

Abstract
Heart failure (HF) is a major cause of morbidity and mortality worldwide. The global burden of HF continues to rise, with prevalence rates estimated at 1–2% and incidence approaching 5–10 per 1000 persons annually. The complex pathophysiology of HF impacts virtually all aspects of normal cardiac function – from structure and mechanics to metabolism and electrophysiology – leading to impaired mechanical contraction and sudden cardiac death. Pharmacotherapy and device therapy are the primary methods of treating HF, but neither is able to stop or reverse disease progression. Thus, there is an acute need to translate basic research into improved HF therapy. Animal model investigations are a critical component of HF research. However, the translation from cellular and animal models to the bedside is hampered by significant differences between species and among physiological scales. Our studies over the last 8 years show that hypotheses generated in animal models need to be validated in human in vitro models. Importantly, however, human heart investigations can establish translational platforms for safety and efficacy studies before embarking on costly and risky clinical trials. This review summarizes recent developments in human HF investigations of electrophysiology remodelling, metabolic remodelling, and β‐adrenergic remodelling and discusses promising new technologies for HF research.

Abbreviations
- 3D‐MIM
3D multifunctional integumentary membrane
- ΔAPD
action potential duration reduction
- AP
action potential
- APD
action potential duration
- APD80
AP duration at 80% repolarization
- β‐AR
β‐adrenergic receptor
- CaTD30
CaT duration at 30% recovery
- CaTD80
CaT duration at 80% recovery
- CaT
calcium transient
- Cx43
connexin 43
- CV
conduction velocity
- DAD
delayed afterdepolarization
- EAD
early after depolarization
- EC
excitation–contraction
- ENDO
endocardial
- EPI
picardial
- ETC
electrophoretic tissue clearing
- Gi
inhibitory regulative G‐protein
- GFP
green fluorescent protein
- Gs
stimulative regulative G‐protein
- HF
heart failure
- I–R
ischaemia and reperfusion
- ICD
implantable cardioverter defibrillator
- LA
left atrium
- LQTS
long‐QT syndrome
- LV
left ventricle
- MID
midmyocardium
- MRI
magnetic resonance imaging
- NF
non‐failing
- NCX
sodium–calcium exchanger
- OAP
optical action potential
- OCT
optical coherence tomography
- PCL
pacing cycle length
- PKC
protein kinase C
- PVC
premature ventricular contraction
- ROS
reactive oxygen species
- SCD
sudden cardiac death
- sub‐ENDO
subendocardium
- sub‐EPI
subepicardium
- VT
ventricular tachycardia
Introduction
Heart failure (HF) is the end stage of many cardiovascular diseases and is characterized by the heart's inability to sustain the metabolic demands of the body. The pathophysiology of HF is complex and often develops amid years of chronic damage and remodelling. Moreover, HF impacts all aspects of cardiac function (i.e. metabolism, mechanics, and electrophysiology), leading to increased morbidity and mortality due to impaired mechanical contraction and sudden cardiac death (SCD).
The global burden of HF continues to rise with the prevalence rates estimated at 1–2% and incidence approaching 5–10 per 1000 persons annually (Mosterd & Hoes, 2007). The average lifetime risk of developing the disease ranges from 20 to 33%, and that risk increases even further with the presence of hypertension or elevated body mass index (Roger, 2013). In addition, HF patients generally have a poor prognosis with 5‐year and 10‐year survival rates reported at 50% and 10%, respectively (Mosterd & Hoes, 2007).
Current pharmacological options for the treatment of HF remain limited and are based on long established ideas: β‐blockers, renin–angiotensin system (RAS) inhibitors, and diuretics. While pharmacotherapy can ameliorate symptoms and slow the progression of HF, mortality rates remain high (Chen et al. 2011). Device therapy is similarly limited. Resynchronization therapy, while effective for some, is ineffectual for many patients (Strickberger et al. 2005). Implantable cardioverter defibrillators are effective at preventing sudden cardiac death (Buxton et al. 1999; Glatter et al. 2006), to which HF patients are highly susceptible, but does nothing to reverse or slow disease progression. Left ventricular assist devices, if placed early enough, are able to reverse the progression of HF (Bruce et al. 2015); however, for many patients they remain a bridge to transplantation, the final option in the event that other therapeutic options fail. For these reasons, there is an acute need for bench to bedside translation.
Animal model investigations remain a critical component of HF research. Indeed, various small and large animal model studies have helped elucidate some of the pathological changes that occur in heart failure (Poelzing & Rosenbaum, 2004; Rose et al. 2005; Valdivia et al. 2005; Tsuji et al. 2006). However, HF is a diverse term for a disorder that can arise from a number of different causes and can be characterized by a variety of symptoms (Houser et al. 2012). As a result, not all clinical HF cases fit in the same mould, and thus not all animal models can recapitulate the specific yet multifaceted forms of chronic HF (Houser et al. 2012). For example, the HF‐like syndrome that can be rapidly induced in rodents, rabbits, and canines with chronic fast pacing has several important differences from human HF. While certain large mammals possess heart rates (HRs), cardiac ion channel expression, and ECG and AP morphologies closer to those of humans (Kaese et al. 2013) these models do not develop significant hypertrophy, and termination of pacing results in phenotype reversal (Hasenfuss, 1998). It is also important to note that many HF patients present with a variety of co‐morbid conditions that can exacerbate cardiac dysfunction and are difficult to replicate in an animal model. These constraints limit the translation of animal research findings to humans and thus it is critically important for researchers to carefully evaluate differences between animal models before making a selection for studying HF and developing new clinical therapies.
Explanted human heart investigations in conjunction with animal model research help bridge the translational gap. Nearly 48% of the donor hearts are rejected for transplantation due to older donor age and medical co‐morbidities, among other factors (Khush et al. 2015). The rejected donor hearts can be a valuable resource for HF research. The recent development of the first stand‐alone organ recovery centre in St Louis, MO, USA, has created a unique opportunity for human heart research (Efimov et al. 2010, Doyle et al. 2014). The data acquired from this resource range from gene and protein expression levels to functional metabolic and electrophysiological measurements conducted in living human heart tissues. In many of our experiments, we have used optical mapping techniques to examine conduction, repolarization and calcium handling properties, and response to β‐adrenergic stimulation in donor vs. failing hearts (Glukhov et al. 2010, 2012; Lou et al. 2011, 2012). In addition, we have begun conducting studies of structural and metabolic remodelling in heart failure in relation to electrophysiological changes.
We focus our review on human HF investigations of electrophysiology remodelling, metabolic remodelling, and β‐adrenergic remodelling. We discuss findings on excitation–contraction (EC) coupling remodelling, including alterations in activation and repolarization: gap junctions, sodium channels, potassium channels, and sodium–calcium exchanger alterations. We review metabolic remodelling, including cellular ultrastructure remodelling and mitochondrial capacity for utilizing different substrates. We also describe remodelling of β‐adrenergic receptor (β‐AR) signalling, and emphasize the differential roles that β1‐AR and β2‐AR play in creating a pro‐arrhythmic substrate. Finally, we highlight novel methodologies such as 3D multifunctional integumentary membranes (3D‐MIMs), CLARITY, and organotypic cardiac tissue slice preparation as alternative but promising experimental methodologies for studying cardiac EC coupling and metabolism in the human heart.
Electrophysiology remodelling
Electrophysiological arrhythmogenic remodelling during HF includes changes in ion channels, cellular uncoupling, altered calcium homeostasis, and altered extracellular matrix (Coronel et al. 2013). These changes, originally uncovered in animal models, are generally applicable to humans. We also highlight gender differences in electrophysiological gene expression in failing and non‐failing human hearts.
Activation remodelling
QRS widening on the body surface ECGs resulting from slowed conduction is a hallmark of HF, which can promote unidirectional conduction block and reentrant arrhythmias (Wit & Rosen, 1983; Jin et al. 2008). As conduction velocity depends on fast activation of sodium channels and proper gap junction coupling, they are both important players in arrhythmogenic remodelling of activation in human hearts (Rohr, 2004).
Voltage gated sodium channels are responsible for the rising phase of the cardiac action potential (AP) and comprise α‐ and β‐subunits, proteins encoded by the SCN5A and SCN1B genes, respectively (Nerbonne & Kass, 2005). Several laboratories have investigated cardiac myocytes in failing and non‐failing human ventricular tissue to elucidate the role of sodium currents in HF. The peak density of transient sodium currents was reported to decrease by 57% in cardiomyocytes isolated from failing hearts in comparison with those acquired from non‐failing hearts (Valdivia et al. 2005). Moreover, mRNA expression for both α‐ and β‐subunits remained the same in both groups, suggesting that the reduction in cardiac sodium current in failing hearts could be due to non‐functional channels or decreased functional expression of sodium channels at the cell surface rather than transcriptional downregulation.
The non‐functional sodium channel hypothesis was further strengthened, when it was demonstrated that non‐functional splice variants of SCN5A are increased in heart failure and that the full‐length mRNA represents only 50% of the total SCN5A mRNA in failing hearts (Shang et al. 2007). In addition, the two splicing factors RBM25 and LUC7L3 that regulate the E28C and E28D non‐functional splice variants of SCN5A were also upregulated in failing hearts (Gao et al. 2011). Thus, the altered sodium channel mRNA splicing is believed to contribute to the downregulation of cardiac sodium current (I Na), which in turn could play a role in the arrhythmogenic conduction slowing in HF. However, no evidence of slowed AP upstroke velocity in microelectrode or optical recordings has been presented so far, which raises questions about translation from ion channel, single cell to tissue level physiology.
Gap junctions serve as electric synapses for intercellular current flow, synchronizing excitation (Rohr, 2004). A gap junction is formed by head‐to‐head docking of two hemichannels (i.e. connexons) from adjacent cells, each composed of six connexin subunits, with connexin 43 (Cx43) being the most prevalent protein subunit in the human ventricle (Smyth et al. 2010). Functional gap junctions are localized predominantly in the intercalated disk region between the cells to allow for fast and efficient spread of cardiac activation (Severs et al. 2004). Gap junction protein expression, phosphorylation, and localization are essential for normal conduction (Rohr, 2004). Cx43 expression is reduced at both mRNA and protein levels in end‐stage human HF due to both ischaemic and dilated cardiomyopathy (Dupont et al. 2001). Gap junctions are also improperly distributed from intercalated disks to lateral cell borders. The disruption of microtubule trafficking caused by oxidative stress is believed to be responsible for the improper localization of Cx43 (Smyth et al. 2010).
An optical mapping investigation of transmural activation in failing human left ventricle (LV) showed significantly reduced conduction velocities in comparison with non‐failing hearts (Fig. 1 A) (Glukhov et al. 2012). Cx43 levels of protein expression and phosphorylation were also uniformly reduced in failing hearts compared with non‐failing hearts (Fig. 1 B); a trend that paralleled the conduction velocity changes. Interestingly, there is a significant gradient of conduction velocity and Cx43 expression from endocardium to epicardium, in both donor and failing hearts. Moreover, the most pronounced differences in Cx43 downregulation and cellular lateralization were found in the subepicardium (Fig. 1 C and D). Thus, the downregulation, lateralization, and de‐phosphorylation of Cx43 may contribute to arrhythmogenic slowing of conduction in failing human hearts.
Figure 1. Downregulation of connexin 43 (Cx43) leads to slowing of conduction velocity in heart failure (HF) .
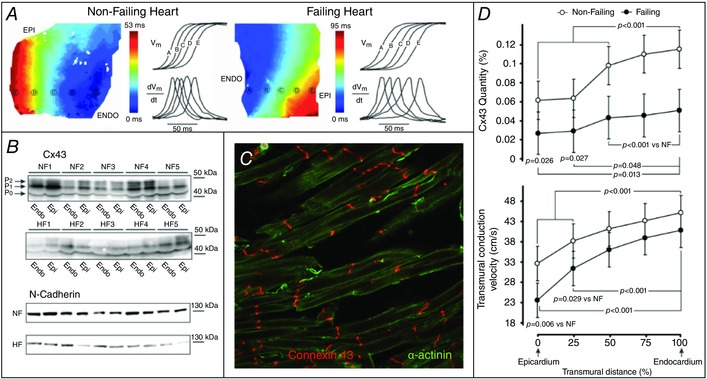
A, typical examples of transmural activation in non‐failing (NF) and failing human wedge preparations. Colour‐coded isochronal maps show activation from endocardial (ENDO) to epicardial (EPI) surfaces, with optical action potential (OAP) upstrokes and their derivatives from equally spaced sites presented next to the maps. B, Western blot analysis of EPI and ENDO expression of Cx43 and N‐cadherin. Different phosphorylated bands for Cx43 protein were observed: the higher‐molecular‐weight and higher phosphorylated isoforms (P1 and P2) and the lower‐molecular‐weight and dephosphorylated isoform (P0). C, reduced expression of Cx43 in failing human cardiomyocytes. D, top: average Cx43 quantity measured from transmural muscle layers spanning from EPI to ENDO. D, bottom: transmural conduction velocity (CV) measured at different transmural layers. Adapted from Glukhov et al. (2012).
Excitation–contraction coupling remodelling
Cardiac EC coupling is a process whereby the electrical excitation of the cardiomyocytes triggers the mechanical contraction of the myocardium (Bers, 2002). The key components of EC coupling include a recyclable pool of intracellular Ca2+, the velocity of Ca2+ recycling, and the mechanisms that trigger Ca2+ release from the sarcoplasmic reticulum (SR) (Sjaastad et al. 2003). Depressed contractility in failing human hearts is associated with altered EC coupling in general and with calcium handling in particular (Piacentino et al. 2003).
Molecular studies have previously reported significantly reduced protein expression of the sarcoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a) in the subendocardium compared with the subepicardium in failing human hearts (Prestle et al. 1999). To investigate this further, we conducted dual optical mapping of membrane potential (V m) and calcium transient (CaT) in LV wedge preparations from both failing and non‐failing human hearts. Our study demonstrated altered transmural heterogeneities of EC coupling and calcium handling in HF characterized by (1) altered gradients in APD and CaT duration, (2) a slow component of upstroke and dome‐shaped plateau in CaT, and (3) a lowered level of SERCA2a expression at the subendocardium in failing human hearts with ischaemic cardiomyopathy (Fig. 2) (Lou et al. 2011). The morphological changes in CaT may be associated with asynchronous Ca2+ release within the cell. Alternately, increased Ca2+ entry during the AP plateau is caused by less calcium‐mediated inactivation of L‐type calcium currents (LTCCs) and increased activity of Na+–Ca2+ exchanger in the reverse mode. Taken together, the heterogeneous remodelling of EC coupling and calcium handling could contribute to arrhythmogenesis in HF by promoting early after depolarizations (EADs), which are especially evident during β2‐adrenergic stimulation (see below).
Figure 2. Left ventricular (LV) wedge preparation and optical recordings of action potentials (APs) and calcium transients (CaTs) .
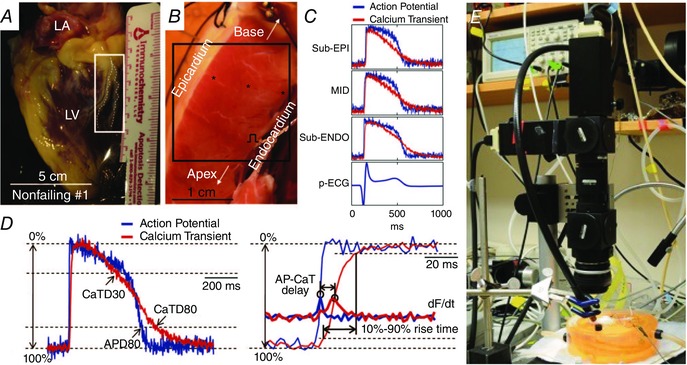
A, an explanted non‐failing human heart. The region indicated by the white rectangle was dissected and cannulated for wedge preparation. B, the left ventricular (LV) wedge preparation from the same heart. C, ECG and representative optical recordings of AP and CaT from locations within subendocardium (sub‐ENDO), midmyocardium (MID), and subepicardium (sub‐EPI), which are indicated by black stars in B. D, left panel: superimposed AP and CaT with illustrations of AP duration at 80% repolarization (APD80) and CaT duration at 30% and 80% recovery (CaTD30 and CaTD80). D, right panel: close‐up view of upstrokes (thin lines) and the derivatives (thick lines, labelled dF/dt) with illustrations of AP–CaT delay and 10% to 90% rise time of CaT. E, setup for simultaneous optical mapping of APs and CaTs. Adapted from Lou et al. (2011).
Repolarization remodelling
Prolongation of APD underlies QT prolongation on body surface ECGs in HF patients (Jin et al. 2008) and promotes arrhythmogenesis by triggered activity (e.g. early or delayed after depolarizations) or, if heterogeneous, by causing enhanced dispersion of repolarization that could lead to dynamic instability, conduction block, and functional reentrant arrhythmia (Wit & Rosen, 1983). Thus, there are many ionic currents and exchangers for which altered regulation can cause APD prolongation.
Optical mapping of human hearts has shown that APD prolongation in failing hearts as compared to non‐failing hearts occurs in a transmurally heterogeneous manner, i.e. dispersion of repolarization, with the greatest prolongation occurring in the subepicardial layer (Glukhov et al. 2010). In non‐failing hearts, as in animal models of HF, we found a significant decrease in APD from endocardium to epicardium. However, in contrast with animal models of HF, we found that the APD gradient was significantly decreased, rather than increased, in failing compared to non‐failing human hearts, leading to transmural APD homogeneity (Fig. 3) (Glukhov et al. 2010). Interestingly, the downregulation of Cx43 in HF (Glukhov et al. 2012) suggests that the reduced gap junctional cell‐to‐cell coupling does not necessarily lead to increased APD heterogeneity, as would have been expected. Thus, the APD homogeneity in failing human hearts may be associated with remodelling of other ion channels or membrane transporters responsible for repolarization.
Figure 3. Optical mapping of activation and repolarization in LV wedge preparation .
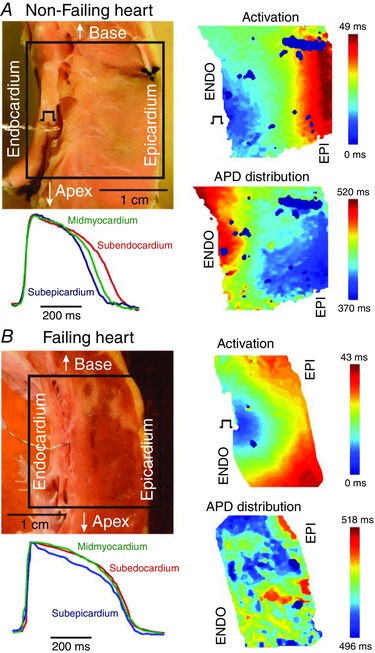
Transmural activation and APD distribution contour maps from non‐failing (A) and failing (B) human hearts are shown at pacing cycle length = 2000 ms. Optical fields of view are denoted by rectangles on corresponding photos. ENDO pacing sites are also marked. Colour scales represent the activation time and APD in corresponding maps. Selected EPI, MID, and ENDO APs are superimposed and demonstrated for each heart. Adapted from Glukhov et al. (2010).
The role of late sodium current activity in APD prolongation remains unclear. Maltsev et al. recorded late sodium current from LV myocytes isolated from donor and failing human hearts and suggested that the cardiac sodium channel isoform underlies this current (Maltsev et al. 1998). In addition, while the late I Na densities and biophysical properties were similar among both failing and non‐failing hearts, EADs from failing ventricular myocytes were observed and could be suppressed using tetrodotoxin. Conversely, Valdivia et al. demonstrated increased late I Na current in failing left ventricular myocytes when compared with normal hearts (Valdivia et al. 2005). Investigation into the biophysical mechanism for late I Na in failing ventricular cells showed that failing ventricular cells demonstrated sodium channel openings with slower inactivation and longer burst durations than those in normal ventricular cells, which are associated with greater late I Na in HF (Undrovinas et al. 2002).
The role of transient outward current (I to) density in determination of APD in the human heart remains an active area of research. Several studies have found I to to be downregulated in human HF (Beuckelmann et al. 1993). Moreover, in non‐failing hearts, I to density was shown to be fourfold lower in subendocardial compared to subepicardial cells, whereas I to in failing human hearts was reduced relative to cells from non‐failing hearts only in the subepicardial region (Wettwer et al. 1994). Even so, the role of I to in the remodelling of APD remains somewhat unclear due to its primary role during phase 1 of the AP. Some have suggested that reduced I to may lead to decreased calcium influx and abbreviated APs (Coraboeuf et al. 1998). Others have suggested that I to would have minimal impact on APD in large mammals (Wang & Hill, 2010).
Delayed rectifier potassium currents have been challenging to evaluate in human isolated cardiomyocytes due to small current amplitudes (Veldkamp et al. 1995). Moreover, the amount of delayed rectifier potassium currents was shown to be dependent on cell isolation procedure (Li et al. 1996, Yue et al. 1996). Previous gene expression studies did not reveal any differences in KCNH2 or KCNQ1 mRNA expression, which encode α‐subunits of rapidly activating potassium current (I Kr) and slowly activating potassium current (I Ks), respectively (Watanabe et al. 2007). However, mRNA expression for the delayed rectifier accessory subunit, KCNE1, was upregulated, which could potentially alter I Ks activation kinetics. We recently investigated I Kr expression pharmacologically in non‐failing and failing human hearts via optical mapping of coronary‐perfused LV wedge preparations (Holzem et al. 2015). Prolongation of transmural APD with I Kr blockade was significantly less in failing hearts compared to non‐failing hearts, suggesting that functional expression of I Kr is reduced in failing hearts (Fig. 4). The functional downregulation may be due to the disruption of the cell surface protein expression (mature hERG1a), contributing to a stoichiometric shift between hERG1a and hERG1b at the protein level. This shift results from a reduction in both mature and immature isoforms of hERG1a, along with an increase in hERG1b. Even so, more detailed investigations, particularly of I Ks are needed in the human heart.
Figure 4. Influence of rapidly activating potassium current (IKr) on repolarization in the human failing heart: action potential duration reduction (ΔAPD) values with IKr blockade .
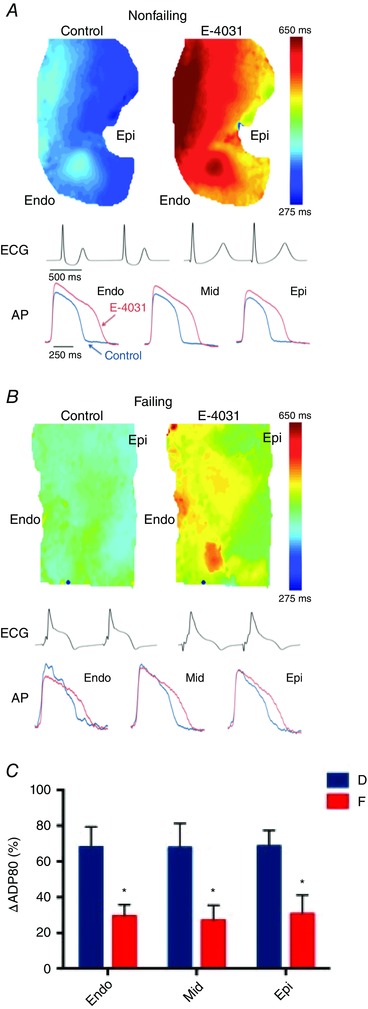
A and B, representative action potential duration (APD) maps, optical recording traces, and ECGs for donor (non‐failing, A) and failing (B) hearts. C, ΔAPDs, expressed as a 1% control for 1000 ms pacing cycle lengths (PCLs). Data are expressed as means ± SEM. Adapted from Holzem et al. (2015).
Inward rectifier potassium current densities are reduced in human HF (Beuckelmann et al. 1993). Moreover, the single‐channel properties were found to be different between inwardly rectifying potassium current (I K1) channels isolated from hearts with ischaemic versus dilated cardiomyopathy suggesting aetiology‐dependent regulation of I K1 (Koumi et al. 1995). Even so, the molecular mechanism for inward rectifier current downregulation in failing human hearts remains unclear. I K1 does not appear to be transcriptionally regulated, as Kir2.1 mRNA, which encodes the I K1 α‐subunit, does not appear to be downregulated in human HF (Wang et al. 1998).
The sodium–calcium exchanger (NCX) serves as an electrogenic pump, allowing three sodium ions to be transported into the cell in exchange for pumping one calcium ion out of the cell, in forward mode. Thus NCX is important for calcium handling and may also regulate APD (Armoundas et al. 2003). NCX protein levels have been reported to either increase or remain unchanged in the failing human hearts (Reinecke et al. 1996, Hasenfuss & Pieske, 2002). It was demonstrated that NCX current direction shifted from forward mode (i.e. calcium efflux and sodium influx) to reverse mode (i.e. calcium influx and sodium efflux) during the AP plateau in failing human heart, due to increased intracellular sodium and reduced intracellular calcium concentration (Weber et al. 2003). In reverse mode, NCX is likely to hyperpolarize the cell, abbreviating the AP. Thus, it is not yet clear what role NCX plays in APD prolongation in human HF. Recently, we reported that remodelling of β2‐adrenergic receptors includes upregulation and dephosphorylation of the receptor, which switches downstream coupling from Gi to Gs isoforms of G‐protein coupled signalling. This switch leads to loss of adrenergic control over NCX, which is coupled to adrenergic system via Gi (see below).
Gender differences in gene expression
Epidemiologically, there are well‐established gender differences in the manifestations of cardiac arrhythmias. Men are more susceptible to atrial fibrillation (Wolbrette et al. 2002), whereas women have higher incidence of long‐QT syndrome (LQTS) and drug‐induced Torsades de Pointes (Abi‐Gerges et al. 2004). In addition, men and women exhibit differences in basic electrophysiological properties during normal sinus rhythm (Burke et al. 1996). However, less is known about the molecular mechanisms underlying the observed gender differences in arrhythmia susceptibility in the human heart. We recently investigated the impact of gender on the transcript expression levels of the major cardiac ion channels, calcium handling proteins, and transcription factors in failing and non‐failing human hearts. We found gender‐specific differences in relative expression levels of ion channel subunits, such as Kv4.3, KChIP2, Kv1.5, and Kir3.1 in the left atrium (LA), but no significant gender specific differences in relative expression levels of these subunits in the LV (Ambrosi et al. 2013). Analyses of transmural heterogeneities in transcript expression levels in the LV revealed a heterogeneity of expression across the LV wall in the I to,slow pore‐forming subunit, Kv1.4, as well as with the I to,fast accessory subunit, KChIP2. In the non‐failing heart, Kv1.4 exhibited higher expression in the epicardium of the non‐failing heart, whereas KChIP2 exhibited stronger expression in the epicardium across gender and disease state.
Metabolic remodelling
Neubauer compared the failing human heart to an engine out of fuel. Each day the heart beats 100,000 times, moving approximately 10 tons of blood and burning through 20 to 30 times its own weight in ATP (Neubauer, 2007). The constant and consistent metabolic requirements of this task make the heart especially sensitive to perturbations in just‐in‐time substrate delivery, energy production, and utilization. While the most immediate effect of such perturbations are associated changes in contractility, an increasing body of research suggests that they have the potential to affect electrophysiological function as well (Ogbaghebriel & Shrier, 1994, Chantawansri et al. 2008, Watanabe et al. 2011).
Metabolic remodelling of conduction
It is well established that cardiac Na+ channels (NaV1.5) are downregulated in response to various types of cardiac injury (Ufret‐Vincenty et al. 2001, Valdivia et al. 2005). Such downregulation results in slowing of conduction velocity, possible conduction block, and increased arrhythmia risk. In similar fashion, it has also been shown that increases in intracellular concentrations of NADH and reactive oxygen species (ROS) are associated with myocardial injury (Di Lisa et al. 2001, Aon et al. 2003). In 2010, Liu et al. hypothesized that elevated NADH can downregulate I Na by activating protein kinase C (PKC), which in turn causes enhanced ROS production and I Na reduction. Administration of NAD+ garnered opposing results through activation of protein kinase A (PKA), inhibition of ROS production, and subsequent increase in I Na. In order to test the implications of this signalling pathway in pathologies like heart failure, similar experiments were conducted in human LV wedge preparations dissected from failing hearts. Treatment of human myopathic LV wedge preparation with NAD+ resulted in significant improvements in conduction velocity measured via optical mapping (Fig. 5) (Liu et al. 2013) Clinically, this type of improvement in HF patients could reduce the risk of conduction block and potential arrhythmias (Chen et al. 1998; Schott et al. 1999; Benson et al. 2003).
Figure 5. Metabolic changes in conduction velocity (CV) via cardiac sodium current (INa) .
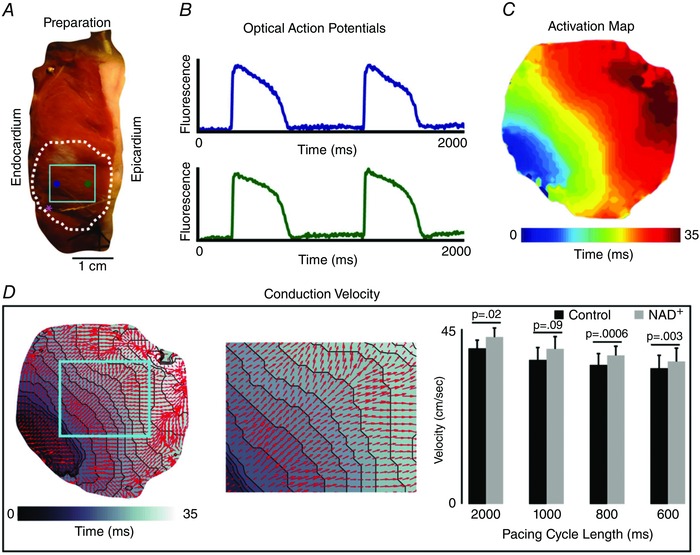
A, representative LV wedge preparation. These preparations provide a transmural view of the LV wall for optical mapping experiments. The dashed white line indicates the region where CV is measured and the solid light blue line indicates the magnified field of view (seen in D). The blue and green dots are the representative optical APs (seen in B). The pink asterisk is the site of the pacing stimulus. B, example optical APs from sub‐ENDO (blue) and sub‐EPI (green). C, a map of activation showing propagation away from the pacing site in panel A. D, left panel: vectors representing magnitude of conduction velocity overlaid on grey scale activation map. D, centre panel: region of conduction velocity used in quantifying changes induced by addition of NAD+. D, right panel: comparison of average conduction velocity of failing hearts before and after application of 500 μm NAD+. Measured at pacing cycle lengths of 2000, 1000, 800, and 600 ms.
Metabolic remodelling of ischaemia–reperfusion
Although the susceptibility of failing hearts to proarrhythmic remodelling induced by ischaemia and reperfusion (I–R) has long been established (Bril et al. 1991; Furukawa et al. 1993), studies of the relationship between I–R and arrhythmia in human failing myocardium have only recently begun. As a result, the mechanism by which I–R enhances arrhythmic risk remains unclear. Recently, we showed that I–R effects in failing hearts are exaggerated, with significantly greater APD shortening and conduction slowing during ischaemia, and incomplete recovery of these measures with subsequent reperfusion (Ng et al. 2014). These changes in electrophysiology have severe implications, especially when viewed in the context of regional ischaemia that would cause increased heterogeneity of conduction and APD across the ischaemic border zone. In conjunction with the electrophysiological studies, 46 genes important in cardiac metabolism were probed, 10 of which were significantly downregulated. These included genes responsible for transport of metabolic substrates, mitochondrial function, and the purine metabolism pathway. While associational, these results contribute to a growing body of research that theorizes that metabolic remodelling could play an important role in arrhythmia susceptibility.
Ultrastructural remodelling of myocytes
The spatial organization of the cardiac myocyte is optimized for just‐in‐time delivery of ATP and Ca2+ to relevant sub‐cellular structures. In this arrangement, transverse tubules (t‐tubules) penetrate the densely packed, intertwined ultrastructure of various organelles to deliver these substrates to the sarcomeres, mitochondria, and sarcoplasmic reticula. While the role of t‐tubules in the delivery of ATP and Ca2+ is well understood, their implication for metabolic function is unknown. Using dual‐beam electron microscopy we were able to create 3D reconstructions of a healthy cardiac myocyte at a nanometer resolution in x, y, and z coordinates (Fig. 6) (Sulkin et al. 2014). Dispersed throughout the ultrastructure are lipid droplets that store fatty acids used in ATP production via glycolysis and β‐oxidation. Upon close inspection, we discovered the majority of lipid droplets located beneath the sarcolemma co‐localized near t‐tubules. Interestingly, therapeutic drugs that target fatty acid metabolism in HF have been largely ineffective (Halbirk et al. 2010; Lionetti et al. 2011). Additionally, t‐tubule degradation (detubulation) is a well‐known side‐effect of HF (Lyon et al. 2009; Ibrahim et al. 2012). The co‐localization of lipid droplets and t‐tubules provides the foundation for our hypothesis that delivery of lipid droplets to the sarcomeric space is dependent upon trafficking through t‐tubules. Thus, detubulation of failing myocytes impedes just‐in‐time delivery of fatty acids to mitochondria located far from the sarcolemma. Therefore, the well‐known metabolic switch from fatty acid to glucose pathways in HF could be explained by the delivery problem rather than mitochondrial degradation (see below). This theory would explain the inability of therapeutic drugs to increase fatty acid metabolism and could also explain the transition of failing hearts from fatty acid metabolism to primarily glucose metabolism (Sack et al. 1996; Osorio et al. 2002).
Figure 6. Ultrastructure nano‐imaging of a human cardiomyocyte .

The sectioned volume is 14.7 μm × 14.8 μm × 2.2 μm and the cardiomyocyte was sectioned in 10 nm slices. A, transverse tubules (blue). B, A‐band (green). C, lipid droplets (yellow). D, mitochondria (red). Adapted from Sulkin et al. (2014).
β‐Adrenergic receptor signalling
From the 1970s onward, β‐AR blockers have become a recommended treatment for most patients with HF (Lindenfeld et al. 2010). In addition to reducing mortality and morbidity, they have been demonstrated to protect against cardiac arrhythmia and arrest, and reverse ventricular remodelling (Lechat et al. 1998; Singh, 2005). Their use is founded on the well‐established observation that β‐AR stimulation is increased in human HF (Lohse, 2003).
β‐ARs are linked to clear changes at the molecular and cellular levels of human HF. In isolated LV myocytes from HF patients, increased β2‐AR stimulation alters normal SR function, enhancing spontaneous SR Ca2+ release and after‐contractions (DeSantiago et al. 2008). At the same time, in comparison to healthy human myocardium, failing human myocardium has a markedly decreased number of β1‐ARs, resulting in relatively higher proportion of β2‐ARs (Bristow et al. 1986). This finding is recapitulated by a statistically significant decrease in β1‐AR density within failing LV subendocardium (Beau et al. 1993). The mechanism behind the relative desensitization β1‐ARs and relative sensitization of β2‐ARs in HF remains unclear. One possibility is that the sensitization of β2‐ARs in HF results from a switch from coupling with inhibitory regulative G‐protein (Gi) to stimulative regulative G‐protein (Gs) (Fig. 7) (Lang et al. 2015). This view is supported by increased phosphorylation of β2‐AR protein, presumably by catalytic (C) subunit cAMP‐dependent protein kinase (PKA), and abrogation of increased NCX activity (Fig. 8 E) (Lang et al. 2015).
Figure 7. Switch from inhibitory regulative G‐protein (Gi) to stimulative regulative G‐protein (Gs) coupling .
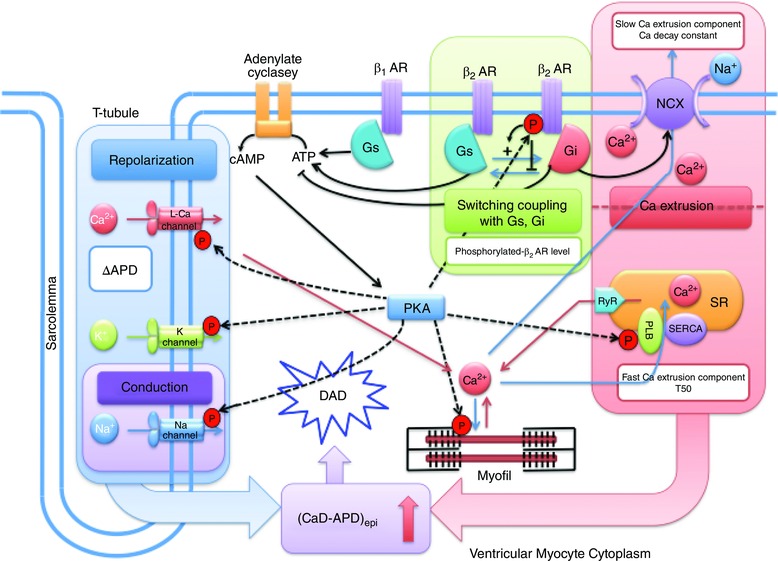
In human HF, stimulation of β2 adrenergic receptors (β2‐ARs) leads to a switch in G‐protein coupled signalling, from Gi to Gs. Loss of control over the sodium–calcium exchanger (NCX) results. Adapted from Lang et al. (2015).
Figure 8. Effect of β‐adrenergic receptor (β‐AR) stimulation on repolarization, protein expression, and protein phosphorylation .
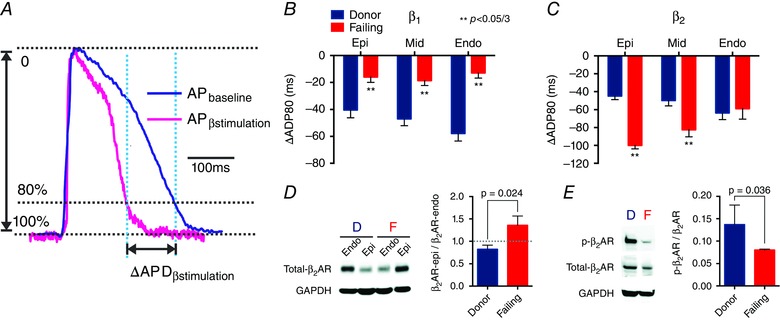
A, AP signals illustrate that the difference between APDbaseline − APDβ‐stimulation is ΔAPD. B, statistical analysis of ΔAPD shows desensitization of β1‐AR at the sub‐EPI, MID, and sub‐ENDO. C, statistical analysis of ΔAPD shows sensitization of β2‐AR at the sub‐EPI and MID but not at the sub‐ENDO. B and C, negative values indicate a reduction in APD. D, Western blot reveals a reversal of the sub‐EPI to sub‐ENDO gradient of β2‐AR protein expression (β2‐AR‐epi/β2‐AR‐endo) in HF. E, phosphorylation of β2‐AR is reduced in HF. D, donor hearts; F, failing hearts. Adapted from Lang et al. (2015).
HF also alters the relative mechanical contributions of β1‐ARs and β2‐ARs. Early studies by Bristow et al. (1986) established that β1‐AR downregulation in failing human myocardium is associated with reduced inotropic response, while β2‐ARs nearly retain their level of inotropic activity in this context. Thus, in conjunction with shifts in abundance of β‐AR subpopulations, the overall contribution to contractile function changes from being predominantly driven by β1‐ARs to β2‐ARs.
Our recent research has revealed differential roles of β1‐ and β2‐ARs in electrophysiological remodelling in human HF, suggesting that increased β2‐AR activity is more arrhythmogenic than increased β1‐AR activity (Lang et al. 2015). This is illustrated by different spatial patterns of action potential duration reduction (ΔAPD). In failing hearts, β1‐AR stimulation leads to a modest, spatially uniform ΔAPD (Fig. 8 B). However, β2‐AR stimulation in failing hearts reversed the endocardial‐to‐epicardial ΔAPD gradient, resulting in increasing ΔAPDs from endocardium to epicardium (Fig. 8 C). Transmural gradients of β2‐AR protein expression in failing hearts mirror this spatial shift, and may well underlie it (Fig. 8 D).
There are also subtype‐dependent shifts in anisotropy (defined as the ratio of longitudinal conduction velocity to transverse conduction velocity), and in the occurrence of arrhythmias, namely premature ventricular contraction (PVC) and ventricular tachycardia (VT) (Lang et al. 2015). While β1‐AR stimulation decreased anisotropy in failing hearts, β2‐AR stimulation increased it, indicating that conduction velocity was much higher in the longitudinal direction than in the transverse direction, and potentially increasing the likelihood of reentrant arrhythmia. Moreover, stimulation of β2‐ARs, not β1‐ARs, increased the number and frequency of spontaneous PVCs via Ca2+ overload and enhanced automaticity. β2‐AR stimulation is also more potent in promoting VT in heart failure by increasing the transmural difference between CaTD and APD (CaTD−APD) period responsible for delayed afterdepolarization (DAD). These findings, in addition to the previously suggested molecular and cellular findings, strongly suggest that selective β2‐AR antagonists may play an important role in future β‐AR blocker therapies.
New methodology
Optical mapping has evolved over the past two decades to become a very powerful and important experimental tool for understanding the spread of electrical activity in normal cardiac rhythm and arrhythmias (Efimov et al. 1994, 2004; Gutbrod et al. 2014). It is the gold standard for cardiac electrophysiological mapping and has revolutionized our understanding of the basic mechanisms of electrical activity, calcium homeostasis, and metabolism (Gutbrod et al. 2014; Lang et al. 2014). Still, while it takes the ‘integrative approach’ of holistically measuring physiological and pathological cardiac electrical activities at the tissue or organ level, optical mapping cannot fully explain the complexities of the heart's dynamic synchrony (Efimov, 2008). The ‘reductionist approach’ is an alternative method which dissects biological systems into their constituent parts and determines the connections (Van Regenmortel, 2004). Through advancements in materials science, 3D visualization techniques, and organotypic cultures, reductionism has created new outlets for strengthening the molecular paradigm of normal and abnormal cardiac cell electrophysiology. In the following section, we discuss three recent technological advances: (1) 3D‐MIMs, (2) CLARITY, and (3) organotypic cardiac cultures. These highlight alternative means for expanding our breadth of biological knowledge and therapeutic options for cardiac arrhythmias beyond optical mapping.
3D‐MIMs
We have established that HF is a condition in which the spatiotemporal synchrony of metabolic, electrophysiological, and mechanical processes is disturbed. In an effort to better understand these processes, extensive research has been done to develop tools for simultaneous spatiotemporal mapping of their underlying components. Various types of devices have been developed to this end, ranging from manually assembled epicardial interfacing sock electrodes (Harrison et al. 1980; Faris et al. 2003) to point‐contact catheters for serially mapping of the endocardial surface (Della Bella et al. 2012; de Chillou et al. 2014). Modalities have also been developed that utilize fluorescence (Efimov et al. 2004), ultrasound (D'hooge et al. 2000), and nuclear magnetic resonance (Chan et al. 2013). However, all of these methods have either lacked adequate spatial and temporal resolution or been unable to interface with the heart in a long‐term or chronic fashion.
3D multifunctional integumentary membranes (3D‐MIMs), developed on the platform of stretchable electronics, allow for multiparametric mapping capabilities in a conformal, high‐resolution manner to overcome such challenges (Kim et al. 2011; Kaltenbrunner et al. 2013). These circuits are lithographically arranged on thin elastic membranes which can conform to the heart surface in a manner similar to the heart's own pericardium (Xu et al. 2014). In this fashion, 3D‐MIMS provide complete surface contact over the entire epicardium. Initial results show excellent performance of sensors designed to measure electrical and mechanical activity, as well as factors like pH, light, and temperature (Fig. 9) (Xu et al. 2014). Devices built on this platform could be used to pinpoint and track the precursors and subsequent activity of pathological conditions like ischaemia, mechanical failure, and arrhythmia (Gutbrod et al. 2014). Future work will focus on development of biocompatibility and wireless data transfer to facilitate the fabrication of chronic diagnostic and therapeutic devices using 3D‐MIMs technology.
Figure 9. 3D multifunctional integumentary membranes (3D‐MIMs) for spatiotemporal measurement and stimulation of the heart .
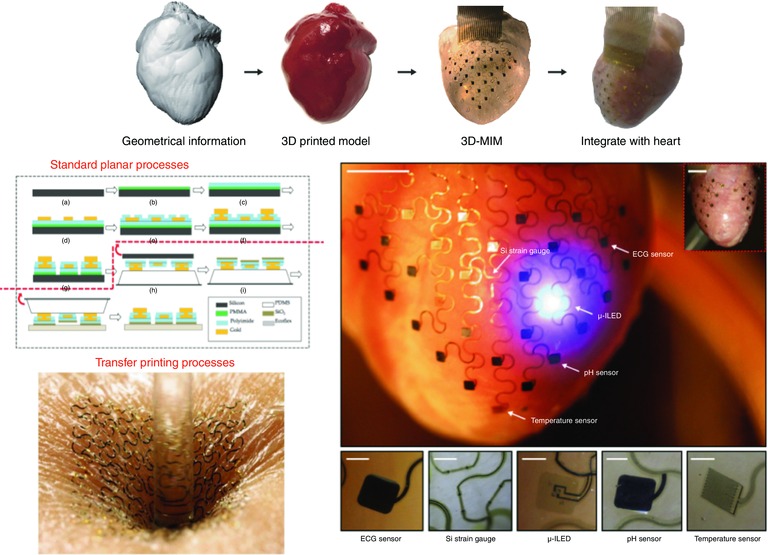
Top panel: graphical depiction of the sequential key steps involved in device design and fabrication. Left middle panel: process of pattern transfer by photolithography Left bottom panel: an ultrathin stretchable electronic circuit is mounted onto the epidermis and can comfortably bend, wrinkle, and stretch with the wearer's skin. Right middle panel: representative image of a 3D‐MIM integrated onto a Langendorff‐perfused rabbit heart, with white arrows pointing to various function elements of the device. Inset image shows how electronics can cover both anterior and posterior surfaces of the heart. Scale bars, 6 mm. Right bottom panel: magnified views of the various function elements in conformal contact with the epicardium. Images are recorded from the back side of the devices. Scale bars, 500 μm. Adapted from Xu et al. (2014).
CLARITY
One of the greatest challenges in biology is the ability to obtain detailed structural and molecular information from complex tissues and organs without losing sight of their functionality (Chung et al. 2013). Conventional methods for imaging intact 3D biological tissues include multiphoton microscopy, confocal microscopy, and optical coherence tomography (OCT). The penetration depths of these imaging modalities are currently limited by the optical scattering in the observed tissues. To overcome these drawbacks, advancements in tissue clearing methods have elucidated new ways to render complex tissues optically transparent while preserving 3D molecular and cellular structures, thereby facilitating deeper light penetration and intact tissue imaging without disassembly (Chung et al. 2013).
CLARITY is a novel optical clearing method developed by researchers at the Stanford University School of Medicine. Originally designed for murine neural tissues, this technique facilitates improved optical and molecular access and offers the ability to reconstruct detailed structural components of intact biological systems with high spatial resolution (Chung et al. 2013; Holzem, 2015). Given that it primarily involves hydrogel‐fixation of biological tissue and electrophoretic extraction of membrane lipids, CLARITY techniques can essentially be adopted for any biological tissue. Our lab has shown CLARITY techniques can be applied to clear whole mouse hearts and human heart tissue specimens of human hearts up to several millimetres thick (Fig. 10 A–C) (Holzem, 2015). Transparent human myocardial tissues were immunofluorescently labelled and successfully imaged with confocal and light sheet microscopy, showing that the structural integrity of cleared cardiac tissues remained intact (Fig. 10 D) (Holzem, 2015). We also demonstrated that the trabecular architecture of the human ventricular endocardium remains intact, with cellular histological differences allowing for identification of fine myofibril networks with high resolution. These findings potentially reveal new mechanisms for the conduction system to generate excitation in the myocardium. Though still in its infancy, CLARITY is a powerful imaging modality that has obvious benefits for further molecular analyses in the heart, thus helping to delineate the molecular and structural changes that occur before and after arrhythmogenesis.
Figure 10. Cardiac application of CLARITY .
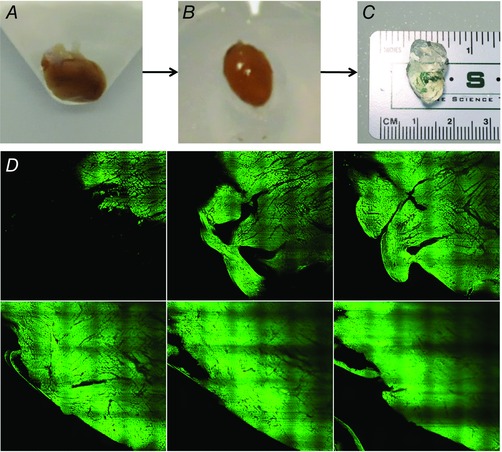
Representative images of whole mouse hearts through the CLARITY clearing process. A, hearts are perfused with a hydrogel monomer solution and polymerized to create a hydrogel–tissue hybrid. B, excess gel is removed as hearts are prepped for electrophoretic tissue clearing (ETC) treatment. C, tissues become fully transparent after the extraction of unbound lipid membranes. D, montage of high resolution light sheet images from human LV highlighting fibre architecture and trabeculation. Autofluorescence is sufficient for imaging.
Organotypic human heart slices
Historically, the investigation of human heart biology was primarily limited to either the molecular and cellular levels or to the clinical bedside, with less focus on physiological mechanisms manifesting only at the intermediate tissue and organ levels. Organotypic culture systems of intact human tissue slices constitute a unique approach to addressing this problem. Automated sectioning methods provide an ex vivo look at 3D cultures of healthy and diseased cardiac slices, and they have proven to be successful at mapping structures of complex tissues in several animal models (Wang et al. 2015).
We have optimized these techniques for human heart studies. In our lab, isolated wedge preparations of human atrial and ventricular slices of failing human intact tissue were cultured for up to 3 weeks. Acute dual (V m–Ca2+) optical mapping showed conduction velocity to be conserved in left ventricular slice preparations as compared to traditional preparations. For the first time, we were able to map AP and CaTs in human atrial slices, shedding new light on calcium dynamics of the human sinus node. Adenovirus‐based gene painting techniques were also performed on cardiac tissue slices as a proof of concept for future pre‐clinical validation of cardiac efficacy and toxicity studies. Virally encoded Green Fluorescent Protein (GFP) was expressed in harvested slices, showing maintenance throughout prolonged culture up to 96 h. Additionally, immunohistochemistry staining for connexin 43, the main gap junction protein isoform expressed in human ventricular tissue, displayed well‐organized gap junctions at the intercalated disc to be conserved during prolonged culture. Altogether, adult human slices effectively serve as a useful platform for providing clinically relevant therapeutic studies in adult human atrial working myocytes, as well as for evaluating heterogeneous physiological responses of the human heart to disease (Camelliti et al. 2011).
Limitations
Despite the knowledge gained from ex vivo studies of human HF, there remain considerable limitations inherent in the way they are currently conducted. End‐stage HF resists existing therapies aiming to slow, stop, or reverse HF progression and associated arrhythmogenic remodelling. Thus, end‐stage failing hearts may not be the best testing ground for novel therapies, which aim alter the course of the disease. Furthermore, management of HF is multifaceted, and depends on lifestyle, pharmacological agents, surgery, and device‐based therapy. The genetic and phenotypic changes as a result of HF management may subsequently compromise the degree to which these studies are able to infer the fundamental mechanisms of HF. Finally, HF progression and treatment involves proliferative signalling mechanisms, which cannot be studied in short‐term in vitro experiments. In part this limitation will be addressed in studies with novel organotypic human cardiac slice preparations and in donor hearts with early stage heart disease.
Conclusions
From the 1950s to the 2010s, we have enjoyed an unprecedented increase in life expectancy by a decade. (Marsh, 2014). Most of this increase is due to a significant decline in mortality from cardiovascular disease. Development of implantable electronic pacemakers and defibrillators, coronary artery imaging and catheterization, and other advances contributed to this dramatic increase in life expectancy. During the last half‐century, basic cardiovascular research has primarily adopted the reductionist approach, focusing on molecular and cellular mechanisms of heart disease. While biological approaches to develop pharmacological, gene, cell, and other biologic therapies are enticing, they have yet to result in viable treatments for heart rhythm, failure, metabolic and other heart disease fields. Translation from cellular and animal models of heart disease to the bedside is hampered by significant differences between species and physiological scales. We know more about the cell physiology of the zebra fish, mouse, rabbit, and guinea‐pig than we know about human cell physiology. The many failures to translate discoveries made in animal species to humans teach us that we need to better understand human cardiac pathophysiology. They show us that we need to establish translational platforms for safety and efficacy studies before we embark on costly and risky clinical trials. Our studies over the last 8 years show that hypotheses generated in animal models must be tested in human in vitro models, such as human tissue preparations.
Additional information
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by National Institutes of Health Grant 7R01HL114395, the Stanley and Lucy Lopata Endowment at Washington University in St Louis, and the Alisann and Terry Collins Endowment at The George Washington University.
Acknowledgements
We thank Maria Efimova for her assistance in preparing the manuscript.
Biography
Igor Efimov earned his MSc and PhD from Moscow Institute of Physics and Technology, and completed postdoctoral training at the University of Pittsburgh. He served on the faculty of the Cleveland Clinic Foundation and Case Western Reserve University in Cleveland, OH, and Washington University in St Louis, MO, prior to recently joining the George Washington University. There, Dr Efimov is the Alisann & Terry Collins Professor and Chairman of the Department of Biomedical Engineering and the Director of the Cardiac Imaging Laboratory. He has developed novel anti‐arrhythmia therapies and in 2008 founded Cardialen to develop low energy AF electrotherapy.

References
- Abi‐Gerges N, Philp K, Pollard C, Wakefield I, Hammond TG & Valentin J‐P (2004). Sex differences in ventricular repolarization: from cardiac electrophysiology to Torsades de Pointes. Fundam Clin Pharmacol 18, 139–151. [DOI] [PubMed] [Google Scholar]
- Ambrosi CM, Yamada KA, Nerbonne JM & Efimov IR (2013). Gender differences in electrophysiological gene expression in failing and non‐failing human hearts. PLoS One e54635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Marbán E & O'Rourke B (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278, 44735–44744. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL & O'Rourke B (2003). Role of sodium–calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 93, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beau SL, Tolley TK & Saffitz JE (1993). Heterogeneous transmural distribution of beta‐adrenergic receptor subtypes in failing human hearts. Circulation 88, 2501–2509. [DOI] [PubMed] [Google Scholar]
- Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH & George AL (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 112, 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Näbauer M & Erdmann E (1993). Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73, 379–385. [DOI] [PubMed] [Google Scholar]
- Bril A, Forest MC & Gout B (1991). Ischemia and reperfusion‐induced arrhythmias in rabbits with chronic heart failure. Am J Physiol Heart Circ Physiol 261, H301–H307. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P & Jamieson S (1986). Beta 1‐ and beta 2‐adrenergic‐receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1‐receptor down‐regulation in heart failure. Circ Res 59, 297–309. [DOI] [PubMed] [Google Scholar]
- Bruce CR, Blumenthal‐Barby JS & Meyers D (2015). Benefits and challenges of early introduction of left ventricular assist device placement: a patient‐centered perspective. J Am Coll Cardiol 66, 1762–1765. [DOI] [PubMed] [Google Scholar]
- Burke JH, Goldberger JJ, Ehlert FA, Kruse JT, Parker MA & Kadish AH (1996). Gender differences in heart rate before and after autonomic blockade: evidence against an intrinsic gender effect. Am J Med 100, 537–543. [DOI] [PubMed] [Google Scholar]
- Buxton AE, Lee KL, Fisher JD, Josephson ME, Prystowsky EN & Hafley G (1999). A randomized study of the prevention of sudden death in patients with coronary artery disease. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med 341, 1882–1890. [DOI] [PubMed] [Google Scholar]
- Camelliti P, Al‐Saud SA, Smolenski RT, Al‐Ayoubi S, Bussek A, Wettwer E, Banner NR, Bowles CT, Yacoub MH & Terracciano CM (2011). Adult human heart slices are a multicellular system suitable for electrophysiological and pharmacological studies. J Mol Cell Cardiol 51, 390–398. [DOI] [PubMed] [Google Scholar]
- Chan KWY, Liu G, Song X, Kim H, Yu T, Arifin DR, Gilad AA, Hanes J, Walczak P, van Zijl PCM, Bulte JWM & McMahon MT (2013). MRI‐detectable pH nanosensors incorporated into hydrogels for in vivo sensing of transplanted‐cell viability. Nat Mater 12, 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantawansri C, Huynh N, Yamanaka J, Garfinkel A, Lamp ST, Inoue M, Bridge JHB & Goldhaber JI (2008). Effect of metabolic inhibition on couplon behavior in rabbit ventricular myocytes. Biophys J 94, 1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Normand S‐LT, Wang Y & Krumholz HM (2011). National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998–2008. JAMA 306, 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz‐Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze‐Bahr E, Keating MT, Towbin JA & Wang Q (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. [DOI] [PubMed] [Google Scholar]
- Chung K, Wallace J, Kim S‐Y, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, Pak S, Bernstein H, Ramakrishnan C, Grosenick L, Gradinaru V & Deisseroth K (2013). Structural and molecular interrogation of intact biological systems. Nature 497, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraboeuf E, Coulombe A, Deroubaix E, Hatem S & Mercadier JJ (1998). [Transient outward potassium current and repolarization of cardiac cells]. Bull Acad Natl Med 182, 325–333. [PubMed] [Google Scholar]
- Coronel R, Wilders R, Verkerk AO, Wiegerinck RF, Benoist D & Bernus O (2013). Electrophysiological changes in heart failure and their implications for arrhythmogenesis. Biochim Biophys Acta 1832, 2432–2441. [DOI] [PubMed] [Google Scholar]
- de Chillou C, Groben L, Magnin‐Poull I, Andronache M, MagdiAbbas M, Zhang N, Abdelaal A, Ammar S, Sellal J‐M, Schwartz J, Brembilla‐Perrot B, Aliot E & Marchlinski FE (2014). Localizing the critical isthmus of postinfarct ventricular tachycardia: the value of pace‐mapping during sinus rhythm. Heart Rhythm 11, 175–181. [DOI] [PubMed] [Google Scholar]
- Della Bella P, Bisceglia C & Tung R (2012). Multielectrode contact mapping to assess scar modification in post‐myocardial infarction ventricular tachycardia patients. Europace 14 (Suppl. 2), ii7–ii12. [DOI] [PubMed] [Google Scholar]
- DeSantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM & Pogwizd SM (2008). Arrhythmogenic effects of 2‐adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res 102, 1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'hooge J, Heimdal A, Jamal F, Kukulski T, Bijnens B, Rademakers F, Hatle L, Suetens P & Sutherland GR (2000). Regional strain and strain rate measurements by cardiac ultrasound: principles, implementation and limitations. Eur J Echocardiog 1, 154–170. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabò R, Canton M, Barile M & Bernardi P (2001). Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276, 2571–2575. [DOI] [PubMed] [Google Scholar]
- Doyle MBM, Vachharajani N, Wellen JR, Lowell JA, Shenoy S, Ridolfi G, Jendrisak MD, Coleman J, Maher M, Brockmeier D, Kappel D & Chapman WC (2014). A novel organ donor facility: a decade of experience with liver donors. Am J Transplant 14, 615–620. [DOI] [PubMed] [Google Scholar]
- Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH & Severs NJ (2001). Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol 33, 359–371. [DOI] [PubMed] [Google Scholar]
- Efimov IR (2008). Nature versus nurture in cardiac conduction: toward integrative paradigm of cardiac tissue engineering. Circ Res 103, 119–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov IR, Fedorov VV, Glukhov A, Lou Q, Ambrosi C, Janks D, Hucker WJ, Kurian T, Schuessler RB & Moazami N (2010). Multiscale imaging of the human heart: Building the foundation for human systems physiology and translational medicine. Conf Proc IEEE Eng Med Biol Soc 2010, 5177–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov IR, Huang DT, Rendt JM & Salama G (1994). Optical mapping of repolarization and refractoriness from intact hearts. Circulation 90, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Efimov IR, Nikolski VP & Salama G (2004). Optical imaging of the heart. Circ Res 95, 21–33. [DOI] [PubMed] [Google Scholar]
- Faris OP, Evans FJ, Ennis DB, Helm PA, Taylor JL, Chesnick AS, Guttman MA, Ozturk C & McVeigh ER (2003). Novel technique for cardiac electromechanical mapping with magnetic resonance imaging tagging and an epicardial electrode sock. Ann Biomed Eng 31, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Bassett AL, Furukawa N, Kimura S & Myerburg RJ (1993). The ionic mechanism of reperfusion‐induced early afterdepolarizations in feline left ventricular hypertrophy. J Clin Invest 91, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Xie A, Huang S‐C, Zhou A, Zhang J, Herman AM, Ghassemzadeh S, Jeong E‐M, Kasturirangan S, Raicu M, Sobieski MA, Bhat G, Tatooles A, Benz EJ, Kamp TJ & Dudley SC (2011). Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation 124, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter KA, Young JN & McElvany MD (2006). Implantable cardioverter‐defibrillators: a new preventive medical option. Prev Cardiol 9, 49–53; quiz 54–5. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, Schuessler RB, Moazami N & Efimov IR (2012). Conduction remodeling in human end‐stage nonischemic left ventricular cardiomyopathy. Circulation 125, 1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N & Efimov IR (2010). Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Re. 106, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutbrod SR, Sulkin MS, Rogers JA & Efimov IR (2014). Patient‐specific flexible and stretchable devices for cardiac diagnostics and therapy. Prog Biophys Mol Biol 115, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbirk M, Nørrelund H, Møller N, Schmitz O, Gøtzsche L, Nielsen R, Nielsen‐Kudsk JE, Nielsen SS, Nielsen TT, Eiskjær H, Bøtker HE & Wiggers H (2010). Suppression of circulating free fatty acids with acipimox in chronic heart failure patients changes whole body metabolism but does not affect cardiac function. Am J Physiol Heart Circ Physiol 299, H1220–H1225. [DOI] [PubMed] [Google Scholar]
- Harrison L, Ideker RE, Smith WM, Klein GJ, Kasell J, Wallace AG & Gallagher JJ (1980). The sock electrode array: a tool for determining global epicardial activation during unstable arrhythmias. Pacing Clin Electrophysiol 3, 531–540. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G (1998). Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res 39, 60–76. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G & Pieske B (2002). Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34, 951–969. [DOI] [PubMed] [Google Scholar]
- Holzem KM (2015). Remodeling of Energetics and Electrophysiology in Cardiac Hypertrophy and Failure: from Function to Structure. Washington University in St. Louis.
- Holzem KM, Gomez JF, Glukhov AV, Madden EJ, Koppel AC, Ewald GA, Trenor B & Efimov IR (2015). Reduced response to IKr blockade and altered hERG1a/1b stoichiometry in human heart failure. J Mol Cell Cardiol DOI: http://dx.doi.org/10.1016/j.yjmcc.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ & Koch W (2012). Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 111, 131–150. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Navaratnarajah M, Siedlecka U, Rao C, Dias P, Moshkov AV, Gorelik J, Yacoub MH & Terracciano CM (2012). Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca2+‐induced Ca2+ release in a rodent model of heart failure. Eur J Heart Fail 14, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Lyon AR & Akar FG (2008). Arrhythmia mechanisms in the failing heart. Pacing Clin Electrophysiol 31, 1048–1056. [DOI] [PubMed] [Google Scholar]
- Kaese S, Frommeyer G, Verheule S, van Loon G, Gehrmann J, Breithardt G & Eckardt L (2013). The ECG in cardiovascular‐relevant animal models of electrophysiology. Herzschrittmacherther Elektrophysiol 24, 84–91. [DOI] [PubMed] [Google Scholar]
- Kaltenbrunner M, Sekitani T, Reeder J, Yokota T, Kuribara K, Tokuhara T, Drack M, Schwödiauer R, Graz I, Bauer‐Gogonea S, Bauer S & Someya T (2013). An ultra‐lightweight design for imperceptible plastic electronics. Nature 499, 458–463. [DOI] [PubMed] [Google Scholar]
- Khush KK, Zaroff JG, Nguyen J, Menza R & Goldstein BA (2015). National decline in donor heart utilization with regional variability: 1995–2010. Am J Transplant 15, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D‐H, Lu N, Ma R, Kim Y‐S, Kim R‐H, Wang S, Wu J, Won SM, Tao H, Islam A, Yu KJ, Kim T, Chowdhury R, Ying M, Xu L, Li M, Chung H‐J, Keum H, McCormick M, Liu P, Zhang Y‐W, Omenetto FG, Huang Y, Coleman T & Rogers JA (2011). Epidermal electronics. Science 333, 838–843. [DOI] [PubMed] [Google Scholar]
- Koumi S, Backer CL & Arentzen CE (1995). Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation 92, 164–174. [DOI] [PubMed] [Google Scholar]
- Lang D, Gutbrod SR, Laughner JI & Efimov IR (2014). Optical mapping of the heart In Manual of Research Techniques in Cardiovascular Research, 1st edn, eds Ardehali H, Bolli R. & Losordo DW, pp. 60–68. John Wiley & Sons, Incorporated, Somerset, NJ, USA. [Google Scholar]
- Lang D, Holzem K, Kang C, Xiao M, Hwang HJ, Ewald GA, Yamada KA & Efimov IR (2015). Arrhythmogenic remodeling of β2 versus β1 adrenergic signaling in the human failing heart. Circ Arrhythm Electrophysiol 8, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechat P, Packer M, Chalon S, Cucherat M, Arab T & Boissel JP (1998). Clinical effects of beta‐adrenergic blockade in chronic heart failure: a meta‐analysis of double‐blind, placebo‐controlled, randomized trials. Circulation 98, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Yue L, Carrier M & Nattel S (1996). Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res 78, 689–696. [DOI] [PubMed] [Google Scholar]
- Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WHW, Teerlink JR & Walsh MN (2010). HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 16, e1–194. [DOI] [PubMed] [Google Scholar]
- Lionetti V, Stanley WC & Recchia FA (2011). Modulating fatty acid oxidation in heart failure. Cardiovasc Res 90, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Gu L, Sulkin MS, Liu H, Jeong E‐M, Greener I, Xie A, Efimov IR & Dudley SC (2013). Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol 54, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ (2003). What is the role of ‐adrenergic signaling in heart failure?. Circ Res 93, 896–906. [DOI] [PubMed] [Google Scholar]
- Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG & Efimov IR (2011). Transmural heterogeneity and remodeling of ventricular excitation‐contraction coupling in human heart failure. Circulation 123, 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Q, Janardhan A & Efimov IR (2012). Remodeling of calcium handling in human heart failure. Adv Exp Med Biol 740, 1145–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE & Gorelik J (2009). Loss of T‐tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA 106, 6854–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M & Undrovinas AI (1998). Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98, 2545–2552. [DOI] [PubMed] [Google Scholar]
- Marsh B, Declining Lethality. The New York Times, 04‐Jan‐2014.
- Mosterd A & Hoes AW (2007). Clinical epidemiology of heart failure. Heart 93, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM & Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol Rev 85, 1205–1253. [DOI] [PubMed] [Google Scholar]
- Neubauer S (2007). The failing heart — an engine out of fuel. N Engl J Med 356, 1140–1151. [DOI] [PubMed] [Google Scholar]
- Ng FS, Holzem KM, Koppel AC, Janks D, Gordon F, Wit AL, Peters NS & Efimov IR (2014). Adverse remodeling of the electrophysiological response to ischemia‐reperfusion in human heart failure is associated with remodeling of metabolic gene expression. Circ Arrhythm Electrophysiol 7, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbaghebriel A & Shrier A (1994). Inhibition of metabolism abolishes transient outward current in rabbit atrial myocytes. Am J Physiol Heart Circ Physiol 266, H182–H190. [DOI] [PubMed] [Google Scholar]
- Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD & Recchia FA (2002). Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor‐alpha in pacing‐induced heart failure. Circulation 106, 606–612. [DOI] [PubMed] [Google Scholar]
- Piacentino V, Weber CR, Chen X, Weisser‐Thomas J, Margulies KB, Bers DM & Houser SR (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res 92, 651–658. [DOI] [PubMed] [Google Scholar]
- Poelzing S & Rosenbaum DS (2004). Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol 287, H1762–H1770. [DOI] [PubMed] [Google Scholar]
- Prestle J, Dieterich S, Preuss M, Bieligk U & Hasenfuss G (1999). Heterogeneous transmural gene expression of calcium‐handling proteins and natriuretic peptides in the failing human heart. Cardiovasc Res 43, 323–331. [DOI] [PubMed] [Google Scholar]
- Reinecke H, Studer R, Vetter R, Just H, Holtz J & Drexler H (1996). Role of the cardiac sarcolemmal Na+‐Ca2+ exchanger in end‐stage human heart failure. Ann N Y Acad Sci 779, 543–545. [DOI] [PubMed] [Google Scholar]
- Roger VL (2013). Epidemiology of heart failure. Circ Res 113, 646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr S (2004). Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res 62, 309–322. [DOI] [PubMed] [Google Scholar]
- Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O'Rourke B, Kass DA, Marbán E & Tomaselli GF (2005). Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol 288, H2077–H2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack MN, Rader TA, Park S, Bastin J, McCune SA & Kelly DP (1996). Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94, 2837–2842. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM & Le Marec H (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23, 20–21. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Coppen SR, Dupont E, Yeh H‐I, Ko Y‐S & Matsushita T (2004). Gap junction alterations in human cardiac disease. Cardiovasc Res 62, 368–377. [DOI] [PubMed] [Google Scholar]
- Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM & Dudley SC (2007). Human heart failure is associated with abnormal C‐terminal splicing variants in the cardiac sodium channel. Circ Res 101, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN (2005). Beta‐Adrenergic blockers as antiarrhythmic and antifibrillatory compounds: an overview. J Cardiovasc Pharmacol Ther 10 (Suppl. 1), S3–S14. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Wasserstrom JA & Sejersted OM (2003). Heart failure – a challenge to our current concepts of excitation–contraction coupling. J Physiol 546, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JW, Hong T‐T, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DYR, Chi NC & Shaw RM (2010). Limited forward trafficking of connexin 43 reduces cell‐cell coupling in stressed human and mouse myocardium. J Clin Invest 120, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickberger SA, Conti J, Daoud EG, Havranek E, Mehra MR, Piña IL & Young J (2005). Patient selection for cardiac resynchronization therapy: from the Council on Clinical Cardiology Subcommittee on Electrocardiography and Arrhythmias and the Quality of Care and Outcomes Research Interdisciplinary Working Group, in collaboration with the Heart Rhythm Society. Circulation 111, 2146–2150. [DOI] [PubMed] [Google Scholar]
- Sulkin MS, Yang F, Holzem KM, Van Leer B, Bugge C, Laughner JI, Green K & Efimov IR (2014). Nanoscale three‐dimensional imaging of the human myocyte. J Struct Biol 188, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Zicha S, Qi X‐Y, Kodama I & Nattel S (2006). Potassium channel subunit remodeling in rabbits exposed to long‐term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed‐rectifier changes. Circulation 113, 345–355. [DOI] [PubMed] [Google Scholar]
- Ufret‐Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE & Santana LF (2001). Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem 276, 28197–28203. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA, Kyle JW, Silverman N & Sabbah HN (2002). Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol 34, 1477–1489. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ & Makielski JC (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38, 475–483. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel MHV (2004). Reductionism and complexity in molecular biology. Scientists now have the tools to unravel biological and overcome the limitations of reductionism. EMBO Rep 5, 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp MW, van Ginneken AC, Opthof T & Bouman LN (1995). Delayed rectifier channels in human ventricular myocytes. Circulation 92, 3497–3504. [DOI] [PubMed] [Google Scholar]
- Wang K, Lee P, Mirams GR, Sarathchandra P, Borg TK, Gavaghan DJ, Kohl P & Bollensdorff C (2015). Cardiac tissue slices: preparation, handling, and successful optical mapping. Am J Physiol Heart Circ Physiol 308, H1112–H1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y & Hill JA (2010). Electrophysiological remodeling in heart failure. J Mol Cell Cardiol 48, 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yue L, White M, Pelletier G & Nattel S (1998). Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation 98, 2422–2428. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Arai M, Koitabashi N, Niwano K, Ohyama Y, Yamada Y, Kato N & Kurabayashi M (2011). Mitochondrial transcription factors TFAM and TFB2M regulate Serca2 gene transcription. Cardiovasc Res 90, 57–67. [DOI] [PubMed] [Google Scholar]
- Watanabe E, Yasui K, Kamiya K, Yamaguchi T, Sakuma I, Honjo H, Ozaki Y, Morimoto S, Hishida H & Kodama I (2007). Upregulation of KCNE1 induces QT interval prolongation in patients with chronic heart failure. Circ J 71, 471–478. [DOI] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, Houser SR & Bers DM (2003). Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108, 2224–2229. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Amos GJ, Posival H & Ravens U (1994). Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res 75, 473–482. [DOI] [PubMed] [Google Scholar]
- Wit AL & Rosen MR (1983). Pathophysiologic mechanisms of cardiac arrhythmias. Am Heart J 106, 798–811. [DOI] [PubMed] [Google Scholar]
- Wolbrette D, Naccarelli G, Curtis A, Lehmann M & Kadish A (2002). Gender differences in arrhythmias. Clin Cardiol 25, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Gutbrod SR, Bonifas AP, Su Y, Sulkin MS, Lu N, Chung H‐J, Jang K‐I, Liu Z, Ying M, Lu C, Webb RC, Kim J‐S, Laughner JI, Cheng H, Liu Y, Ameen A, Jeong J‐W, Kim G‐T, Huang Y, Efimov IR & Rogers JA (2014). 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat Commun 5, 3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Feng J, Li GR & Nattel S (1996). Transient outward and delayed rectifier currents in canine atrium: properties and role of isolation methods. Am J Physiol Heart Circ Physiol 270, H2157–H2168. [DOI] [PubMed] [Google Scholar]