.1216613110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soskine M., Tawfik D.S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 2010;11:572–582. doi: 10.1038/nrg2808. [DOI] [PubMed] [Google Scholar]
- 38.de Vos M.G.J., Poelwijk F.J., Tans S.J. Environmental dependence of genetic constraint. PLoS Genet. 2013;9:e1003580. doi: 10.1371/journal.pgen.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinberg B., Ostermeier M. Environmental changes bridge evolutionary valleys. Sci. Adv. 2016;2:e1500921. doi: 10.1126/sciadv.1500921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rockah-Shmuel L., Tóth-Petróczy Á., Tawfik D.S. Systematic mapping of protein mutational space by prolonged drift reveals the deleterious effects of seemingly neutral mutations. PLOS Comput. Biol. 2015;11:e1004421. doi: 10.1371/journal.pcbi.1004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suckow J., Markiewicz P., Müller-Hill B. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- 42.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firnberg E., Labonte J.W., Ostermeier M. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol. Biol. Evol. 2014;31:1581–1592. doi: 10.1093/molbev/msu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tungtur S., Skinner H., Beckett D. In vivo tests of thermodynamic models of transcription repressor function. Biophys. Chem. 2011;159:142–151. doi: 10.1016/j.bpc.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye K., Vriend G., IJzerman A.P. Tracing evolutionary pressure. Bioinformatics. 2008;24:908–915. doi: 10.1093/bioinformatics/btn057. [DOI] [PubMed] [Google Scholar]
- 46.Lee Y., Mick J., Beamer L.J. A coevolutionary residue network at the site of a functionally important conformational change in a phosphohexomutase enzyme family. PLoS One. 2012;7:e38114. doi: 10.1371/journal.pone.0038114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunn S.D., Wahl L.M., Gloor G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- 48.Kann M.G., Shoemaker B.A., Przytycka T.M. Correlated evolution of interacting proteins: looking behind the mirrortree. J. Mol. Biol. 2009;385:91–98. doi: 10.1016/j.jmb.2008.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livesay D.R., Kreth K.E., Fodor A.A. A critical evaluation of correlated mutation algorithms and coevolution within allosteric mechanisms. Methods Mol. Biol. 2012;796:385–398. doi: 10.1007/978-1-61779-334-9_21. [DOI] [PubMed] [Google Scholar]
- 50.Martínez Cuesta S., Rahman S.A., Thornton J.M. The classification and evolution of enzyme function. Biophys. J. 2015;109:1082–1086. doi: 10.1016/j.bpj.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishwar A., Tang Q., Fenton A.W. Distinguishing the interactions in the fructose 1,6-bisphosphate binding site of human liver pyruvate kinase that contribute to allostery. Biochemistry. 2015;54:1516–1524. doi: 10.1021/bi501426w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhan H., Taraban M., Swint-Kruse L. Subdividing repressor function: DNA binding affinity, selectivity, and allostery can be altered by amino acid substitution of nonconserved residues in a LacI/GalR homologue. Biochemistry. 2008;47:8058–8069. doi: 10.1021/bi800443k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peña M.I., Davlieva M., Shamoo Y. Evolutionary fates within a microbial population highlight an essential role for protein folding during natural selection. Mol. Syst. Biol. 2010;6:387. doi: 10.1038/msb.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhan H., Swint-Kruse L., Matthews K.S. Extrinsic interactions dominate helical propensity in coupled binding and folding of the lactose repressor protein hinge helix. Biochemistry. 2006;45:5896–5906. doi: 10.1021/bi052619p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meinhardt S., Manley M.W., Jr., Swint-Kruse L. Novel insights from hybrid LacI/GalR proteins: family-wide functional attributes and biologically significant variation in transcription repression. Nucleic Acids Res. 2012;40:11139–11154. doi: 10.1093/nar/gks806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Connaghan K.D., Miura M.T., Bain D.L. Analysis of a glucocorticoid-estrogen receptor chimera reveals that dimerization energetics are under ionic control. Biophys. Chem. 2013;172:8–17. doi: 10.1016/j.bpc.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perica T., Kondo Y., Teichmann S.A. Evolution of oligomeric state through allosteric pathways that mimic ligand binding. Science. 2014;346:1254346. doi: 10.1126/science.1254346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gray V.E., Kukurba K.R., Kumar S. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics. 2012;28:2093–2096. doi: 10.1093/bioinformatics/bts336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meinhardt S., Manley M.W., Jr., Swint-Kruse L. Rheostats and toggle switches for modulating protein function. PLoS One. 2013;8:e83502. doi: 10.1371/journal.pone.0083502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeBartolo J., Dutta S., Keating A.E. Predictive Bcl-2 family binding models rooted in experiment or structure. J. Mol. Biol. 2012;422:124–144. doi: 10.1016/j.jmb.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McLaughlin R.N., Jr., Poelwijk F.J., Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Starita L.M., Pruneda J.N., Klevit R.E. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc. Natl. Acad. Sci. USA. 2013;110:E1263–E1272. doi: 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hietpas R.T., Jensen J.D., Bolon D.N.A. Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci. USA. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pál G., Kouadio J.L., Sidhu S.S. Comprehensive and quantitative mapping of energy landscapes for protein-protein interactions by rapid combinatorial scanning. J. Biol. Chem. 2006;281:22378–22385. doi: 10.1074/jbc.M603826200. [DOI] [PubMed] [Google Scholar]
- 65.Jonson P.H., Petersen S.B. A critical view on conservative mutations. Protein Eng. 2001;14:397–402. doi: 10.1093/protein/14.6.397. [DOI] [PubMed] [Google Scholar]
- 66.Meinhardt S., Swint-Kruse L. Experimental identification of specificity determinants in the domain linker of a LacI/GalR protein: bioinformatics-based predictions generate true positives and false negatives. Proteins. 2008;73:941–957. doi: 10.1002/prot.22121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tungtur S., Egan S.M., Swint-Kruse L. Functional consequences of exchanging domains between LacI and PurR are mediated by the intervening linker sequence. Proteins. 2007;68:375–388. doi: 10.1002/prot.21412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melero C., Ollikainen N., Kortemme T. Quantification of the transferability of a designed protein specificity switch reveals extensive epistasis in molecular recognition. Proc. Natl. Acad. Sci. USA. 2014;111:15426–15431. doi: 10.1073/pnas.1410624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kryazhimskiy S., Dushoff J., Plotkin J.B. Prevalence of epistasis in the evolution of influenza A surface proteins. PLoS Genet. 2011;7:e1001301. doi: 10.1371/journal.pgen.1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dellus-Gur E., Elias M., Tawfik D.S. Negative epistasis and evolvability in TEM-1 β-lactamase—the thin line between an enzyme’s conformational freedom and disorder. J. Mol. Biol. 2015;427:2396–2409. doi: 10.1016/j.jmb.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothman S.C., Kirsch J.F. How does an enzyme evolved in vitro compare to naturally occurring homologs possessing the targeted function? Tyrosine aminotransferase from aspartate aminotransferase. J. Mol. Biol. 2003;327:593–608. doi: 10.1016/s0022-2836(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 72.Dellus-Gur E., Toth-Petroczy A., Tawfik D.S. What makes a protein fold amenable to functional innovation? Fold polarity and stability trade-offs. J. Mol. Biol. 2013;425:2609–2621. doi: 10.1016/j.jmb.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 73.Couñago R., Wilson C.J., Shamoo Y. An adaptive mutation in adenylate kinase that increases organismal fitness is linked to stability-activity trade-offs. Protein Eng. Des. Sel. 2008;21:19–27. doi: 10.1093/protein/gzm072. [DOI] [PubMed] [Google Scholar]
- 74.Bahar I., Cheng M.H., Zhang S. Structure-encoded global motions and their role in mediating protein-substrate interactions. Biophys. J. 2015;109:1101–1109. doi: 10.1016/j.bpj.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marsh J.A., Teichmann S.A. Parallel dynamics and evolution: protein conformational fluctuations and assembly reflect evolutionary changes in sequence and structure. BioEssays. 2014;36:209–218. doi: 10.1002/bies.201300134. [DOI] [PubMed] [Google Scholar]
- 76.Siltberg-Liberles J., Grahnen J.A., Liberles D.A. The evolution of protein structures and structural ensembles under functional constraint. Genes (Basel) 2011;2:748–762. doi: 10.3390/genes2040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bloom J.D., Labthavikul S.T., Arnold F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tóth-Petróczy A., Tawfik D.S. The robustness and innovability of protein folds. Curr. Opin. Struct. Biol. 2014;26:131–138. doi: 10.1016/j.sbi.2014.06.007. [DOI] [PubMed] [Google Scholar]