

Summary
Embryonic stem cells (ESCs), which are derived from a peri-implantation embryo, are routinely cultured in medium containing diabetic glucose (Glc) concentrations. While pregnancy in women with pre-existing diabetes may result in small embryos, whether such high Glc levels affect ESC growth remains uncovered. We show here that long-term exposure of ESCs to diabetic Glc inhibits their proliferation, thereby mimicking in vivo findings. Molecularly, Glc exposure increased oxidative stress and activated Forkhead box O3a (FOXO3a), promoting increased expression and activity of the ROS-removal enzymes superoxide dismutase and catalase and the cell-cycle inhibitors p21cip1 and p27kip1. Diabetic Glc also promoted β-catenin nuclear localization and the formation of a complex with FOXO3a that localized to the promoters of Sod2, p21cip1, and potentially p27kip1. Our results demonstrate an adaptive response to increases in oxidative stress induced by diabetic Glc conditions that promote ROS removal, but also result in a decrease in proliferation.
Key words: glucose, oxidative stress, proliferation, FOXO, β-catenin, embryonic stem cell, metabolism, AKT, c-jun kinase
Graphical Abstract
Highlights
-
•
Exposure of ESCs to diabetic glucose (Glc) induces oxidative stress
-
•
ESCs fight oxidative stress via FOXO3a-mediated transcription of Sod2
-
•
FOXO3a activation promotes p21cip1 and p27kip1 expression and cell-cycle inhibition
-
•
Glc regulates FOXO3a/β-catenin co-occupation of the p21 and Sod2 promoters
zur Nieden and colleagues demonstrate that exposure to high glucose stimulates an oxidative stress response in embryonic stem cells leading to the expression and activity of ROS-removal enzymes, preventing cellular damage. However, the activation of the underlying molecular cascades simultaneously causes increased expression of cell-cycle regulatory genes, inhibiting proliferation.
Introduction
Because of the many similarities to a pre-implantation embryo, embryonic stem cells (ESCs) are a powerful model of early embryogenesis that can be used to examine the molecular mechanism underlying cell-fate specification. Furthermore, and especially due to their reliance on glycolysis to produce energy (Cho et al., 2006), which they share with cancer cells, any insight into their extensive proliferative potential may provide novel clues to understanding cellular responses in cells that proliferate indefinitely, but do not become transformed.
ESCs were first discovered in 1981 independently by two groups (Martin, 1981, Evans and Kaufman, 1981) and, while they were initially cultivated in medium containing physiological glucose (Glc) concentrations of 5.5 mM, descriptions of their routine cultivation in 25 mM Glc can be traced back to reports published as early as 1987 (Doetschman et al., 1987). It is unclear why this switch in culture conditions occurred, especially since newer reports suggest that ESC derivation is more successful in physiological Glc by increasing the pool of proliferating cells (Wang et al., 2006). The mechanism by which normoglycemia supports proliferation is currently unknown, although the same study illustrating that exposure to hyperglycemia leads to an increase in reactive oxygen species (ROS) may provide an important first step in elucidating the connection between hyperglycemia and complications during ESC establishment and growth.
The beneficial effect of physiological Glc concentrations on ESC proliferation is in line with in vivo findings from diabetic pregnancies, in which poorly controlled blood Glc levels result in embryos that are small in size, possibly due to a decrease in proliferation mediated by hyperglycemia-induced overactivation of p21cip1 (Zanetti et al., 2001, Varma et al., 2005, Scott-Drechsel et al., 2013).
These in vivo data and the newer studies suggesting a beneficial effect of physiological Glc on ESC growth are in contrast to a previous study, in which a high Glc environment resulted in an increase in ESC proliferation (Kim et al., 2006). Molecularly, Glc may increase proliferation of cells through various mechanisms, including increased phosphorylation and inhibition of forkhead box O transcription factors (FOXOs). As FOXO proteins have been suggested to transcriptionally regulate the cell-cycle inhibitory genes p21cip1 and p27kip1 (Dijkers et al., 2000, Hauck et al., 2007), the inhibition of these transcription factors and the consequent inhibition of cell-cycle inhibitors would contribute to extensive cellular proliferation. In line with this notion, Kim et al. (2006) found that hyperglycemia activated the serine/threonine kinase AKT in ESCs, a known upstream regulator of FOXO nuclear exclusion (Dickson et al., 2001).
Due to this apparent controversy, we sought to examine exposure of ESCs to high Glc levels for longer than 12 hr, as was done by Kim et al. (2006), to investigate whether this longer exposure would be able to mimic the in vivo effects of Glc on the early embryo and represent the conditions found during ESC derivation. Indeed, in acute exposures (5 days), we found decreased proliferation. We further suggest that a molecular cascade involving oxidative stress, inhibition of AKT, activation of c-jun NH2-terminal kinase (JNK), and transcriptional regulation of p21cip1 and p27kip1 through FOXO1, FOXO3a, and β-catenin (βcat) produces the proliferation inhibition caused by hyperglycemia.
Results
Exposure to Varying Glc Concentrations Modulates Proliferation
We hypothesized that prolonged exposure to a hyperglycemic environment (25 mM) would be able to better mimic the effects of Glc on the early embryo than short-term treatment (Kim et al., 2006). To test this hypothesis, we cultured ESCs in four different Glc concentrations (1, 5.5, 25, and 55 mM) and compared their phenotypes. Cultures exposed to 25 mM Glc for 24 hr appeared more densely populated compared with cells cultured in all other Glc concentrations (Figure 1A), supporting the previously described highly proliferative nature of short-term Glc-challenged cells (Kim et al., 2006). However, as the cells continued in the hyperglycemic environment, the pattern appeared to reverse, with cultures in physiological Glc (5.5 mM) containing more colonies. Cell counts and doubling times (Figures 1B, S1A, and S1B) confirmed that cells in 25 mM Glc initially proliferated more, while fewer cells were counted after longer treatment. Taken together, these data demonstrate that murine ESCs treated with Glc for longer periods in vitro do exhibit similar growth defects as found during ESC derivation.
Figure 1.

Hyperglycemia Leads to a Decrease in Cell Number and Is Coupled with an Increase in Oxidative Stress
(A) Micrographs of D3 ESCs exposed to varying Glc concentrations.
(B) Cell counts demonstrated that brief hyperglycemic exposure led to an initial increase in cell numbers, but these numbers were decreased after 5 days of exposure. n = 3 independent replicates ± SD.
(C) Superoxide anion content normalized to cell number. n = 5 independent replicates ± SD.
(D) Percentage of cells positive for reacted dihydrorhodamine was recorded on a flow cytometer. n = 5 independent replicates ± SD.
(E) qPCR for the determination of Sod2 and Cat mRNA levels after 5 days of Glc exposure. n = 3 independent replicates ± SD.
(F) SOD activity was measured after 5 days and normalized to protein content. n = 5 independent replicates ± SD.
(G) CAT activity is also increased in a Glc-dependent manner. n = 5 independent replicates ± SD.
∗p < 0.05, one-way ANOVA versus 5.5 mM Glc at 24 hr; Δp < 0.05, one-way ANOVA versus 5.5 mM Glc at 5 days. Glc, glucose; SOD, superoxide dismutase; CAT, catalase; RLU, relative light units; DHR, dihydrorhodamine.
Hyperglycemia Results in Increases in Oxidative Stress and Induces Activation of ROS-Removal Enzymes
After confirming that exposure to varying Glc did not alter expression of advanced glycation end products and their cognate receptors (Figure S1B), we sought to determine whether the Glc-induced generation of ROS (Zhang et al., 2010) and downstream cell-cycle inhibition were a potential mechanism for the Glc-induced growth defect. When cells were exposed to hyperglycemic conditions, their levels of superoxide anion (O2∙−) and hydrogen peroxide (H2O2) initially increased in a Glc-dependent fashion, but over time ROS levels declined, in a pattern similar to the one observed for proliferation (Figures 1C and 1D).
In concordance with the switch in ROS regulation during acute Glc exposure, the mRNA expression levels and enzyme activities of two ROS-removal enzymes, superoxide dismutase (SOD) and catalase (CAT), were elevated in the hyperglycemic condition (Figures 1E–1G and S1D), suggesting that cells in the hyperglycemic environment adapted to Glc-induced increases in ROS by activating enzymes responsible for their removal.
Increases in ROS Levels Promote FOXO1 and FOXO3a Expression and Nuclear Localization
After observing that oxidative stress pathways were altered by culture in hyperglycemia, we hypothesized that an upstream regulator may be altered by the increased Glc levels and that this may be leading to changes in downstream oxidative stress pathways. Initial PCR screens of possible regulators revealed that FoxO1 and FoxO3a, which encode for proteins that are known to be involved in ROS removal and cell-cycle control (Essers et al., 2004), showed increased transcription upon exposure of cells to 25 mM Glc while the FoxO4 isoform showed an inverse pattern (Figure 2A). Because FOXO1 and FOXO3a are important for maintaining ESC phenotypes (Zhang et al., 2011) and because their mRNA expression was stimulated under hyperglycemic conditions, we focused on these isoforms in subsequent analyses. Western blot analysis and immunostaining confirmed increased nuclear shuttling and localization of FOXO3a in 25 mM Glc, while increased FOXO1 nuclear levels seem to be at least partially explainable by Glc-mediated increases in overall protein levels (Figures 2B, 2C, and S2A).
Figure 2.

Hyperglycemia Promotes FOXO Activation
ESCs were exposed to various Glc concentrations for 5 days.
(A) qPCR analysis of FoxO genes. n = 3 independent replicates ± SD.
(B) Western blot on nuclear cell lysates.
(C) Immunocytochemistry demonstrating an increase in nuclear FOXO1 and FOXO3a localization with 25 mM Glc treatment.
(D and E) qPCR analysis of Sod2 (D) and Cat (E) expression following exposure of the indicated cell lines to Glc with or without antioxidants. n = 3 independent replicates ± SD.
(F and G) SOD (F) and CAT (G) activity. n = 5 independent replicates ± SD.
(H) Superoxide anion and hydrogen peroxide content in the above conditions. n = 5 independent replicates ± SD.
∗p < 0.05, one-way ANOVA compared with 5.5 mM Glc; Δp < 0.05, Student's t test compared with WT. CAT, catalase; DHR, dihydrorhodamine; Glc, glucose; GSH-OEt, glutathione reduced ethyl ester; RLU, relative light units; SOD, superoxide dismutase; TBP, TATA binding protein; VitE, vitamin E; WT, wild-type.
Because FOXO nuclear shuttling is initiated by elevated levels of ROS, we treated cells with glutathione reduced ethyl ester (GSH-OEt) and another antioxidant, vitamin E (VitE), and measured nuclear levels of the two FOXOs as well as levels of ROS-removal enzymes to determine whether increases in Sod2 and Cat expression were due to ROS-induced nuclear shuttling of FOXO. Antioxidant treatment attenuated Glc regulation of FOXO nuclear levels (Figure 2B) and reversed the pattern of Glc regulation of Sod2 and Cat (Figures 2D and 2E). GSH-OEt treatment also prevented the Glc regulation of SOD and CAT activity, illustrating that Glc-dependent differences in the activity of these proteins are mediated through changes in ROS levels (Figures 2G and 2H).
To confirm FOXO involvement in regulation of the oxidative stress response, we used a FoxO1- and FoxO3a-targeting short hairpin RNA (shRNA) to knock down both FoxO1 and FoxO3a (Figure S2B). Similar to the antioxidant treatment, FoxO1/3 knockdown reversed or attenuated the trend of Glc regulation of Sod2 and Cat mRNA expression and SOD and CAT activity (Figures 2D–2G).
Congruent with a role for FOXOs in Sod2 and Cat transcription, sh-FoxO1/3 cells showed lower overall mRNA levels than wild-type (WT) cells. However, this only translated into overall decreased SOD activity levels while CAT activity was globally increased. This global increase was also noted in Cat mRNA levels when cells were treated with GSH-OEt. Together, these data could suggest that Glc-mediated oxidative stress, in addition to activating nuclear FOXOs, may lead to the activation of one or more transcription factors repressive on the Cat promoter and that this repression is lifted under antioxidant treatment. We have preliminary evidence that the identity of this transcription factor could be TCF7L1, formerly known as TCF3. TCF7L1 nuclear levels are increased in 25 mM Glc, and an overexpression of a dominant negative form of this transcription factor inhibits Cat mRNA expression in all Glc concentrations (Figure S3). The combined action of FOXO and TCF7L1 may culminate in the increased CAT activity observed. Consistent with these findings, sh-FoxO1/3 cells exhibited a significant decrease in H2O2 levels (Figure 2H).
FOXO1/3a Inhibit Proliferation in Hyperglycemia by Regulating Gene Expression of Cell-Cycle Inhibitors
In addition to Sod2 and Cat, FOXO proteins are known to influence mRNA expression encoding for many other proteins. To determine whether Glc-induced activation of FOXOs would alter expression and activity of other known FOXO downstream targets, we used qPCR analysis to measure mRNA levels of PA26, Sirt1, and Gadd45 (Figure 3A) as well as the cyclin-dependent kinase inhibitors (CKIs) p21cip1 and p27kip1 (Figures 3B and 3C) in different Glc concentrations. Expression of all five FOXO targets measured increased with increasing Glc concentration, indicating that Glc-induced changes in nuclear FOXOs may lead to differential expression of all these target genes.
Figure 3.

Cell Cycle Is Inhibited in Response to Long-Term Exposure to High Glc
(A) qPCR analysis demonstrates a Glc-dependent increase in expression of the FOXO targets PA26, Sirt1, and Gadd45. n = 3 independent replicates ± SD.
(B and C) qPCR demonstrating a Glc-dependent increase in the cell-cycle regulators p21cip1and p27kip1. n = 3 ± SD.
(D) p21-luc reporter activity is increased in 25 mM Glc conditions. n = 3 independent replicates ± SD.
(E) Cell numbers in WT and sh-FoxO1/3 ESCs. n = 3 independent replicates ± SD.
(F) Mitotic index.
(G) Embryoid body formation assay. n = 3 independent replicates ± SD.
(H) S/G1 ratio calculated from cell-cycle analysis using propidium iodide. All analyses were performed after 5 days of Glc exposure.
∗p < 0.05, one-way ANOVA compared with 5.5 mM Glc. Δp < 0.05, one-way ANOVA compared with p21-luc. Glc, glucose; WT, wild-type; VitE, vitamin E; GSH-OEt, glutathione reduced ethyl ester; EB, embryoid body.
To more closely monitor whether hyperglycemia may be affecting cell-cycle regulation through p21cip1, we transfected ESCs with a p21-luciferase reporter construct and measured luciferase activity in cells from different Glc concentrations (Figure 3D). Luciferase activity was Glc-dependently increased in p21-luc cells while no effect was seen in cells transfected with a mutated p21 promoter (p21mut-luc) or mock-transfected cells, indicating that hyperglycemia may increase p21cip1 promoter activity leading to inhibition of the cell cycle and thus a decrease in overall cell numbers (Figure 3E), seemingly independent of p53 activation, another known regulator of p21cip1 (el-Deiry et al., 1993). In line with these findings, the mitotic index and the embryoid body-forming ability of ESCs exposed to 25 mM Glc was decreased when compared with cells exposed to physiological Glc concentrations (Figures 3F and 3G). An analysis of the cell cycle revealed that fewer of the cells cultured in 25 mM Glc were in S phase (Figure 3H). Together, these data demonstrate that Glc-dependent increases in expression of CKIs inhibit cell-cycle progression at the G1-S phase transition, leading to a decrease in cell number.
To determine whether Glc-induced increases in ROS were upstream of the observed changes in cell-cycle regulation, we next treated cells with the antioxidants VitE or GSH-OEt. qPCR analysis revealed that antioxidant treatment reversed Glc-dependent CKI expression patterns, indicating that Glc-induced changes in their expression correlated with higher ROS levels (Figures 3B and 3C). In addition, antioxidant treatment rescued the S/G1 ratio of cells cultured in 25 mM Glc to levels found in 5.5 mM Glc (Figure 3H), further supporting the conclusion that a Glc-mediated increase in ROS decreases cellular proliferation.
To examine whether FOXO activation linked the Glc-induced increases in ROS to p21cip1 and p27kip1 expression and cell-cycle inhibition, we repeated experiments with sh-FoxO1/3 ESCs. FoxO1/3 knockdown caused a decrease in p21cip1 and p27kip1 expression with increasing Glc levels, a pattern similar to the one seen in antioxidant-treated cells (Figures 3B and 3C), but reversed from WT, untreated cells. Cell-count experiments confirmed that sh-Foxo1/3 ESCs exposed to hyperglycemic conditions were more proliferative than WT cells exposed to hyperglycemia or sh-FoxO1/3 ESCs cultured in physiological Glc (Figure 3E). Furthermore, the increased S/G1 ratio in sh-FoxO1/3 cells indicates a rescue of proliferation in hyperglycemic sh-Foxo1/3 ESCs (Figure 3H). In summary, these data demonstrate the importance of FOXOs in regulating cell-cycle progression downstream of ROS through regulation of p21cip1 and p27kip1 mRNA expression.
The FOXO-Mediated Glc Response Is Dependent on Suppression of AKT and Activation of JNK
In adult stem and somatic cells, FOXO nuclear import and export is mediated by AKT and JNK 1 and 2 (Liang and Slingerland, 2003, Oh et al., 2005, Wang et al., 2012). While nuclear AKT levels are positively regulated by Glc to support FOXO nuclear exclusion (Dickson et al., 2001), JNK1/2 are activated in response to oxidative stress to promote FOXO nuclear import (Martindale and Holbrook, 2002). Western blot analysis of ESCs demonstrated that the activating phosphorylation on the S473 residue of AKT was reduced in 25 mM Glc and correlated with lower levels of nuclear AKT (Figure 4A). In contrast, the activating T183/Y185 phosphorylation of JNK1/2 was increased in 25 mM glucose (Figure 4B). Thus, in response to a hyperglycemic environment, cells increase activation of JNK1/2 while decreasing activation and nuclear localization of AKT, which may result in altered expression of FOXO proteins and, thus, downstream effects on ROS removal and cell-cycle progression. Phosphorylation of both proteins was either normalized or abolished upon antioxidant treatment, suggesting that their phosphorylation levels were regulated by the oxidative stress status of the cell (Figures 4A and 4B).
Figure 4.

AKT and JNK Oppositely Regulate the Long-Term Glc Response in ESCs
(A–D) Western blots on whole cell lysates or nuclear fractions as indicated.
(E) Cells were immunostained for FOXO3a and counterstained with DAPI.
(F) qPCR analysis for FOXO target genes determined with the 2−ΔΔCt method and correction for PCR efficiency using Tbp as the housekeeper. n = 3 independent replicates ± SD.
(G) Percentage of cells in cell-cycle stages were measured with flow cytometry using propidium iodide-treated cells and S/G1 ratios calculated. n = 3 independent replicates ± SD.
(H and I) SOD (H) and CAT (I) activity is increased in AKT-inhibited cells, similar to cells exposed to 25 mM Glc alone. n = 5 independent replicates ± SD.
(J) Superoxide anion levels. n = 5 ± SD.
(K) Hydrogen peroxide levels. n = 5 independent replicates ± SD.
∗p < 0.05, one-way ANOVA versus 5.5 mM Glc; Δp < 0.05, one-way ANOVA versus 25 mM Glc. 124005, AKT inhibitor; CAT, catalase; DHR, dihydrorhodamine; Glc, glucose; GSH-OEt, glutathione reduced ethyl ester; RLU, relative light units; SOD, superoxide dismutase; TBP, TATA binding protein; UT, untreated.
To examine the effects of altered AKT and JNK activation on FOXO nuclear localization, FOXO-mediated transcription, and cellular response on AKT and JNK, we used a Jnk1/2 null ESC line and a small-molecule AKT inhibitor, compound 124005. Western blot analysis confirmed that treatment with compound 124005 resulted in a decrease in phosphorylation of AKT at S473 (Figure 4D), a phosphorylation site that is critical for its activity (Sarbassov et al., 2005). At the same time, there was a noted decrease in the inhibitory phosphorylation of FOXO3a at S253 in 25 mM Glc and AKT-inhibited 5.5 mM Glc, the residue known to be phosphorylated by active AKT (Figure 4D). In addition, an increase in nuclear localization of FOXO3a was observed upon AKT inhibition in physiological Glc (Figure 4E), and FOXO3a levels were similar in both 5.5 mM and 25 mM Glc in the Jnk1/2−/− ESCs (Figure 4C). Similarly, AKT inhibition also decreased phosphorylated FOXO1 levels over non-treated 5.5-mM Glc cells. However, contrary to what was expected from the western blot results of nuclear 25-mM Glc-challenged cells, which had shown a nuclear increase in FOXO1 levels, the levels of phosphorylated FOXO1 were higher in the 25-mM Glc condition.
To demonstrate that the changes mediated by AKT inhibition and Jnk1/2 knockout resulted in downstream changes in FOXO target gene expression, we used qPCR analysis to measure the expression of Sod2, Cat, p21cip1, and p27kip1 (Figure 4F). Treatment with 124005 in 5.5 mM Glc increased expression of these FOXO targets, mimicking expression in the 25-mM Glc condition. At the same time we observed a decrease in cellular proliferation, which may result from the observed increase in expression of the cell-cycle inhibitors p21cip1 and p27kip1 (Figure 4G). Concomitant with the reduction in mRNA levels of Sod2 and Cat, AKT inhibition led to a loss of Glc-mediated increases of O2∙− and H2O2 levels (Figures 4E and 4F), which could be mediated by increased SOD and CAT activity (Figures 4G and 4H), an effect independent of phosphoinositide 3-kinase (see Figure S3C). In contrast, Jnk1/2 null ESCs exposed to 25 mM Glc showed increased levels of p21cip1 and p27kip1, increased proliferation and decreased levels of Sod2 mRNA, a reduction in SOD activity, and increased O2∙− levels; these changes are similar to the phenotype seen in 5.5 mM Glc and in cells treated with antioxidant (Figures 4K and 4L). In summary, both AKT inhibition and JNK activation appear to be upstream of the Glc-mediated nuclear presence of FOXOs, specifically FOXO3a, and its regulation of target gene promoters.
The only pattern that did not match the hypothesis was the differential regulation of Cat mRNA and activity levels, which showed increased levels instead of the expected reduction and caused a decrease in H2O2 production (Figures 4F, 4I, and 4L). We conclude that, although JNK may generally contribute to the FOXO-mediated regulation of Cat, additional factors may play roles in the regulation of Cat during oxidative stress response of ESCs that counteract FOXOs.
FOXO3a/CTNNB1 Interaction Is Increased in Hyperglycemia
Although FOXO3a regulation of Sod2 and p27kip has been well studied for many years, recent reports suggest that FOXO3a binding to the βcat protein, CTNNB1, is necessary for its role as a transcriptional activator of downstream target genes (Hoogeboom et al., 2008), and roles for CTNNB1 in regulating oxidative stress and the cell cycle have been described (Behrens and Lustig, 2004, Boo et al., 2009, Essers et al., 2005). However, their co-occupation of target gene promoters has not been conclusively shown. To determine whether CTNNB1 may be binding to FOXO3a and mediating the Glc-dependent changes in cell-cycle regulation and oxidative stress management we observed, we first investigated the expression and activity levels of CTNNB1 in different Glc concentrations. Phosphorylation of the CTNNB1 residues Y142 and Y654 that promote nuclear translocation were increased in hyperglycemic conditions (Figure 5A), which coincided with a Glc-dependent increase in nuclear CTNNB1 levels (Figures 5A, 5B, and S2A).
Figure 5.

Glc Increases CTNNB1 Activity and Interaction with FOXO1 and FOXO3a
(A) Western blots demonstrating an increase in the active and phosphorylated form of CTNNB1, and an increase in its nuclear localization in cells exposed to 25 mM Glc for 5 days.
(B) Cells were immunostained with anti-CTNNB1 and nuclei counterstained with DAPI.
(C) Western blots demonstrating the effect of AKT inhibition on CTNNB1 phosphorylation.
(D) Immunoprecipitation studies demonstrating an increase in FOXO/CTNNB1 interaction with increasing Glc. Top panel shows representative images of densitometry studies shown in bottom panel (n = 3 independent replicates ± SD). ∗p < 0.01, one-way ANOVA versus 5.5 mM Glc.
(E) FOXO3a/CTNNB1 occupation of promoters of FOXO target genes increased in response to Glc, an effect not seen in sh-FoxO1/3 ESCs. WT, wild-type; Glc, glucose; TBP, TATA binding protein; IgG, immunoglobulin G; ChIP, chromatin immunoprecipitation; IP, immunoprecipitation; IB, immunoblot.
AKT Is Upstream of FOXO, but Not CTNNB1
In addition to its role in regulating FOXO nuclear localization, active AKT has been positively correlated with nuclear accumulation of CTNNB1 by both direct and indirect mechanisms. AKT has been shown to directly phosphorylate CTNNB1 on S552, which leads to stabilization, nuclear accumulation, and activation of CTNNB1 target genes (Taurin et al., 2006, Fang et al., 2007, He et al., 2007). Furthermore, AKT may phosphorylate and thereby inactivate GSK3β, a member of the CTNNB1 destruction complex, resulting in CTNNB1 accumulation and nuclear shuttling (Lawlor and Alessi, 2001). To test whether AKT was responsible for the noted increase in nuclear CTNNB1 by either mechanism, we next determined the levels of specific phospho forms of CTNNB1 with and without AKT inhibition. CTNNB1 phosphorylation on S552 was observed in 5.5 mM Glc but not in 25 mM Glc (Figure 5C), which correlates with the increased AKT activation observed in 5.5 mM Glc (Figure 4A). Because nuclear accumulation of CTNNB11 was increased in 25 mM rather than 5 mM Glc and because inhibition of AKT did not alter CTNNB1 phosphorylation at S552, AKT does not appear to be responsible for this phosphorylation and subsequent nuclear accumulation.
To determine whether AKT regulates nuclear accumulation of CTNNB1 indirectly through GSK3β, we measured levels of CTNNB1 phosphorylated at residues S33/37/T41, the GSK3β phosphorylation sites, in control and AKT inhibitor-treated cells. AKT inhibition did not alter CTNNB1 phosphorylation at S33/37/T41, which demonstrates that AKT does not regulate CTNNB1 through GSK3β (Figure 5D). However, there was an overall decrease in CTNNB1 phosphorylation at residues S33/37/T41 in 25 mM Glc, illustrating that in the hyperglycemic condition less CTNNB1 is tagged for degradation, which may allow the protein to accumulate in the nucleus in 25 mM Glc (Figure 5C, compare with Figure 5A).
These data indicate that AKT inhibition in physiological Glc conditions does not alter CTNNB1 phosphorylation on residues S552 and S33/37/T41 or its cellular localization, but that decreases in GSK3β inhibition possibly contribute to the higher levels of nuclear CTNNB1 in 25 mM Glc.
FOXO3a/CTNNB1 Complex Is Present on the Promoters of Genes that Regulate ROS Removal and Cell-Cycle Inhibition
Because both FOXOs and CTNNB1 are recruited to the nucleus in hyperglycemic conditions, we next investigated whether these proteins associate in complexes. Co-immunoprecipitation analysis demonstrated that CTNNB1/FOXO1 and CTNNB1/FOXO3a complexes were increased in 25 mM Glc (Figure 5D), suggesting that these proteins may work together to affect transcription of target genes. However, the FOXO3a/CTNNB1 complex was more abundant than the FOXO1/CTNNB1 complex, suggesting that the transcriptional responses observed may have primarily been mediated by FOXO3a. To determine whether the observed nuclear FOXO3a/CTNNB1 complexes were in fact altering transcription of target genes, we used sequential chromatin immunoprecipitation (ChIP) to quantify FOXO3a/CTNNB1 binding to the promoters of p21cip1, p27kip1, and Sod2. We observed a Glc-dependent increase in localization of this complex to the promoters of these genes (Figure 5E), which correlates with the increase in interaction between these two proteins in 25 mM Glc. This increase in localization of transcription factor complexes to target gene promoters may be responsible for the Glc-dependent changes in expression observed in the cells. This binding pattern is abolished in sh-FoxO1/3 cells, further confirming the importance of the FOXO3a/CTNNB1 interaction in regulating ROS removal and cell-cycle regulation (Figure 6). However, it appears that there is still binding at the p27kip1 promoter, either suggesting different affinities for binding to different promoters or that the ChIP antibody employed may also be recognizing another protein. Thus, these results provide direct evidence that the promoters of p21cip1 and Sod2 are regulated by a FOXO3a/CTNNB1 complex, but we cannot ultimately conclude that FOXO3a and CTNNB1 are co-binding at the p27kip1 locus.
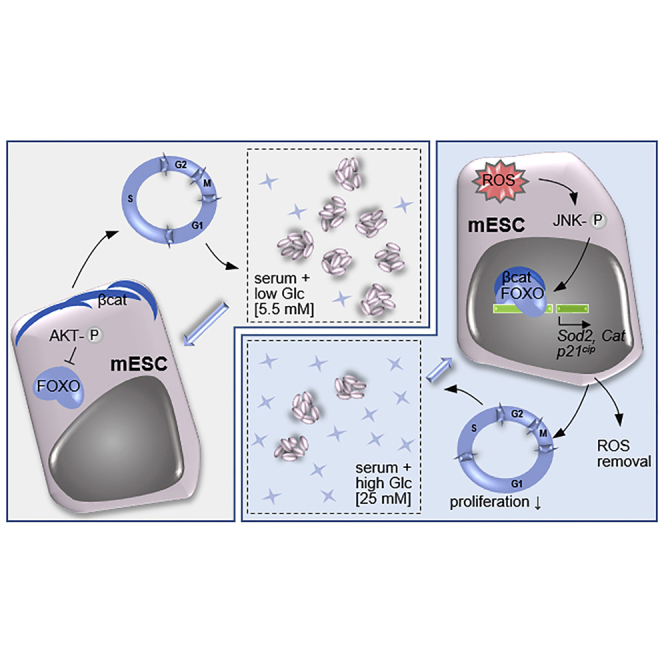
Figure 6.

Proposed Mechanism of Glc Action
Exposure to diabetic Glc leads to an immediate increase in ROS generation and subsequent JNK activation and results in the nuclear activation of FOXO3a/CTNNB1 by 5 days of exposure. This leads to the localization of this complex to the promoters of genes that regulate ROS removal and the cell cycle. In addition, AKT remains inactive in these conditions, rendering it unable to promote FOXO3a removal from the nucleus. CKI, cyclin-dependent kinase inhibitor; Glc, glucose; ROS, reactive oxygen species.
Discussion
Our results have shown that exposure of ESCs to high Glc levels leads to an oxidative stress response, resulting in repression of AKT, phosphorylation of JNK, and subsequent activation of FOXO1 and FOXO3a. Simultaneously, elevated levels of CTNNB1 are found in the nucleus, but this increase in nuclear accumulation is independent of AKT. The increased nuclear presence of FOXOs and CTNNB1 increases the levels of nuclear FOXO/CTNNB1 complexes, which co-occupy target gene promoters responsible for ROS removal and cell-cycle inhibition.
Molecularly, we suggest FOXO transcription factors as the mediators for the noted reduction in proliferation and the upregulation of ROS-removal enzymes (Figure 6). Previous studies have identified FOXO3a as a key regulator of Sod2 (Kops et al., 2002), p27kip1 (Dijkers et al., 2000), and p21cip1 expression (Hauck et al., 2007). Here, an shRNA-mediated knockdown of FoxO1/3 led to a major decline in FOXO3a binding to these promoters. Interestingly, there was still FOXO3a binding observed on the promoters of these genes, which may be due to the fact that the knockdown was not 100% effective.
The combined regulation of p27kip1 and Sod2 by FOXO4 and CTNNB1 has been suggested through promoter reporter studies (Essers et al., 2004), but direct evidence of FOXO/CTNNB1 binding to these promoters through ChIP has so far been lacking; however, such evidence is provided here for Sod2 and also additionally for p21cip1. In addition, we show here that this binding is dependent on Glc-induced ROS levels, which has widespread implications not only for the routine culture of pluripotent stem cells but also for the field of diabetes and aging.
There are multiple ways whereby oxidative stress may promote FOXO activation. For example, FOXO proteins can directly sense ROS through the ability of ROS to induce disulfide bridges between cysteine residues on the FOXO protein, resulting in FOXO activation and transcription of ROS-removal enzymes (Dansen, 2011). However, while this would explain the noted increase in FoxO1 and FoxO3a nuclear localization, it does not explain the Glc-dependent increase in FoxO1 and FoxO3a mRNA expression that was also observed. Nonetheless, the Glc-mediated FoxO1 and FoxO3a mRNA induction could potentially be due to a previously described relationship between ROS, phosphorylated JNK, and FoxO transcription (Essers et al., 2004) that goes beyond the protein level investigated here. Furthermore, treatment with antioxidants in this study reversed the Glc-dependent increase in FOXO3a nuclear localization and Sod2 and Cat expression, suggesting that the generation of excess ROS is indeed upstream of FOXO3a target expression. Interestingly, treatment with antioxidants led to a global increase in CAT activity that was unexpected, but could be due to the alleviation of TCF7L1-regulated repression following antioxidant treatment, leading to an increase in gene expression (Solberg et al., 2012).
An unanswered question remains the identity of the kinase responsible for nuclear CTNNB1 accumulation. Our data suggested that it is not AKT. Treatment with the AKT inhibitor 124005 in physiological Glc conditions resulted in an increase in nuclear FOXO3a, but did not affect CTNNB1 nuclear localization, demonstrating that nuclear CTNNB1 localization in hyperglycemia is independent of AKT inhibition. These data are inconsistent with previous findings, as AKT has been shown to promote an increase in CTNNB1 transcriptional activity through the phosphorylation of S552 (Fang et al., 2007) and also to inhibit GSK3β, which normally phosphorylates CTNNB1 on S33/37/T41, tagging it for degradation (Shiojima and Walsh, 2006, Srivastava and Pandey, 1998). One possible explanation for this could be the existence of different pools of CTNNB1 that are regulated independently of one another (Mbom et al., 2013), or perhaps that CTNNB1 is regulated differently in stem cells than in other cell types.
Our findings also raise questions regarding current ESC culture techniques. Routinely used media contain 25 mM Glc, and it is often observed that there is a tremendous amount of spontaneous differentiation and that ESCs behave differently from passage to passage. In fact, one study demonstrated that the derivation and culture of ESCs in physiological Glc reduced oxidative stress and improved proliferative capacity (Wang et al., 2006) while a different study confirmed that exposure to oxidative stress promoted cell-cycle arrest and senescence (Guo et al., 2010). Our contribution here is to have provided a molecular consequence of oxidative stress on ESCs, mediated through FOXO3a activation and AKT inhibition. Recent studies have demonstrated that FOXOs are important in ESC identity, with FOXO1 being a positive regulator in the maintenance of pluripotency, while the role of FOXO3a remains elusive (Zhang et al., 2011) and stimulation of AKT in ESCs promotes proliferation (Heo and Han, 2006). Our results are consistent with these reports, demonstrating that hyperglycemia results in an increase in oxidative stress that leads to changes in the proliferative capacity of pluripotent cells mediated partly through the activation of FOXO3a downstream of AKT and JNK regulation.
Conclusion
Our findings demonstrate that early exposure to high Glc levels stimulates an oxidative stress response that promotes the expression and activity of ROS-removal enzymes, protecting the embryonic cells from ROS-induced damage. At the same time this leads to an increase in expression of cell-cycle regulatory genes, inhibiting the proliferative capacity of these cells. By extension, our data are relevant for early pre-implantation embryos of mothers with pre-existing diabetes developing proliferative defects.
Experimental Procedures
Cell Culture
Mouse ESC lines D3 and CCE were maintained in the undifferentiated state on BD Falcon Primaria tissue culture flasks (BD Biosciences) in medium containing 1,000 U/ml Leukemia Inhibitory Factor (Chemicon). Cell-culture medium contained 15% fetal bovine serum (FBS) (PAA), 0.1 mM non-essential amino acids, 50 U/ml streptomycin and 50 U/ml penicillin, and 0.1 mM β-mercaptoethanol (Invitrogen) diluted in DMEM (Invitrogen). Cells were passaged every 48 hr using 0.25% trypsin-EDTA (Invitrogen). Cell numbers were determined using a CASYcell counter (Innovates).
Reporter and Knockdown ESC Lines
The p21-luc and p21mut-luc ESC lines were created by transfecting luciferase reporter constructs containing various portions of the human p21 promoter (−2.15 kb [p21Δp53] and −93 bp [p21P93-S]) (Datto et al., 1995) at the passage of analysis using Effectene (zur Nieden et al., 2005, Ding et al., 2012). For creation of shRNA-mediated knockdown FoxO1/3 ESCs, a pSuperior-FoxO1/3 plasmid (de Keizer et al., 2010) was linearized with HindIII, gel purified, and 1 μg transfected into ESCs using Effectene. Clones were chosen after 72 hr of puromycin selection, and plasmid integration was confirmed using PCR for the puromycin gene (forward primer 5′-TGCAAGAACTCTTCCTCACG-3′, reverse primer 5′-AGGCCTTCCATCTGTTGCT-3′) with an annealing temperature of 65°C. Western blotting was used to estimate the percentage of knockdown. The Jnk1/2−/− ESC line was a kind gift of Rodger J. Davis (University of Massachusetts Medical School).
Glc and Compound Treatment
Culture medium was made from no-Glc DMEM (Invitrogen) containing the above cell-culture supplements with the addition of the appropriate amount of Glc. Stock solutions of 10 mM AKT Inhibitor I (Calbiochem, 124005) dissolved in DMSO were diluted for a final treatment concentration of 5 μM. A solvent negative control was tested alongside inhibitor treatment. Antioxidant treatment consisted of glutathione reduced ethyl ester (GSH-OEt, 250 μmol/l) or vitamin E (VitE, 1 μg/ml).
Protein Extraction and Western Blotting
Sodium orthovanadate-pretreated cells (30 min) were trypsinized and lysed in radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in 1× PBS [pH 7.4]) containing a protease inhibitor cocktail (104 mM AEBSF, 80 μM aprotinin, 4 mM bestatin, 1.4 mM E-64, 2 mM leupeptin, 1.5 mM pepstatin A [Sigma-Aldrich]). Fractionated protein samples were prepared with an NE-PER cell fractionation kit (Pierce, 78833) and protein quantified using the DC protein assay (Bio-Rad). Protein (25 μg) was loaded on an SDS-PAGE gel and transferred to a polyvinylidene fluoride membrane overnight at 30 V and 4°C. Membranes were blocked in 5% BSA diluted in 1× TBST (Tris-buffered saline with Tween 20) for 1 hr at room temperature, and incubated with primary antibody at 4°C. Membranes were washed, incubated with secondary antibody, and developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce). The following antibodies were used: ACTINB (#8456), pan AKT (#4691), phospho AKTS473 (#4060), anti-rabbit HRP (#7074), and anti-mouse HRP (#7076) (all from Cell Signaling Technology); pan CTNNB1 (#ab16051), phospho CTNNB1Y142 (#ab27798), phospho CTNNB1Y654 (#ab59430), pan FOXO3a (#ab47409), phospho FOXO3aS253 (#ab47285), phospho JNK1/2T183/Y185 (#ab4821), pan JNK1/2 (#ab112501), and TBP (#ab818) (all from Abcam).
Immunocytochemistry
Cells were fixed in 7:3 acetone/methanol for 5–10 min at −20°C. Cell membranes were permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. Non-specific binding was blocked with 10% FBS, 0.5% BSA in PBS for 30 min at 37°C. Primary antibody was diluted in blocking buffer and incubated overnight at 4°C. Plates were incubated with secondary antibody and DAPI for 2 hr at room temperature. The following antibodies were used: pan CTNNB1 (Life Technologies, #71-2700), pan FOXO3a (Abcam, #ab47409), anti-mouse Alexa Fluor 488 (Life Technologies, #MHZAP7020), and anti-rabbit Alexa Fluor 546 (Life Technologies, #A-11035).
Superoxide Anion and Hydrogen Peroxide Quantification Assays
Superoxide anion and hydrogen peroxide content were measured using dihydrorhodamine 123 (DHR; Cayman Chemical) and the Superoxide Anion Detection Kit (Agilent) as per manufacturer's instructions. Light emission for superoxide anion content was recorded with a Lucetta luminometer (Lonza) and data were normalized to the total cell number collected.
Luciferase Activity Assay
Luciferase activity was assayed with the luciferase assay system from Promega. Cells were lysed in Luciferase Cell Culture Lysis 5× Reagent. Lysates were cleared by centrifugation (12,000 × g for 2 min at 4°C) and luminescence read in a Lucetta Luminometer (Lonza).
SOD and CAT Activity Assays
RIPA protein lysates were assessed for SOD and CAT activity according to the manufacturer's instructions (Cayman Chemical). Protein content was quantified with a Lowry assay and used to normalize enzyme activity.
Flow Cytometry
The percentage of marker-positive cells in single-cell suspensions were gated based on WT or non-reacted cells. Ten thousand events were measured in a Beckman Coulter flow cytometer and mean percentages of GFP/marker-positive cells were calculated from three independent treatments.
RNA Extraction and cDNA Synthesis
RNA was extracted using the Nucleospin RNA extraction kit (Macherey Nagel) following the manufacturer's instructions, and quantified using a Nanodrop spectrophotometer (ND-1000; Nanodrop). 625 ng of mRNA was used for cDNA synthesis in a reaction containing 0.5 mM deoxynucleoside triphosphates, 0.3 μg random hexamers, 100 mM DTT, 5 mM MgCl2, 50 U of RNase inhibitor, and 80 U of reverse transcriptase (Fermentas).
RT-PCR and qPCR
Gene-expression analysis was done from 25–50 ng of cDNA using RT-PCR as described previously (zur Nieden et al., 2007). Final PCR products were run on an agarose gel containing ethidium bromide. qPCR was performed using an iCycler iQ system and SYBR Green PCR master mix (Bio-Rad) with post-run melting curves. For the initial 5-min denaturation step, the temperature was maintained at 95°C and the machine cycled between 30 s at 95°C and 45 s at the appropriate annealing temperature for 40 rounds. Data were then analyzed according to the 2−ΔΔCt method with correction for PCR efficiency (Pfaffl, 2001) and normalization to Tbp, and expressed as n-fold change over 25 mM Glc (if not noted otherwise). Primers used are listed in Table S1.
Cell-Cycle Analysis
Ethanol (70%) was added to trypsinized cells in a dropwise fashion. Following a 60-min incubation in ethanol, samples were incubated in propidium iodide for 3 hr at 4°C, spun down, washed, and analyzed by flow cytometry.
Co-immunoprecipitation
Nuclear protein (60 μg) was mixed with an equal volume of Ag/Ab buffer (50 mM Tris [pH 8.0], 1 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 10% glycerol) and mixed with 4 μg of anti-CTNNB1 antibody (Abcam, #ab2365) with gentle agitation for 1 hr at 4°C. Fifty microliters of Protein A Sepharose beads (1:1 slurry in PBS) was then added for an additional 3 hr at 4°C with gentle agitation. Beads were washed three times in 0.01% NP-40 buffer (1 mM Tris-Cl [pH 8.0], 2.4 mM NaCl, 0.01% NP-40), followed by two additional washes in PBS. Proteins were eluted adding SDS-PAGE loading buffer and subjected to 95°C for 10 min. The 6%/10% SDS-PAGE gel was run for approximately 3 hr at 100 V and the western blot for FOXOs was continued as described above.
Sequential Chromatin Immunoprecipitation
Crosslinking was performed in 1% paraformaldehyde (15 min, room temperature) and stopped with 0.125 M glycine (5 min). Cells were lysed in PBS containing phosphatase inhibitor mix (PI, Sigma-Aldrich) and 1 mM PMSF, and the pellet was resuspended in 1 ml of ChIP sonication buffer (50 mM HEPES [pH 7.9], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) also containing PI and PMSF. DNA was sheared by sonication (10 rounds of [10 × 0.5 s on, 0.6 s off, 27% amplitude]) and a sample was used for determining fragment size. DNA (50 μg) was then used for two rounds of immunoprecipitation (round 1: FOXO3a [Santa Cruz, sc-11351]; round 2: CTNNB1 [BD Transduction Laboratories, 610154]), protein was digested using proteinase K, and the resulting sample was used for PCR analysis as described above with primers as listed in Table S2.
Statistics
p Values were calculated with a standard weighted-means ANOVA when independent treatment groups were compared. Post hoc Tukey honest significant difference tests were performed when more than two groups were compared, and the ANOVA yielded a significant F ratio to determine statistical differences among two individual groups (http://faculty.vassar.edu/lowry/anova1u.html). A p value equal to or less than 0.05 was considered statistically significant.
Acknowledgments
The authors thank Dr. Xiao-Fan Wang (Duke University Medical Center, Durham, NC) for generously providing the p21 luciferase reporter constructs, Dr. Boudewijn Burgering for the pSuperior-FoxO1/3 plasmid, and Dr. Irving Weissman (Stanford University) for the LEF/TCF-GFP ESCs. We further express gratitude to Susann Horvat, Aashima Singhal, and Beatrice Kuske for excellent technical assistance. This study was supported by start-up funds from the University of California Riverside.
Published: July 12, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.06.006.
Supplemental Information
References
- Behrens J., Lustig B. The Wnt connection to tumorigenesis. Int. J. Dev. Biol. 2004;48:477–487. doi: 10.1387/ijdb.041815jb. [DOI] [PubMed] [Google Scholar]
- Boo J.H., Song H., Kim J.E., Kang D.E., Mook-Jung I. Accumulation of phosphorylated beta-catenin enhances ROS-induced cell death in presenilin-deficient cells. PLoS One. 2009;4:e4172. doi: 10.1371/journal.pone.0004172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y.M., Kwon S., Pak Y.K., Seol H.W., Choi Y.M., Park D.J., Park K.S., Lee H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Dansen T.B. Forkhead box O transcription factors: key players in redox signaling. Antioxid. Redox Signal. 2011;14:559–561. doi: 10.1089/ars.2010.3778. [DOI] [PubMed] [Google Scholar]
- Datto M.B., Yu Y., Wang X.F. Functional analysis of the transforming growth factor β responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- de Keizer P.L., Packer L.M., Szypowska A.A., Riedl-Polderman P.E., van den Broek N.J., de Bruin A., Dansen T.B., Marais R., Brenkman A.B., Burgering B.M. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer Res. 2010;70:8526–8536. doi: 10.1158/0008-5472.CAN-10-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson L.M., Lingohr M.K., McCuaig J., Hugl S.R., Snow L., Kahn B.B., Myers M.G., Jr., Rhodes C.J. Differential activation of protein kinase B and p70(S6)K by glucose and insulin-like growth factor 1 in pancreatic beta-cells (INS-1) J. Biol. Chem. 2001;276:21110–21120. doi: 10.1074/jbc.M101257200. [DOI] [PubMed] [Google Scholar]
- Dijkers P.F., Medema R.H., Cornelieke P., Banerji L., Thomas N.S.B., Lam E.W.F., Burgering B.M.T., Raaijmaker J.A.M., Lammers J.W.J., Koenderman L., Coffer P.J. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27KIP1. Mol. Cell. Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H., Keller K.C., Martinez I.K.C., Geransar R.M., zur Nieden K.O., Nishikawa S.G., Rancourt D.E., zur Nieden N.I. NO/beta-catenin crosstalk modulates primitive streak formation prior to embryonic stem cell osteogenic differentiation. J. Cell. Sci. 2012;125(Pt 22):5564–5577. doi: 10.1242/jcs.081703. [DOI] [PubMed] [Google Scholar]
- Doetschman T., Gregg R.G., Maeda N., Hooper M.L., Melton D.W., Thompson S., Smithies O. Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature. 1987;330:576–578. doi: 10.1038/330576a0. [DOI] [PubMed] [Google Scholar]
- el-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Essers M.A., Weijzen S., de Vries-Smits A.M., Saarloos I., de Ruiter N.D., Box J.L., Burgering B.M. FoxO transcription factor activation by oxidative stress mediated by small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers M.A., de Vries-Smits L.M., Barker N., Polderman P.E., Burgering B.M., Korswagen H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- Evans M.J., Kaufman M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G.B., Kobayashi R., Hunter T., Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.L., Chakraborty S., Rajan S.S., Wang R., Huang F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-renewal. Stem Cells Dev. 2010;19:1321–1331. doi: 10.1089/scd.2009.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck L., Harms C., Grothe D., An J., Gertz K., Kronenberg G., Dietz R., Endres M., von Harsdorf R. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ. Res. 2007;100:50–60. doi: 10.1161/01.RES.0000254704.92532.b9. [DOI] [PubMed] [Google Scholar]
- He X.C., Yin T., Grindley J.C., Tian Q., Sato T., Tao W.A., Dirisina R., Porter-Westpfahl K.S., Hembree M., Johnson T. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 2007;39:189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J.S., Han H.J. ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathways. Stem Cells. 2006;24:2637–2648. doi: 10.1634/stemcells.2005-0588. [DOI] [PubMed] [Google Scholar]
- Hoogeboom D., Essers M.A., Polderman P.E., Voets E., Smits L.M., Burgering B.M. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J. Biol. Chem. 2008;283:9224–9230. doi: 10.1074/jbc.M706638200. [DOI] [PubMed] [Google Scholar]
- Kim Y.H., Heo J.S., Han H.J. High glucose increase cell cycle regulatory proteins level of mouse embryonic stem cells via PI3-K/AKT and MAPKs signal pathways. J. Cell Physiol. 2006;209:94–102. doi: 10.1002/jcp.20706. [DOI] [PubMed] [Google Scholar]
- Kops G.J., Dansen T.B., Polderman P.E., Saarloos I., Wirtz K.W., Coffer P.J., Huang T.T., Bos J.L., Medema R.H., Burgering B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Lawlor M.A., Alessi D.R. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001;114(Pt 16):2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- Liang J., Slingerland J.M. Multiple roles of the PI3K/PKB (Akt) pathways in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Martin G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale J.L., Holbrook N.J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Mbom B.C., Nelson W.J., Barth A. Beta-catenin at the centrosome: discrete pools of beta-catenin communicate during mitosis and may co-ordinate centrosome functions and cell cycle progression. Bioessays. 2013;35:804–809. doi: 10.1002/bies.201300045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.W., Mukhopadhyay A., Svrzikapa N., Jiang F., Davis R.J., Tissenbaum H.A. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of AKT/PKB by the rictor/mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Scott-Drechsel D.E., Rugyoni S., Marks D.L., Thornburg K.L., Hinds M.T. Hyperglycemia slows embryonic growth and suppresses cell cycle via cyclin D1 and p21. Diabetes. 2013;62:234–242. doi: 10.2337/db12-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I., Walsh K. Regulation of cardiac growth and coronary angiogenesis by the AKT/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- Solberg N., Machon O., Machonova O., Krauss S. Mouse Tcf3 represses canconical Wnt signaling by either competing for beta-catenin binding or through occupation of DNA binding sites. Mol. Cell. Biochem. 2012;365:53–63. doi: 10.1007/s11010-012-1243-9. [DOI] [PubMed] [Google Scholar]
- Srivastava A.K., Pandey S.K. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol. Cell. Biochem. 1998;182:135–141. [PubMed] [Google Scholar]
- Taurin S., Sandbo N., Qin Y., Browning D., Dulin N.O. Phosphorylation of beta catenin by cyclic-AMP-dependent protein kinase. J. Biol. Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- Varma S., Lal B.K., Zheng R., Breslin J.W., Saito S., Pappas P.J., Hobson R.W., 2nd, Duran W.N. Hyperglycemia alters PI3K and Akt signaling and leads to endothelial cell proliferative dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H1744–H1751. doi: 10.1152/ajpheart.01088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Thirumangalathu S., Loeken M.R. Establishment of new mouse embryonic stem cell lines is improved by physiological glucose and oxygen. Cloning Stem Cells. 2006;8:108–116. doi: 10.1089/clo.2006.8.108. [DOI] [PubMed] [Google Scholar]
- Wang X., Chen W.R., Xing D. A pathway from JNK through decreased ERK and Akt activities for FOXO3a nuclear translocation in response to UV irradiation. J. Cell Physiol. 2012;227:1168–1178. doi: 10.1002/jcp.22839. [DOI] [PubMed] [Google Scholar]
- Zanetti M., Zwacka R., Engelhardt J., Katusic Z., O’Brien T. Superoxide anions and endothelial cell proliferation in normoglycemia and hyperglycemia. Arterioscler. Thromb. Vasc. Biol. 2001;21:195–200. doi: 10.1161/01.atv.21.2.195. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Liew C.W., Handy D.E., Zhang Y., Leopold J.A., Hu J., Guo L., Kulkami R.N., Loscalzo J., Stanton R.C. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. 2010;24:1497–1505. doi: 10.1096/fj.09-136572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yalcin S., Lee D.F., Yeh T.Y., Lee S.M., Su J., Mungamuri S.K., Rimmelé P., Kennedy M., Sellers R. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 2011;13:1092–1099. doi: 10.1038/ncb2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Nieden N.I., Kempka G., Rancourt D.E., Ahr H.J. Induction of chondro-, osteo-, and adipogenesis in embryonic stem cells by bone morphogenetic protein-2: effect of cofactors on differentiating lineages. BMC Dev. Biol. 2005;5:1. doi: 10.1186/1471-213X-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Nieden N.I., Cormier J.T., Rancourt D.E., Kallos M.S. Embryonic stem cells remain highly pluripotent following long term expansion as aggregates in suspension bioreactors. J. Biotechnol. 2007;129:421–432. doi: 10.1016/j.jbiotec.2007.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.