

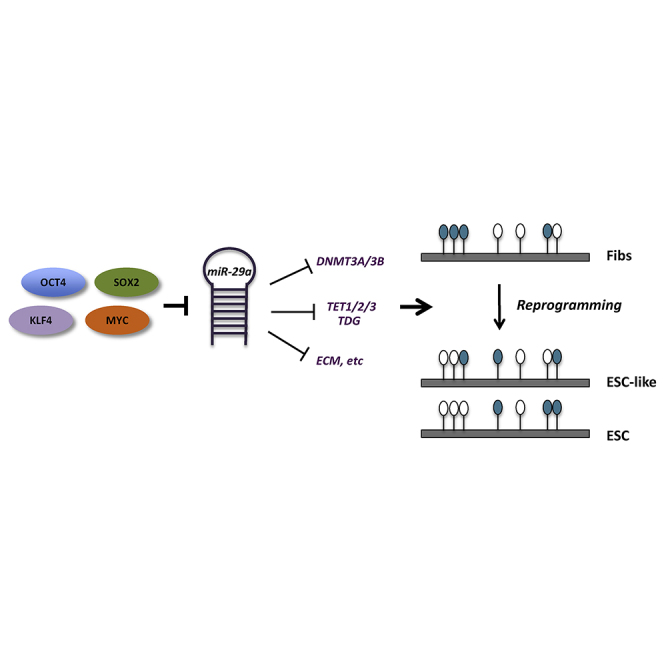
Summary
Reprogramming to pluripotency after overexpression of OCT4, SOX2, KLF4, and MYC is accompanied by global genomic and epigenomic changes. Histone modification and DNA methylation states in induced pluripotent stem cells (iPSCs) have been shown to be highly similar to embryonic stem cells (ESCs). However, epigenetic differences still exist between iPSCs and ESCs. In particular, aberrant DNA methylation states found in iPSCs are a major concern when using iPSCs in a clinical setting. Thus, it is critical to find factors that regulate DNA methylation states in reprogramming. Here, we found that the miR-29 family is an important epigenetic regulator during human somatic cell reprogramming. Our global DNA methylation and hydroxymethylation analysis shows that DNA demethylation is a major event mediated by miR-29a depletion during early reprogramming, and that iPSCs derived from miR-29a depletion are epigenetically closer to ESCs. Our findings uncover an important miRNA-based approach to generate clinically robust iPSCs.
Graphical Abstract
Highlights
-
•
Depletion of the miR-29a family improves reprogramming efficiency
-
•
DNA demethylation is a major change induced by miR-29a depletion
-
•
Methylation at the CpG island shore is highly regulated by the miR-29a family
-
•
iPSC derived by depletion of the miR-29a family is epigenetically close to ESCs
In this article, Park and colleagues provide robust DNA methylome change induced by change in expression of the miR-29a family. In somatic cells, miR-29a depletion highly induced DNA demethylation and improved reprogramming efficiency. iPSC derived in the presence of miR-29a depletion acquire an epigenetic state close to ESCs. The data provide an important approach to improve the quality of iPSCs.
Introduction
Overexpression of four transcription factors (OCT4, SOX2, KLF4, and MYC) reprograms differentiated cells to become induced pluripotent stem cells (iPSCs). The global epigenomic changes that accompany reprogramming include histone modification, DNA methylation, expression of non-coding RNAs, and reactivation of the inactive X chromosome (Kim et al., 2014, Papp and Plath, 2013). iPSCs maintain the genetic composition of donor cells, and thus have been proposed to model human diseases in vitro through differentiation into target cell types. In addition, iPSCs can provide autologous cells for cell replacement therapy (Wu and Hochedlinger, 2011). However, studies have shown that iPSCs contain localized aberrant epigenetic states compared with human embryonic stem cells (hESCs) despite their high similarity (Bock et al., 2011, Lister et al., 2011). Understanding the reprogramming mechanisms and developing novel reprogramming technologies to minimize the abnormality of iPSCs are critical for the future use of iPSCs.
Among the epigenetic aberrations of iPSCs, DNA methylation is of particular importance. Previous studies showed that unique de novo differentially methylated (DMR) or hydroxymethylated regions (hDMR) are present in iPSCs compared with hESCs (Lister et al., 2011, Wang et al., 2013). Furthermore, the retention of the epigenetic memory of donor cell types via cell-type-specific methylation affects the differentiation potential of iPSCs (Kim et al., 2011). There are three major enzymes that mediate DNA methylation. De novo DNA methyltransferases (DNMT3A and DNMT3B) are responsible for transferring a methyl moiety from S-adenosyl-methionine to cytosine to make 5-methylcytosine (5mC). DNMT1 together with hemi-methylated DNA-binding protein UHRF1 maintain 5-mC during cell-cycle progression (Jones, 2012). DNA demethylation, on the other hand, is either passive or indirect in mammalian cells. It has been shown to be mediated by enzymes recruited during base or nucleotide excision DNA repair responses, as well as by cytidine deaminases (Wu and Zhang, 2010). Ten-eleven translocation proteins (TET1, TET2, and TET3) belonging to the family of 2-oxoglutarate- and iron (II)-dependent dioxygenases were also identified as DNA demethylation proteins (Kriaucionis and Heintz, 2009, Tahiliani et al., 2009). TETs were shown to catalyze the oxidation of 5mC into 5-hydroxymethylcytosine (5hmC) (Kriaucionis and Heintz, 2009, Tahiliani et al., 2009). TETs further convert 5-hmC to formylcytosine (5fC) and carboxycytosine (5caC), which undergo base excision repair by thymine-DNA glycosylase (TDG) (Ito et al., 2011, Shen and Zhang, 2013). Whereas 5mC is enriched in promoter regions of silent genes, 5mC in the gene body is positively correlated with gene expression (Ball et al., 2009, Lister et al., 2009). In contrast, 5hmC in both the promoter and gene body is associated with promoting gene expression (Song et al., 2011).
MicroRNAs, or miRNAs, are a family of small ∼22 nt RNAs that regulate gene expression at the mRNA or protein level, and with functional implications in a wide range of biological processes (Bartel, 2004). miRNAs are extensively studied for their cell- and tissue-specific roles in cancer where they are significant contributors to epigenetic landscaping (Croce, 2009). The function of miRNAs was also explored in the context of somatic cell reprogramming. It was found that the miRNA 290–295 cluster is highly expressed in ESCs (Marson et al., 2008), and could enhance reprogramming efficiency in combination with Oct4, Sox2, and Klf4 (Judson et al., 2009, Nakagawa et al., 2008). It was also shown that miRNA cluster 302–367 (Anokye-Danso et al., 2011), or the cocktail miR-200c, miR-302, and miR-369 (Miyoshi et al., 2011) alone, could successfully reprogram both human and mouse cells to pluripotency, although efficiency is low (Lu et al., 2012). A diverse number of miRNA targeting processes such as mesenchymal-epithelial transition, apoptosis, and senescence, have been characterized and shown to modulate reprogramming in combination with the classical transcription factors (Bao et al., 2013). The miR-29 family, comprising miR-29a, miR-29b1, and miR-29c, is aberrantly expressed in various cancers, plays a role in extracellular matrix (ECM) production and fibrosis, and has also been shown to target DNA methylation enzymes Dnmt3a and Dnmt3b (Fabbri et al., 2007, Roderburg et al., 2011, Suh et al., 2012, Yang et al., 2013). More recently, with the help of our collaborators and others, we have shown that miR-29a also targets the TET protein family and TDG that convert 5mC to 5hmC and C (Cheng et al., 2013, Yang et al., 2013). Furthermore, miR-29 levels are high in senescent cells (Martinez et al., 2011) and repressed in the presence of Myc (Chang et al., 2008). Downregulation of miR-29a also showed some improvement of reprogramming efficiency in mouse fibroblasts, but its role in human reprogramming remains unexplored (Yang et al., 2011).
Given the significance of methylation/demethylation during reprogramming, we set out to investigate the function of miR-29a in regulating the iPSC methylome. Although miR-29a was shown to directly regulate both de novo DNA methyltransferases and demethylases, we found that depletion of miR-29a resulted in DNA demethylation in fibroblasts, suggesting that the major targets of miR-29a in somatic cells are DNA demethylases. Interestingly, and in support of our study, miR-29a targets TETs and TDG were recently shown to impair reprogramming of murine fibroblasts when downregulated, due to the block of miRNA-mediated mesenchymal-to-epithelial transition that is required for reprogramming (Hu et al., 2014). More importantly, iPSC lines derived from miR-29a depletion partly overcome the aberrant DNA methylation status observed in control-derived iPSC and resemble hESCs in their methylome. Our study facilitates the understanding of the role of small RNAs in modulating the iPSC epigenome.
Results
The miR-29 Family Regulates Proteins Involved in DNA Methylation
Because our previous works and others have shown that iPSCs undergo global DNA methylation/hydroxymethylation changes, we reasoned that miR-29 plays critical roles in regulating epigenetic changes during reprogramming (Doi et al., 2009, Lister et al., 2011). First, we confirmed the in silico targeting of 3′ UTRs of DNMTs, TETs, and TDG by miR-29 (Figure S1). Previous studies found that a large number of miRNAs show differences in expression between pluripotent and differentiated cells (Morin et al., 2008, Stadler et al., 2010). We found that the miR-29 family is highly expressed in the human primary fibroblast line Detroit 551 (D551), but low in human pluripotent cells, including ESCs (H1, H9), iPSCs (BJ-iPSC, PGP1-iPSCs, and D551-iPSCs), and human embryonic carcinoma cells (NCCIT) (Figure 1A). Most of its known targets (DNMT3A/3B and TET1/3) involved in DNA methylation show an inverse expression pattern to that of miR-29 expression: low in fibroblasts (D551, MRC-5) and high in pluripotent cells (Figure S2A). The luciferase reporter containing the 3′ UTR of TET1 further confirmed the direct repression by miR-29a (Figure S2B). When the reporter was used for unbiased screening to identify miRNAs directly regulating TET1, we found miR-29a and miR-29b were among the miRNAs that most strongly reduced the activity of the reporter (Table S1). When examining cells undergoing reprogramming, miR-29 expression decreased while its targets (TET1/3, DNMT3A/B, TDG) increased as the cells became pluripotent (Figures 1B and S2C). Furthermore, treating fibroblasts with miR-29a inhibitor increased, while miR-29a mimic decreased total genomic levels of 5hmC, 5fC, and 5caC (Figure S2D). Although Guo et al. (2013) reported that miR-29 family expression increased during reprogramming and that miR-29b enhances iPSCs generation in mice, our data showed reduction of miR-29 family expression in humans. In addition, we found that the expression level of miR-29b is significantly lower than the other miR-29 family in fibroblasts (Figure S2E), indicating the species difference of miRNA-mediated iPSC reprogramming. These data suggest that predicted and known targets of miR-29a may be tightly regulated by this miRNA in pluripotency, reprogramming, and DNA methylation. We employed RNA sequencing (RNA-seq) to examine the global gene expression changes in cells depleted of miR-29. D551 fibroblasts were transfected with miR-29a antagomir or infected with miR-29 sponge, and subsequently used for processing and analysis (Figure S2F). miR-29 sponge and miR-29a antagomir similarly affected global gene expression (Figure 1C). Genes with predicted miR-29a target sequences at 3′ UTR were significantly upregulated (Table S2, p = 4.01 × 10−33 by hypergeometric test). Gene ontology (GO) analysis showed that ECM genes are highly induced after miR-29a depletion (Figures 1C and 1D). The predicted and tested targets of miR-29a, including DNMT3A/B, TETs, and TDG, showed upregulation upon miR-29a depletion, whereas DNMT1 level did not change significantly (Figures 1E and 1F). The expression of pluripotency genes OCT4, SOX2, PRDM14, LIN28A, NANOG, and REX1 were not induced, suggesting no effect in the core pluripotency network by miR-29a depletion (Figures S2G and S2H). However, KLF4 showed a slight increase after miR-29a depletion, with increasing levels of 5hmC and 5mC in the promoter and gene body, respectively (Figure S2I). Because miRNA regulates protein expression via mRNA degradation or translation arrest, we performed proteomics analysis to determine regulation by miR-29a at the protein level. We confirmed that the overall global protein expression is well correlated with the mRNA expression change upon miR-29a depletion (Figure 1G and Table S3). Furthermore, mRNA and protein expression changes increase as the number of target sites on the 3′ UTR of target genes increases for several seed matches, suggesting that the observed gene expression changes come from the direct regulation by miR-29a (Figure S2J). Proteomics data identified the DNA methylation regulator TDG as an upregulated protein after miR-29a depletion (Figure 1G). Protein levels of other DNA methylation regulators appear to be under the detection limit of this proteomics assay. Taken together, these data suggest that miR-29a regulates the expression of genes involved in DNA methylation regulation.
Figure 1.

Functional Regulation of DNA Methylation Proteins by the miR-29 Family
(A) qPCR reveals that miR-29 family members are not expressed in pluripotent stem cells (ESCs H1, H9; iPSCs BJ1, PGP1, D551; embryonal carcinoma cell line NCCIT) but highly expressed in fibroblasts (D551) (n = 3, independent experiments).
(B) The miR-29a family shows a decrease of expression during three- (OSK) and four-factor (OSKM) reprogramming. w1-w4 represent samples collected for week 1 to week 4 after infection of reprogramming factors (n = 3, independent experiments).
(C) Overexpression of miR-29a antagomir (29ai) and miR-29 sponge (29a sp) increases expression of 141 genes and decrease expression of 100 genes (total RNA collected 3 days post-infection/transfection) (>1.5-fold change in both 29ai and 29a sp). Orange and blue bars represent the false discovery rate (FDR) of GO terms in miR-29a-depleted and control cells, respectively. Dashed line represents the 0.05 FDR cutoff.
(D and E) Genes for (D) ECM proteins and (E) de novo DNA methylation and demethylation are among the top genes showing upregulation in miR-29a-depleted cells. Ratios of log2-scaled expression values of miR-29a knockdown over control cells are shown by colors from blue to red. + represents miR-29a targets predicted by TargetScan.
(F) Inhibition of miR-29 by a miR-29 sponge construct induces TET1/2/3, DNMT3A, and -3B, but not DNMT1, as validated by qRT-PCR (n = 3, independent experiments).
(G) Comparative analysis of protein and RNA expression. The X and Y axes represent log2(29ai/Ctrli) of protein and RNA expression, respectively. Genes showing differential expression between protein and RNA for ECM proteins (blue) and TDG (red) are shown.
∗p < 0.05 by one-sided t test. Error bars represent the SD.
Depletion of the miR-29 Family Coordinates DNA Methylation and Demethylation
To investigate the effect of miR-29a depletion on DNA methylation regulation, we performed global DNA methylome and hydroxymethylome using methylated and hydroxylmethylated DNA immunoprecipitation sequencing ((h)MeDIP-seq) in D551 cells transfected with miR-29a antagomir. Overall, fibroblasts showed a limited number of 5hmC peaks, in contrast to the large number of 5hmC peaks in human ESCs (Figure 2A). In addition, only a small number of genes have overlapping hDMRs between miR-29a-depleted fibroblasts and ESCs (Figure S3A). Surprisingly, we found that miR-29a depletion significantly increased the formation of 5hmC in SOX2 binding sites but not in other reprogramming factor binding sites (Figure S3B) (Soufi et al., 2012). Chromatin immunoprecipitation (ChIP)-qPCR analysis revealed that the increasing level of SOX2 binding with 5hmC is caused by miR-29a depletion (Figure S3C). These data suggest that changes of the methylation status in SOX2-binding sites upon miR-29a depletion may regulate the binding strength of SOX2 to its targets during early reprogramming.
Figure 2.

DNA Methylation Changes in Fibroblasts after miR-29a Depletion
(A) The number of hyper- and hypo-DMRs in D551 fibroblasts depleted of miR-29a (antagomir-mediated, samples collected for MeDIP and hMeDIP 3 days post-transfection) and human ESCs.
(B) Venn diagrams show overlap of hyper- and hypo-DMRs of 5hmC between miR-29a-depleted cells and human ESCs.
(C) The distribution of hyper- and hypo-DMRs of 5hmC and 5mC around the gene body and CpGI in miR-29a-depleted cells and human ESCs.
(D) GO analysis of 5mC- and 5hmC-enriched regions specific to control and miR-29ai. Venn diagrams show the number of genes regulated by 5hmC and 5mC in miR-29a depleted and control cells. We did not detect any significant GO terms in genes regulated by 5hmC.
(E) NANOG undergoes demethylation and CDH2 undergoes methylation in miR-29a-depleted cells. These loci become similar to those in human ESCs.
5mC displayed a more dramatic change after miR-29a depletion. Whereas around 1,000 regions of hyper-differentially methylated regions (hyper-DMRs) were detected, over 7,000 regions of hypo-differentially methylated regions (hypo-DMRs) were observed in miR-29a-depleted cells (Figure 2A). Interestingly, 92.8% (6,565) of hypo-DMRs in miR-29a-depleted cells overlapped with those in human ESCs and are enriched in developmental genes (Figure S3D), suggesting that miR-29a depletion modulates the fibroblast methylation pattern primarily via DNA demethylation (Figure 2B). Changes in gene body methylation were predominant in hyper- and hypo-DMRs for both 5mC and 5hmC (Figure 2C). Through GO analysis for the DMRs between control and miR-29a-depleted cells, we found that genes involved in histone modification and development of several lineages have lower 5mC levels in miR-29a-depleted cells compared with controls, reflecting epigenetic dynamics associated with miR-29a modulation (Figure 2D). Although promoter methylation marks gene suppression, gene body methylation is highly correlated with gene activation (Ball et al., 2009, Lister et al., 2009). Induction of hypo-methylation in the gene body of developmental genes by miR-29a depletion suggests that miR-29a depletion represses the developmental genes and thus facilitates the cell fate change. We also found slightly higher 5hmC levels in miR-29a-depleted loci compared with controls, but we could not identify significant GO categories in the loci associated with these 5hmC gains. Interestingly, when we compared the methylation profiles of control versus miR-29a-depleted fibroblasts, many loci resemble the methylation tracks found in the H1 human ESC line (Figure 2E). A short 3-day depletion of miR-29a appears to be sufficient to initiate more demethylation in the promoter of the critical pluripotency marker NANOG. Bisulfite sequencing showed that DNA methylation in the NANOG locus was diminished by the miR-29a inhibitor (Figure S3E, 34%–0%). In contrast, Cadherin 2 (CDH2), a developmental gene not expressed in pluripotent stem cells, gains de novo methylation peaks in its promoter (Figure 2E) (Su et al., 2013). Our results suggest that one major role of miR-29a in differentiated cells is to maintain the cell fate by DNA methylation.
Next, we transduced fibroblasts individually with virus expressing each DNA methylation-related protein, miR-29 sponge, OSKM, or individual O, S, K, M, and carried out methylation analysis using Illumina's 450K BeadChip. We found a high similarity of methylated (higher ratio to control sample) and demethylated (low ratio to control sample) regions between miR-29 sponge cells and OSKM and TET1/2/3 transduced cells (Figures S3F and S3G). These data support the idea that the DNA demethylation upon miR-29a depletion has a strong correlation with the regulation by TET family proteins at an early time point in miR-29a-depleted cells, even though the overall pattern is most likely a result of synergistic regulation of all miR-29 targets.
Depletion of the miR-29 Family Contributes to ESC-Specific Transcriptome and Epigenetic Profile in iPSCs
Because depletion of the miR-29a family contributes to the demethylation of fibroblast-specific DNA marks (Figures 2A and 2B), and a previous report showed that reprogramming of murine somatic cells is improved via miR-29a depletion (Yang et al., 2011), we tested whether depletion of miR-29a affects the efficiency of reprogramming in human somatic cells, and whether it influences the epigenetic states in iPSCs. We transfected human fibroblast cells with miR-29a antagomir or mimic, or infected them with retrovirus expressing the miR-29 sponge, and subsequently initiated reprogramming. Suppression of miR-29a by either antagomir or sponge increased reprogramming efficiency, whereas overexpression of miR-29a mimic reduced it (Figure 3A), as assessed by quantifying alkaline-phosphatase-stained colonies. We further scored the reprogramming efficiency by staining colonies with pluripotency cell surface marker TRA-160 (Figure S4A). The overexpression of TET1 and DNMT3B also significantly increased reprogramming efficiency (Figures S4B–S4D), suggesting that positive regulation of reprogramming by depletion of miR-29a may be due to the upregulation of these of DNA methylation-related proteins. We found that DNMT3B and TET1 display only demethylation (Figure S3G), whereas TET2/3 and TDG introduce both methylation and demethylation in developmental genes. Since it was previously reported that miR-29a suppresses p53 (Yang et al., 2011), we also tested whether the reprogramming effect by miR-29a involves the p53 pathway. To this end, we reprogrammed human fibroblasts in the presence of miR-29a mimic alone, p53 shRNA alone, or miR-29a mimic + p53 shRNA. We found that reprogramming increases when p53 is depleted, decreases when miR-29a is overexpressed, and does not change in presence of both p53 depletion and miR-29a mimic (Figure S4E). This suggests that either p53 is upstream of miR-29a and its depletion is canceling the miRNA effect, or the pathways are separate and their effects annul each other.
Figure 3.

Regulation of Reprogramming by miR-29a Inhibition
(A) Inhibition of miR-29a using antagomir increases the reprogramming efficiency of human fibroblasts (D551) (n = 6, ∗∗∗p < 0.003, independent experiments), overexpression using miR-29a mimic decreases reprogramming (n = 6, ∗∗∗p < 0.005), and sponge-dependent miR-29 inhibition (retrovirus multiplicity of infection = 2.5) increases reprogramming efficiency (n = 3, ∗p < 0.02, independent experiments). Left: quantification. Right: alkaline phosphatase (AP) staining of two representative wells. Reprogramming with OSKM and miR-29a depletion or overexpression.
(B) Principal component analysis shows the similarity of ESCs (H1 and H9) with miR-29a-depleted D551 fibroblasts and iPSCs, and FM-1 fetal-myoblast-derived iPSCs.
(C) Left: Differentially expressed genes separating control versus miR-29 knockdown cells irrespective of parental cell of origin (factor loading PC2 < −0.2 and PC3 > 0.2). Right: GO overrepresentation of upregulated genes in miR-29a-depleted iPSCs. The dashed red line denotes FDR = 0.05.
(D and E) Average reads per kilobase per million values across all control and miR-29a-depleted iPSCs for (D) genes high in miR-29a-depleted cells and (E) genes high in control iPSCs. ESC-specific genes FAM19A5, TMEM132D, and TCERG1L increased in miR-29-depleted iPSCs (miR29-D denotes miR-29 depletion) (n = 6, 8, 2 in Ctrl iPSC, miR29a-D iPSC, and ESC, respectively, independent experiments).
Error bars represent the SD.
Next, we asked whether miR-29a depletion plays a role in genetic and epigenetic regulation in reprogramming. We derived human iPSC clones from D551 or fetal myoblast line (FM-1) depleted of miR-29a through antagomir or sponge construct, and performed transcriptome analysis in these iPSC clones. 3D principal component analysis of over 14,000 genes showed closer localization of miR-29a-depleted iPSCs to the H1 and H9 ESC groups than the control counterparts (Figure 3B). Furthermore, we observed separate clustering of control and miR-29a-depleted iPSCs independently of parental cell origin (fibroblasts versus myoblasts) and depletion method (antagomir versus sponge) (Figure 3C). GO analysis of differentially expressed genes revealed ECM organization genes overrepresented in miR-29a-depleted iPSCs, as expected due to many miR-29a targets falling under this category (Figure 3C). Interestingly, neuronal lineage regulators and the WNT pathway are also enriched networks in miR-29a-depleted cells, suggesting both an increase in the pluripotent program pathways (WNT) and modulation of the differentiation potential (neuronal/glial) of the iPSCs by this miRNA, despite the non-neuronal cell of origin. Our group and others have compared gene expression changes between human ESCs and iPSCs and identified aberrantly expressed genes in iPSCs (Huang et al., 2014, Lister et al., 2011, Ruiz et al., 2012, Tanaka et al., 2013, Wang et al., 2013). We found that most iPSC lines derived under miR-29a-depleted conditions showed suppression of some iPSC-specific genes and recovery of several ESC-specific genes (Figure 3D). The most commonly identified ESC-specific genes TCERG1L, FAM19A5, and TMEM132D, and others like ZDHHC19 and HTR6, showed significant recovery in iPSC lines derived from miR-29a depletion (Figures 3D and S4F). Some iPSC-specific genes such as FBP2, CCR8, FAM82A1, MGC16121, and SLC22A2, but not others like ZNF454 and ZNF572, were suppressed in miR-29-depleted cells (Figures 3E and S4G). These data suggest that the major effect of miR-29a depletion is demethylation and leads to high similarity with ESC state in hypo-DMRs (Figures 2A and 2B).
In order to determine the methylation change in iPSC lines derived from miR-29a depletion, we performed global DNA methylation analysis through MeDIP in ESCs and iPSCs. Surprisingly, we found that iPSC lines with miR-29 depletion cluster with ESC lines for the transcription start sites (TSS), CpG shore, and gene body methylation patterns (Figure 4A). CpG island methylation showed no discernible pattern, with the majority of iPSCs clustering separately from ESCs. GO analysis for TSS and gene bodies revealed methylation enrichment in loci involved in metabolic and catabolic processes for ESCs and miR-29-depleted iPSCs compared with control lines (Figures 4B and 4C). Interestingly, at the CpGI shore level, we found depletion of methylation in loci involved in lineage specification in ESCs and miR-29-depleted iPSCs compared with control iPSCs (Figure 4D). Gene expression shows significant correlation with methylome changes in TSS, gene body, and CpGI shore, suggesting a functional relevance (Figure 4E). Our finding is noteworthy, as DMRs that are present particularly at CpGI shores were distinct between different somatic, cancer, and reprogrammed cell types, possibly acting as cell identity marks (Doi et al., 2009).
Figure 4.

miR-29a Depletion Promotes an ESC-Specific Epigenetic Profile in iPSCs
(A) Hierarchical clustering of global DNA methylation profiles (MeDIP). TSS, gene body, and CpGI shore in -29abc sponge D551 iPSCs are closer to those in ESCs compared with control sponge and non-treated iPSCs.
(B–D) Heatmaps show DMRs in (B) TSS, (C) gene bodies, and (D) CpGI shore. GO analyses to genes neighboring DMRs show that CpGI shores in developmental genes are hyper-methylated in Ctrl-sponge and non-treated iPSCs.
(E) DMRs between ESC/29a-iPSC and control iPSC show enrichment in gene expression. ∗Denotes significance p < 0.05 by one-sided t test. Pearson correlation coefficients between gene expression and DNA methylation level are TSS, 0.180 (p < 2.2 × 10−16); gene body, 0.173 (p < 2.2 × 10−16); CpGI, 0.020 (p = 0.038); and CpGI shore, 0.113 (p < 2.2 × 10−16).
Discussion
In this study, we set out to investigate the function of miRNAs that regulate the epigenetic landscape in somatic cells and thus may have a function in somatic cell reprogramming. We found that the miR-29 family is highly expressed in somatic cells and decreases in expression during somatic cell reprogramming (Figures 1A and 1B). Depletion of miR-29 in fibroblasts dramatically changed the DNA methylation status, suggesting that miR-29a is one of the major miRNAs that maintains the DNA methylation status in fibroblasts (Figure 2). Previous studies showed that DNMT3A and DNMT3B, as well as TET proteins, are targets of the miR-29a family (Cheng et al., 2013, Hu et al., 2015). Our data also demonstrated a strong inverse correlation of the expression of the miR-29a family and the expression of DNMTs and TETs. It is not yet clear which proteins are direct targets of miR-29a in maintaining the DNA methylation status in fibroblasts. However, the global hypo-methylation upon miR-29a depletion suggests that proteins involved in the DNA demethylation including TETs and TDGs may be strong candidate targets (Figures 2A and 2B).
Previous studies implicated the miR-29a family in mouse somatic cell reprogramming through different pathways (Guo et al., 2013, Yang et al., 2011). Yang et al. (2011) showed that suppression of miR-29a via Myc improves the reprogramming by increasing p85alpha and CDC42, thus suppressing the p53 pathway. Guo et al. (2013) demonstrated that Sox2 is a critical factor inducing miR-29b, and that ectopic overexpression of miR-29b improves the reprogramming efficiency by suppressing Dnmt3a and Dnmt3b. In reprogramming human somatic cells, we found that suppression of the miR-29a family increased reprogramming efficiency. We also found that overexpression of DNMT3B as well as TET1 improves reprogramming efficiency. These seemingly contrasting results may be due to the bidirectional global epigenetic changes accompanying reprogramming. Not only DNA demethylation but also de novo DNA methylation is found in reprogrammed iPSCs (Lister et al., 2011). In light of the bidirectional epigenetic regulation, the miR-29a seems to be a unique regulator that potentially suppresses both de novo DNA methyltransferases and demethylases, although miR-29a depletion seems to have a more pronounced effect on DNA demethylation in fibroblasts (Figures 2A and 2B).
miR-29a depletion could not replace MYC in a three- factor reprogramming attempt (data not shown), although it improves reprogramming moderately but significantly with four-factor reprogramming. MYC is a potent transcription factor inducing and repressing a large number of genes; suppression of miR-29a is one of many functions of MYC during reprogramming (Yang et al., 2011). iPSC lines, for which aberrant DNA methylation was reported, were those derived from four-factor reprogramming (Lister et al., 2011). Most likely, MYC expedites reprogramming independent of its role in suppressing miR-29a. Expression of DNA (de)methylation proteins via suppression of miR-29a then becomes supplementary to the function of MYC during reprogramming. When miR-29a targets are upregulated at later stages of reprogramming, most loci are already reprogrammed. Thus, the accessibility of TETs or DNMT3A/B to target loci becomes limited, and thus epigenetic reprogramming is limited in four-factorbased reprogramming.
miRNAs are known to regulate the expression of multiple targets via binding to 3′ UTR. Our finding that the miR-29 family regulates global DNA methylation and demethylation adds another role to the list of common regulatory roles of miRNAs. The miR-29 family was known to regulate ECM proteins in physiological responses, such as systemic sclerosis, hepatic fibrogenesis, and cardiac fibrosis (Hubmacher and Apte, 2013, Kriegel et al., 2012, Zhang et al., 2012). The ECM proteins are also important in pluripotent stem cells. Laminin promotes stem cell renewal of ESCs in feeder-free conditions (Xu et al., 2001), and a mixture of human collagen, fibronectin, and laminin has been used as animal-free culture conditions for ESCs (Ludwig et al., 2006). Our transcriptome analysis in miR-29a-depleted cells found changes in ECM proteins. Because the process of mesenchymal-epithelial transition and the dynamic changes in ECM proteins are highly coordinated during reprogramming, the miR-29 family seems likely to regulate both the extracellular and intracellular epigenetic responses during reprogramming.
iPSC lines derived by miR-29a depletion showed a similar DNA methylation status to that of ESCs (Figure 4). TSS, gene body, and CpGI shore revealed the clustering of miR-29a depleted iPSCs with ESCs, suggesting that miR-29a depletion has a global impact on DNA methylation changes. Gene expression showed a high correlation with DNA methylation status (Figure 4E). Among the genes that express low in iPSCs compared with ESCs are some subtelomericly located genes such as TCERG1L, TMEM132D, and FAM19A5 (Lister et al., 2011). We found that the expression these genes, particularly TCERG1L, is partially reactivated by suppressing miR-29a. Investigation of the functional relevance of miR-29a depletion on iPSCs, such as developmental potential or epigenetic memory (Kim et al., 2011), will be important in demonstrating the utility of regulating miR-29a to derive clinically useful iPSCs.
Taken together, we identified miR-29a as an important epigenetic regulator for somatic cells. The acquisition of proper DNA methylation and hydroxymethylation states in iPSCs is not optimal with current reprogramming methods. We provide evidence that DNA methylation undergoes rapid changes upon miR-29a depletion during progression to pluripotency (Figure S4H). We believe that the coordinated regulation of multiple DNA methylation proteins by a miRNA family is a tool that will perfect the epigenetic reprogramming of iPSCs in future clinical utilities.
Experimental Procedures
Cell Culture, Reprogramming, and Modulation of miR-29 Expression
Normal primary fibroblast D551 were maintained in DMEM high glucose (Gibco) supplemented with 10% fetal bovine serum and penicillin/streptomycin. Human ESCs and iPSCs were cultured on irradiated murine embryonic feeder cells in medium containing DMEM/F12, 20% knockout serum replacement, and 4 ng/ml basic fibroblast growth factor. Reprogramming was carried out using the four human transcription factors OCT4, SOX2, KLF4, and MYC in murine stem cell virus (pMSCV) retrovirus backbone as previously described (Park et al., 2008). miR-29a mimic and antagomir were purchased from Dharmacon. miR-29 sponge was generated as described before (Cheng et al., 2013). Transfection was performed using Lipofectamine.
Gene Expression Analysis
Cells were lysed and RNA was isolated using an RNeasy Mini Kit (Qiagen). Reverse transcription was performed using iScript (Bio-Rad) according to the manufacturer's protocol with primer sets in Table S4A.
ChIP, RNA-Seq, hMeDIP-Seq, and Proteomics
Three days after infection with pMSCV retrovirus expression reprogramming factors, cells were harvested for ChIP, RNA-seq, hMeDIP-seq, bisulfite sequencing, or proteomics analysis. Total RNA was extracted using a miRNeasy Mini Kit (Qiagen), and used for qRT-PCR and RNA-seq. Genomic DNA was isolated from D551 fibroblasts transfected with control and miR-29a antagomir and processed for MeDIP and hMeDIP. RNA-seq, ChIP, hMeDIP-seq, dot blot, and proteomics were performed as described in Supplemental Experimental Procedures. Total reads and mapped reads are listed in Table S4B.
Author Contributions
E.H. designed and conducted most of the experiments. Y.T. performed all bioinformatics analysis. Y.T., J.S., K.-Y.K., T.Z., X.-L.Z., R.J., and Y.-W.J. performed experiments. J.C. and J.L. designed and performed the reporter assay. X.P., M.Z., and S.M.W. were involved in designing and generating high-throughput sequencing data. L.G. and C.Q. designed the hESC cultures. I.-H.P. conceived and coordinated the project. E.H., Y.T., and I.-H.P. wrote the manuscript.
Acknowledgments
We thank Dr Anjana Rao for wild-type and mutant TET1 constructs. We thank all the Park Lab members for helpful comments and discussion. I.-H.P. was supported in part by NIH (GM0099130-01, GM111667-01), CSCRF (12-SCB-YALE-11, 13-SCB-YALE-06), KRIBB/KRCF (NAP-09-3), and a CTSA grant UL1 RR025750 from the National Center for Advancing Translational Science (NCATS), a component of the NIH, and NIH roadmap for Medical Research. The contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. The sequencing service was conducted at Yale Stem Cell Center Genomics Core facility, which was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation. Computation time was provided by Yale University Biomedical High Performance Computing Center.
Published: June 30, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.05.014.
Accession Numbers
All data are deposited in the Gene Expression Omnibus (GEO) repository under accession number GEO: GSE81794.
Supplemental Information
References
- Anokye-Danso F., Trivedi C.M., Juhr D., Gupta M., Cui Z., Tian Y., Zhang Y., Yang W., Gruber P.J., Epstein J.A. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M.P., Li J.B., Gao Y., Lee J.H., LeProust E.M., Park I.H., Xie B., Daley G.Q., Church G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X., Zhu X., Liao B., Benda C., Zhuang Q., Pei D., Qin B., Esteban M.A. MicroRNAs in somatic cell reprogramming. Curr. Opin. Cell Biol. 2013;25:208–214. doi: 10.1016/j.ceb.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bock C., Kiskinis E., Verstappen G., Gu H., Boulting G., Smith Z.D., Ziller M., Croft G.F., Amoroso M.W., Oakley D.H. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T.C., Yu D., Lee Y.S., Wentzel E.A., Arking D.E., West K.M., Dang C.V., Thomas-Tikhonenko A., Mendell J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Guo S., Chen S., Mastriano S.J., Liu C., D'Alessio A.C., Hysolli E., Guo Y., Yao H., Megyola C.M. An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A., Park I.H., Wen B., Murakami P., Aryee M.J., Irizarry R., Herb B., Ladd-Acosta C., Rho J., Loewer S. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M., Garzon R., Cimmino A., Liu Z., Zanesi N., Callegari E., Liu S., Alder H., Costinean S., Fernandez-Cymering C. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Liu Q., Wang G., Zhu S., Gao L., Hong W., Chen Y., Wu M., Liu H., Jiang C. microRNA-29b is a novel mediator of Sox2 function in the regulation of somatic cell reprogramming. Cell Res. 2013;23:142–156. doi: 10.1038/cr.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Zhang L., Mao S.Q., Li Z., Chen J., Zhang R.R., Wu H.P., Gao J., Guo F., Liu W. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell. 2014;14:512–522. doi: 10.1016/j.stem.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Hu W., Dooley J., Chung S.S., Chandramohan D., Cimmino L., Mukherjee S., Mason C.E., de Strooper B., Liston A., Park C.Y. miR-29a maintains mouse hematopoietic stem cell self-renewal by regulating Dnmt3a. Blood. 2015;125:2206–2216. doi: 10.1182/blood-2014-06-585273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K., Shen Y., Xue Z., Bibikova M., April C., Liu Z., Cheng L., Nagy A., Pellegrini M., Fan J.B. A panel of CpG methylation sites distinguishes human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014;2:36–43. doi: 10.1016/j.stemcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher D., Apte S.S. The biology of the extracellular matrix: novel insights. Curr. Opin. Rheumatol. 2013;25:65–70. doi: 10.1097/BOR.0b013e32835b137b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Judson R.L., Babiarz J.E., Venere M., Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009;27:459–461. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Zhao R., Doi A., Ng K., Unternaehrer J., Cahan P., Huo H., Loh Y.H., Aryee M.J., Lensch M.W. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.Y., Hysolli E., Tanaka Y., Wang B., Jung Y.W., Pan X., Weissman S.M., Park I.H. X Chromosome of female cells shows dynamic changes in status during human somatic cell reprogramming. Stem Cell Rep. 2014;2:896–909. doi: 10.1016/j.stemcr.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S., Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel A.J., Liu Y., Fang Y., Ding X., Liang M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genomics. 2012;44:237–244. doi: 10.1152/physiolgenomics.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O'Malley R., Castanon R., Klugman S. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D., Davis M.P., Abreu-Goodger C., Wang W., Campos L.S., Siede J., Vigorito E., Skarnes W.C., Dunham I., Enright A.J. MiR-25 regulates Wwp2 and Fbxw7 and promotes reprogramming of mouse fibroblast cells to iPSCs. PLoS One. 2012;7:e40938. doi: 10.1371/journal.pone.0040938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T.E., Levenstein M.E., Jones J.M., Berggren W.T., Mitchen E.R., Frane J.L., Crandall L.J., Daigh C.A., Conard K.R., Piekarczyk M.S. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006;24:185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I., Cazalla D., Almstead L.L., Steitz J.A., DiMaio D. miR-29 and miR-30 regulate B-Myb expression during cellular senescence. Proc. Natl. Acad. Sci. USA. 2011;108:522–527. doi: 10.1073/pnas.1017346108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi N., Ishii H., Nagano H., Haraguchi N., Dewi D.L., Kano Y., Nishikawa S., Tanemura M., Mimori K., Tanaka F. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011;8:633–638. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Morin R.D., O'Connor M.D., Griffith M., Kuchenbauer F., Delaney A., Prabhu A.L., Zhao Y., McDonald H., Zeng T., Hirst M. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M., Koyanagi M., Tanabe K., Takahashi K., Ichisaka T., Aoi T., Okita K., Mochiduki Y., Takizawa N., Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Papp B., Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–1343. doi: 10.1016/j.cell.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Zhao R., West J.A., Yabuuchi A., Huo H., Ince T.A., Lerou P.H., Lensch M.W., Daley G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Roderburg C., Urban G.W., Bettermann K., Vucur M., Zimmermann H., Schmidt S., Janssen J., Koppe C., Knolle P., Castoldi M. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- Ruiz S., Diep D., Gore A., Panopoulos A.D., Montserrat N., Plongthongkum N., Kumar S., Fung H.L., Giorgetti A., Bilic J. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012;109:16196–16201. doi: 10.1073/pnas.1202352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr. Opin. Cell Biol. 2013;25:289–296. doi: 10.1016/j.ceb.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.X., Szulwach K.E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C.H., Zhang W., Jian X. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Donahue G., Zaret K.S. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler B., Ivanovska I., Mehta K., Song S., Nelson A., Tan Y., Mathieu J., Darby C., Blau C.A., Ware C. Characterization of microRNAs involved in embryonic stem cell states. Stem Cells Dev. 2010;19:935–950. doi: 10.1089/scd.2009.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Wang L., Huang W., Qin D., Cai J., Yao X., Feng C., Li Z., Wang Y., So K.F. Immediate expression of Cdh2 is essential for efficient neural differentiation of mouse induced pluripotent stem cells. Stem Cell Res. 2013;10:338–348. doi: 10.1016/j.scr.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Suh E.J., Remillard M.Y., Legesse-Miller A., Johnson E.L., Lemons J.M., Chapman T.R., Forman J.J., Kojima M., Silberman E.S., Coller H.A. A microRNA network regulates proliferative timing and extracellular matrix synthesis during cellular quiescence in fibroblasts. Genome Biol. 2012;13:R121. doi: 10.1186/gb-2012-13-12-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Kim K.Y., Zhong M., Pan X., Weissman S.M., Park I.H. Transcriptional regulation in pluripotent stem cells by methyl CpG-binding protein 2 (MeCP2) Hum. Mol. Genet. 2013;23:1045–1055. doi: 10.1093/hmg/ddt500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wu H., Li Y., Szulwach K.E., Lin L., Li X., Chen I.P., Goldlust I.S., Chamberlain S.J., Dodd A. Subtelomeric hotspots of aberrant 5-hydroxymethylcytosine-mediated epigenetic modifications during reprogramming to pluripotency. Nat. Cell Biol. 2013;15:700–711. doi: 10.1038/ncb2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.M., Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011;13:497–505. doi: 10.1038/ncb0511-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.C., Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Inokuma M.S., Denham J., Golds K., Kundu P., Gold J.D., Carpenter M.K. Feeder-free growth of undifferentiated human embryonic stem cells. Nat. Biotechnol. 2001;19:971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- Yang C.S., Li Z., Rana T.M. microRNAs modulate iPS cell generation. RNA. 2011;17:1451–1460. doi: 10.1261/rna.2664111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Liang Y., Lin Q., Liu J., Luo F., Li X., Zhou H., Zhuang S., Zhang H. miR-29 mediates TGFbeta1-induced extracellular matrix synthesis through activation of PI3K-AKT pathway in human lung fibroblasts. J. Cell. Biochem. 2013;114:1336–1342. doi: 10.1002/jcb.24474. [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhao X., Fiskus W., Lin J., Lwin T., Rao R., Zhang Y., Chan J.C., Fu K., Marquez V.E. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22:506–523. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.