

Abstract
The nervous system and cardiovascular system develop in concert and are functionally interconnected in both health and disease. This white paper focuses on the cellular and molecular mechanisms that underlie neural–cardiac interactions during development, during normal physiological function in the mature system, and during pathological remodelling in cardiovascular disease. The content on each subject was contributed by experts, and we hope that this will provide a useful resource for newcomers to neurocardiology as well as aficionados.
Abbreviations
- APD
action potential duration
- β‐AR
β‐adrenergic receptor
- AT II
angiotensin II
- CaM
calmodulin
- CaMKII
calmodulin‐dependent protein kinase II
- CGRP
calcitonin gene related peptide
- ChR2
channelrhodopsin‐2
- ICG
intrinsic cardiac ganglia
- NA
noradrenaline
- nNOS
nitric oxide synthase
- NGF
nerve growth factor
- NPY
neuropeptide Y
- PKA
protein kinase A
- RyR
ryanodine receptor
- SHR
spontaneously hypertensive rat
- SR
sarcoplasmic reticulum
- TH
tyrosine hydroxylase
- VIP
vasoactive intestinal peptide
Neural–cardiac interactions: co‐maturation in development
Autonomic control of the heart occurs through transmission by sympathetic and parasympathetic neurons, which have a common neural crest origin. The anlagen of cardiac parasympathetic ganglia first appear around the middle of embryonic development in rodents, and vagal input is already present as the immature neurons migrate to positions at the dorsal atrium (Hildreth et al. 2009). Parasympathetic neurons and surrounding satellite glial cells mature postnatally in rodents, and the neurons double in size as they develop connections with the myocardium (Fregoso & Hoover, 2012). The rodent sympathetic–cardiac circuit is established in the late prenatal and early postnatal period as growing sympathetic fibres innervate the developing heart (Hildreth et al. 2009). This can be considered as a co‐maturation system in which signals from cardiac tissue regulate the growth, patterning and transmission properties of the innervating sympathetic neurons, while reciprocal signalling from the nerve fibres influence the maturation of cardiac myocytes and adult cardiac properties.
The role of cardiac target‐derived factors as regulators of neuronal maturation has been well established. Cardiac‐derived neurturin is essential for the survival of cardiac parasympathetic neurons (Hiltunen et al. 2000; Mabe & Hoover, 2009), and cardiac signals control multiple events in sympathetic neuron maturation. Trophic signals that include nerve growth factor, neurotrophin‐3, and cytokines regulate outgrowth of sympathetic axons within the heart, continued survival upon reaching the target (Chun & Patterson, 1977; Landis & Keefe, 1983; Lockhart et al. 2000; Glebova & Ginty, 2005), regulation of neurotransmitter phenotypes (Yamamori et al. 1989; Slonimsky et al. 2003), and synaptic transmission (Lockhart et al. 1997; Luther & Birren, 2006, 2009; Hasan et al. 2012). Additional signals from cardiac tissue regulate developmental transitions between different growth states in sympathetic neurons. For example, contact with cardiac tissue results in a change in the neuronal response to bone morphogenetic proteins (BMPs), providing a stop‐growth signal for sympathetic axons that have reached their target (Moon & Birren, 2008). Interacting actions of neurotrophic factors (Habecker et al. 2008; Lorentz et al. 2010) and Sema3a signalling (Ieda et al. 2007) are also likely to act in the patterning of sympathetic fibres within the heart. Overall, a rich array of heart‐derived signals drives the maturation and function of the postganglionic sympathetic system.
While less is known of the effects of innervating sympathetic fibres on cardiac development, the development of a number of cardiac properties are regulated by sympathetic signals. Co‐culture with sympathetic neurons regulates the expression of angiotensin II and atrial natriuretic peptide (ANP) in developing cardiomyocytes (Hunt et al. 1995; Hansson et al. 2001). Additionally, sympathetic innervation has been linked to functional alterations in a number of important cardiac ionic currents, including Na+, L‐type Ca2+, pacemaker, inward rectifier and transient outward K+ currents (Qu & Robinson, 2004). Recent studies also suggest that sympathetic interactions play an important developmental role in regulating cardiomyocyte maturation during the postnatal period.
Questions and controversies
Can we exploit the fact that myocyte proliferation in pathological situations is influenced by the autonomic nervous system with new therapeutics or interventional treatments in man?
Is there a component of inherited structural and electrophysiological heart conditions that arises from dysfunctional autonomic control during cardiac development?
Does abnormal autonomic cardiovascular development contribute to human essential hypertension?
One way that early sympathetic innervation regulates cardiomyocyte development, and the properties of the adult heart, is through the regulation of cell cycle withdrawal and determination of cardiomyocyte cell number (Kreipke & Birren, 2015). While some organs are able to replenish cell number throughout life, the regenerative capacity of cardiomyocytes is, at best, limited (Zak, 1973). Cardiomyocytes withdraw from the cell cycle shortly after birth and, for the most part, remain quiescent throughout an organism's life. Following cell cycle withdrawal, heart size increases predominantly via cellular hypertrophy, or the enlargement of individual cardiomyocytes (Li et al. 1996; Ahuja et al. 2007). Thus, the number of postnatal cardiomyocyte divisions, and the timing of the developmental transition between proliferative and hypertrophic growth in the young animal are significant factors for determining the total number of cardiomyocytes in the adult heart.
The rapid transition of cardiomyocytes from a proliferative to a hypertrophic growth state (Li et al. 1996) corresponds to the postnatal period of sympathetic ingrowth into the heart (Hildreth et al. 2009). Recent evidence has shown that neonatal sympathectomy results in smaller hearts in developing and adult rats, demonstrating that early sympathetic innervation has long‐term effects on heart development (Kreipke & Birren, 2015). In vitro experiments using neonatal cardiomyocytes grown in the presence or absence of innervating sympathetic fibres demonstrated that sympathetic β‐adrenergic signalling prolonged the period of postnatal cardiomyocyte proliferation, delaying the period of hypertrophic growth. While, ultimately, the size of individual cardiomyocytes was not altered in adult animals that were sympathectomized at birth, the total number of these cells was reduced in the sympathectomized animals (Kreipke & Birren, 2015). This work shows that early sympathetic signalling leads to the development of larger hearts with more cardiomyocytes in vivo and suggests that sympathetic signalling is part of a homeostatic system that promotes cardiomyocyte proliferation and acts to set the final number of cardiomyocytes in the adult heart.
Sympathetic regulation of postnatal cardiomyocyte proliferation may also have implications for heart regeneration following cardiac damage. The neonatal mouse heart undergoes regeneration following tissue damage, a transient capability that is greatly diminished in older animals (Porrello et al. 2011; Bryant et al. 2015). Recent studies have shown that disruption of peripheral nerves blocks this regeneration. In one study, pharmacological inhibition of cholinergic signalling prevented cardiomyocyte proliferation following neonatal resection of the mouse ventricle (Mahmoud et al. 2015). This suggests a role for parasympathetic innervation in heart regeneration, a model supported by the finding that vagal nerve ablation also decreased cardiomyocyte proliferation following cardiac injury via resection or myocardial infarction (Mahmoud et al. 2015). Interestingly, another study showed that chemical sympathectomy in young animals also blocked early regeneration of damaged ventricular tissue, while increasing scarring in the tissue (White et al. 2015). While additional work defining the relative roles of cholinergic and noradrenergic signalling in the regeneration process is needed, together this work defines a new role for cardiac innervation in regenerative processes. Genetic programmes associated with proliferative cardiomyocytes are reactivated during pathological hypertrophy (Komuro et al. 1988; Ahuja et al. 2007), suggesting that these reciprocal interactions have implications for later processes in the cardiac system.
Questions and controversies
What are the best molecular targets to ensure optimal neural remodelling to promote myocyte survival and minimize the risk of arrhythmia post myocardial infarction and during chronic heart failure?
Normal neurotransmission and injury‐induced plasticity in cardiac nerves
The mature heart is densely innervated, with parasympathetic, sympathetic and sensory neurons innervating regions of the myocardium and cardiac conduction system. Each of these populations of neurons has a distinct distribution and phenotype that is critical to its function. Cardiac nerves undergo significant changes in morphology and phenotype in disease. Changes in growth factor expression, oxidative stress, and inflammatory cytokines within the heart and vasculature contribute to neuronal remodelling. These issues have been reviewed in detail recently (Kimura et al. 2012; Fukuda et al. 2015; Gardner et al. 2016), and will be summarized here briefly.
Sensory nerves
Sensory innervation of the heart includes both mechanosensitive and chemosensitive nerve endings (reviewed by Hainsworth, 1991; Schultz, 2001). Mechanosensitive afferents transduce pressure changes in the atria and ventricles during the cardiac cycle, while chemosensitive nerve endings respond to endogenous substances including adenosine and H+ and chemicals like capsaicin (Longhurst et al. 2001). Most of the sensory neurons innervating the heart reside in the nodose–petrosal ganglia and dorsal root ganglia, although some neurons within the intrinsic cardiac ganglion are sensory afferents (Armour, 2008). Many cardiac sensory fibres that arise in dorsal root ganglia produce nitric oxide synthase (nNOS), substance P and/or calcitonin gene related peptide (CGRP), distinguishing them from nodose–petrosal neurons which lack all three (Hoover et al. 2008). CGRP and substance P levels are decreased in coronary artery disease and diabetes mellitus, and end stage heart failure (Taquet et al. 1992; Wang et al. 2012). The loss of neuropeptide content in diabetes corresponds with CGRP‐immunopositive cardiac sensory denervation and atrophic changes in the dorsal root ganglia (Ieda et al. 2006).
Parasympathetic nerves
Cardiac parasympathetic neurons reside in intrinsic cardiac ganglia (ICG), which have a diffuse distribution on the surface of the heart, are connected by an extensive plexus of nerve fibre bundles (O'Shea & Evans, 1985; Pauza et al. 2000; Ardell, 2001; Hoover et al. 2004, 2009; Mabe et al. 2006), and project to distinct regions of myocardium (Quan et al. 1999, 2002; Zarzoso et al. 2013). Cholinergic nerve fibres are most abundant in sinoatrial and atrioventricular nodes, atrial myocardium and the ventricular conducting system (Hoover et al. 2004). Stimulation of cardiac parasympathetic nerves projecting to these regions leads to acetylcholine (ACh) release, which can evoke prominent bradycardia, negative dromotropic responses and decreases in atrial contractility via activation of postjunctional M2 muscarinic receptors. Cholinergic innervation in less abundant in the ventricles, but studies in humans and other large mammals have shown that these nerve fibres can trigger significant negative inotropic responses mediated by muscarinic receptors (DeGeest et al. 1965; Xenopoulos & Applegate, 1994; Ardell, 2001; Lewis et al. 2001; Coote, 2013). Only two studies have evaluated vagal parasympathetic effects on ventricular contractility in small mammals (Takahashi et al. 2003; Nalivaiko et al. 2010). While the first study failed to detect a negative inotropic response to vagal stimulation, the second study provided definitive evidence for a small decrease in contractility in a majority of experimental preparations. Based on the slow onset of these responses, it was suggested that cholinergic inhibition might occur indirectly though attenuation of noradrenaline (norepinephrine) release and/or postjunctional signalling (Nalivaiko et al. 2010). Cardiac parasympathetic ganglia integrate information from a variety of sources, including cholinergic preganglionic neurons (McAllen & Spyer, 1976; Hoover et al. 2004, 2009; Mabe et al. 2006), presumptive noradrenergic nerve fibres (Leger et al. 1999; Hoard et al. 2008; Hoover et al. 2009) and peptidergic fibres (Calupca et al. 2000, 2001).
Studies of human ICG have demonstrated that detrimental changes can occur in cardiac disease. Morphological studies of ganglia from ischaemic hearts revealed pathological changes in about 35% of the neurons evaluated (Hopkins et al. 2000). Abnormalities included various inclusions and vacuoles in somata, neuronal enlargement, loss of dendrites, and neuronal degeneration with the infiltration of phagocytes. Recently, another study found significant hypertrophy of intrinsic cardiac neurons from patients with autopsy‐confirmed heart failure (Singh et al. 2013). In contrast, mouse models of diabetes, which have significantly impaired vagal control of the heart, do not exhibit any changes within cardiac parasympathetic neurons of the ICG (Mabe & Hoover, 2011). Decreased parasympathetic transmission in the heart, and its role in cardiovascular pathology, is discussed further in the section on ‘Neuromodulation and autonomic imbalance’.
Sympathetic nerves
The sympathetic neurons innervating the heart reside predominantly in the stellate ganglia (Norris et al. 1974; Kostreva et al. 1977; Pardini et al. 1990) and produce the transmitter noradrenaline (NA) and the co‐transmitter neuropeptide Y (NPY). Sympathetic axons innervate the atria, cardiac conduction system and ventricles, where they stimulate increased heart rate (chronotropy), conduction velocity (dromotropy), and contractility (inotropy) via NA activation of β1‐adrenergic receptors. Sympathetic nerves are most dense in the atria and near the base of the ventricles with fewer nerves near the apex of the heart. Stimulation of the stellate increases contractility throughout the heart (Szentivanyi et al. 1967), but a transmural gradient of axons accompanies an epicardial to endocardial gradient in cardiac action potential duration that is important for normal activation and repolarization of the left ventricle (Antzelevitch et al. 1991; Nabauer et al. 1996; Brunet et al. 2004). Activation of cardiac β‐adrenergic receptors (β‐ARs) modulates myocyte repolarization by altering transmembrane currents and Ca2+ homeostasis (Thomas et al. 2004; Bers, 2008; Cutler et al. 2011), while it increases contractility by modulating calcium handling proteins so that Ca2+ release from the sarcoplasmic reticulum (SR) is increased (Valdivia et al. 1995; Marx et al. 2000; Shannon et al. 2000). The effects of sympathetic stimulation allow myocytes to meet increased cardiac demands during stress or exercise, but changes to sympathetic transmission in pathological conditions can contribute to cardiac dysfunction.
Cardiac sympathetic neurons can change their function in cardiovascular disease. Sympathetic hyperinnervation (Zhou et al. 2004; Hasan et al. 2006; Meloni et al. 2010) and denervation (Barber et al. 1983; Stanton et al. 1989; Dae et al. 1995; Li et al. 2004 b; Gardner & Habecker, 2013) have both been observed after myocardial infarction. Hyperinnervation has also been observed in compensatory cardiac hypertrophy during the transition to overt heart failure (Kimura et al. 2007; Lu et al. 2012), but NA synthesis is paradoxically reduced due to downregulation of tyrosine hydroxylase (TH), the rate‐limiting enzyme in NA synthesis (Kimura et al. 2007, 2010). In heart failure highly polysialylated neural cell adhesion molecule (PSA‐NCAM), which is a marker of immature neurons, is highly expressed in cardiac sympathetic neurons (Kimura et al. 2007). Further, many neurons in the stellate ganglia and left ventricle have increased expression of cholinergic markers such as choline transporter and choline acetyltransferase during heart failure. This is accompanied by decreased TH expression and thought to represent cholinergic transdifferentiation of cardiac adrenergic neurons into cholinergic neurons, which is observed in animal models of heart failure and in autopsy specimens from patients with heart failure (Kanazawa et al. 2010). Emerging evidence suggests that a similar cholinergic transdifferentiation occurs transiently after acute myocardial infarction (Olivas et al. 2016). Leukemia inhibitory factor (LIF) and other members of the interleukin‐6 (IL‐6) family, which can induce fetal gene expression (so‐called rejuvenation) in adult cardiomyocytes, are upregulated during heart failure (Kimura et al. 2007) and after myocardial infarction (Aoyama et al. 2000; Gritman et al. 2006). IL‐6 family cytokines secreted from the damaged myocardium act as negative modulators of sympathetic function by suppressing noradrenergic function (Kimura et al. 2007; Parrish et al. 2009) and inducing cholinergic differentiation (Kanazawa et al. 2010; Olivas et al. 2016) via a gp130 signalling pathway.
Nerve growth factor
Many of the injury‐induced changes in cardiac sympathetic and sensory nerves are driven by alterations in cardiac nerve growth factor (NGF). NGF supports the development and maintenance of cardiac sympathetic axons as well as the CGRP‐immunoreactive cardiac sensory nerves that richly innervate the epicardium and the ventricular myocardium (Ieda et al. 2006). Increased cardiac NGF is observed after myocardial infarction (Zhou et al. 2004; Hasan et al. 2006; Meloni et al. 2010; Gardner & Habecker, 2013) and in compensated cardiac hypertrophy (Ieda et al. 2004; Kimura et al. 2007), where it is associated with regional sympathetic hyperinnervation (Cao et al. 2000 b; Li et al. 2004 b; Zhou et al. 2004; Hasan et al. 2006; Kimura et al. 2012). Conversely, decreased cardiac NGF in experimental and clinical heart failure (Kaye et al. 2000; Qin et al. 2002; Kimura et al. 2010) and diabetes mellitus (Hellweg & Hartung, 1990; Schmid et al. 1999; Ieda et al. 2006) is associated with the loss of sympathetic and sensory fibres and reduced cardiac NA and CGRP levels (Taquet et al. 1992; Qin et al. 2002; Kristen et al. 2006; Kimura et al. 2007, 2010; Ieda & Fukuda, 2009; Kuehl & Stevens, 2012). Restoring NGF to the heart prevents sensory dysfunction in diabetes mellitus (Ieda et al. 2006). The regulation of NGF in the heart is not completely understood, but the hypertrophic factor endothelin‐1 increases NGF expression in cardiomyocytes (Ieda et al. 2004; Kimura et al. 2007), while mechanical stretch and α1‐adrenergic stimulation attenuate myocyte NGF expression (Rana et al. 2009).
Neuromodulation and autonomic imbalance
Emerging evidence suggests that factors produced within the microenvironment of the heart, its vasculature and between neuronal populations can influence sympatho‐vagal balance. These neuromodulators can be intrinsic to sympathetic ganglia or vagal neurons (such as neuronal nitric oxide and its adaptor protein, CAPON), or may act as either autoinhibitory or crosstalk communicating messengers between neuronal populations (for example the co‐transmitters neuropeptide Y (NPY), galanin and vasoactive intestinal peptide (VIP)). Substances released from cardiac myocytes and the coronary vasculature, including natriuretic peptides and angiotensin II (AT II), may also influence neuronal function acting in a paracrine manner (see Fig. 1). These signalling pathways converge on neuronal calcium handling, transmitter release and/or re‐uptake mechanisms. The plasma levels and protein expression of many of these neuromodulators are increased in both animal models and patients with cardiovascular disease. However, their functional significance and the potential to exploit these pathways pharmacologically in disease states is yet to be established.
Figure 1. Schematic representation of the neuromodulatory pathways influencing the release of noradrenaline (NA) and acetylcholine (ACh) from postganglionic sympathetic and parasympathetic neurons .
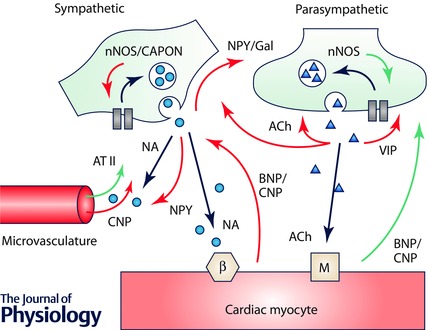
Neuromodulators can arise from the coronary microvasculature (e.g. angiotensin II (AT II) and C‐type natriuretic peptide (CNP)), myocytes (B‐type natriuretic peptide (BNP)) as well as between neurons (neuropeptide Y (NPY), galanin (Gal), acetylcholine (ACh) and vasoactive intestinal peptide (VIP)) and within neurons (such as neuronal nitric oxide synthase (nNOS) and its activator protein, CAPON). Stimulatory pathways are represented by green arrows, and inhibitory pathways by red arrows. β, beta adrenergic receptor; M, muscarinic receptor.
Studying the actions of cholinergic neuromodulators is particularly challenging. It should be noted that whilst stimulation of the cervical vagus activates efferent preganglionic parasympathetic neurons, up to 70% of the fibres are sensory afferents (Berthoud & Neuhuber, 2000). Proximal stimulation of the cut cervical vagus in vitro will therefore cause retrograde stimulation of sensory afferents. Stimulation of the intact vagus nerve in vivo will cause both anterograde and retrograde stimulation of both afferent and efferent fibres. Recent work has shown that this has the potential to elicit a complex profile of mediators and modulators from both efferent and afferent fibres engaging multiple levels of the cardiac neuraxis to alter peripheral neural processing and central–peripheral neural interactions, including central drive (Ardell et al. 2015). With the introduction of implantable cardiac vagal nerve stimulators in patients with heart failure, it is important to understand the differences in cellular mechanisms by which this intervention may influence the heart, compared to application of exogenous neurotransmitters, or stimulation of the cervical vagus removed from the central nervous system, as these approaches may yield different results.
Intrinsic neuromodulators
Neuronal nitric oxide and its activator protein, CAPON
Neuronal nitric oxide synthase (nNOS) is localized in both intrinsic cardiac vagal neurons and postganglionic sympathetic neurons of the stellate ganglia. Nitric oxide acts via stimulation of soluble guanylate cyclase, to generate cGMP, leading in parasympathetic neurons to inhibition of phosphodiesterase 3 (PDE3) and an increase in cAMP–protein kinase A (PKA) dependent phosphorylation of N‐type calcium channels and release of acetylcholine (Herring et al. 2000, 2001 b; Herring & Paterson, 2001), whilst in sympathetic neurons stimulation of PDE2 and/or protein kinase G (PKG) leads to inhibition of calcium influx and NA release (Schwarz et al. 1995; Wang et al. 2007). Increasing nNOS expression using viral vectors can reverse impaired vagal (Heaton et al. 2007) and exaggerated sympathetic drive (Li et al. 2007) in the spontaneously hypertensive rat (SHR), and improve acetylcholine release and mortality acutely following myocardial infarction in the guinea pig (Dawson et al. 2008).
Genome wide association studies implicate a variant in the neuronal nitric oxide synthase adaptor protein (CAPON) in electrocardiographic QT variation and sudden cardiac death, both of which are strongly influenced by cardiac autonomic balance (Arking et al. 2006; Kao et al. 2009). Recently it was shown that CAPON resides in cardiac sympathetic neurons (Lu et al. 2015) and its expression, along with nNOS (Li et al. 2013 a), is decreased in the pre‐hypertensive SHR. Moreover, increasing its expression using a novel noradrenergic specific adenoviral vector increases nNOS activity and normalizes sympathetic neurotransmission. Many questions relating to the role of nNOS/CAPON in cardiac autonomic control remain.
Questions and controversies
How is nNOS localized within postganglionic neurons and is its activity influenced by calcium influx triggering neurotransmitter release at individual varicosities?
Is the main role of CAPON intracellular targeting of nNOS and/or to increase nNOS enzyme activity?
Are CAPON single nucleotide polymorphisms in neurons the trigger for sudden cardiac death in patients with and without QT disturbances?
Convergence of neuromodulators on calcium induced transmitter release and re‐uptake
The main trigger for the exocytotic release of neurotransmitter is the binding of calcium to synaptotagmin to activate the SNARE complex and cause fusion of the vesicular and cellular membranes (Sudhof & Rothman, 2009). The local calcium signal is a function of the opening of neuronal calcium channels and may also be influenced by calcium induced calcium release from the endoplasmic reticulum (ER) and/or mitochondrial buffering of intracellular calcium. The contribution of these calcium sources has been studied in postganglionic sympathetic neurons from the stellate ganglia at the whole cell level through patch clamping and fluorescence imaging. The pre‐hypertensive SHR for example has a larger intracellular calcium transient, associated with a higher ER calcium content, lower mitochondrial membrane potential and reduced mitochondrial buffering of intracellular calcium (Li et al. 2012). The whole cell calcium current is also larger in the pre‐hypertensive SHR (Lu et al. 2015). Moreover increased NA release (Shanks et al. 2013 b) and impaired NA re‐uptake (Shanks et al. 2013 a) is also observed in the SHR at this developmental stage which may contribute to a larger tachycardia during stimulation of the stellate ganglia and increased heart rate observed in vivo (Shanks et al. 2013 b).
Co‐transmitters as neuromodulators
Sympathetic co‐transmitters
Sympathetic nerves throughout the autonomic nervous system contain co‐transmitters such as ATP, neuropeptide‐Y (NPY) and galanin. Both NA and ATP have relatively short half‐lives as NA is taken back into the nerve terminal via the NA transporter and ATP is rapidly metabolized. Conversely whilst the half‐lives of NPY and galanin are significantly longer, they are released in much smaller quantities (Burnstock, 2009). The role of co‐transmitters may therefore be more important during conditions associated with high levels of adrenergic drive. For example, plasma NPY levels are elevated following myocardial infarction (Cuculi et al. 2013) and during left ventricular failure in humans where they correlate positively with one‐year mortality (Hulting et al. 1990; Ullman et al. 1994). In the presence of β‐blockers, which improve mortality in these conditions, both sympathetic release of NPY and galanin can produce crosstalk inhibition of vagal neurotransmission via the Y2 and GALR1 receptors in animal models resulting in a shift in autonomic balance (Herring et al. 2008, 2012). Moreover, NPY may also have a direct action on ventricular myocytes to increase susceptibility to ventricular arrhythmias (Herring, 2015) and also limit myocardial perfusion through vasoconstriction of the coronary vasculature (Shanks & Herring, 2013).
Parasympathetic co‐transmitters
Many parasympathetic neurons within the heart also co‐stain for different co‐transmitters including NPY, somatostatin, VIP and endogenous opioids such as dynorphins (Steele & Choate, 1994). Whether these co‐transmitters are released during physiological activation of the vagus nerve or change their expression profile in cardiovascular disease is not known. For example, inhibitors of VIP enhance vagally mediated bradycardia in the rat although high frequency stimulation is required to observe these effects (Hogan & Markos, 2006). Several cardiac opioid analogues have also been identified, including the heptapeptide methionine‐enkephalin‐arginine‐phenylalanine (MEAP) (Younes et al. 2000). MEAP has been shown to be vagolytic (Farias et al. 2001), probably via δ2 receptors (Jackson et al. 2001), whilst low doses of MEAP can augment vagal bradycardia, via δ1 receptors (Farias et al. 2003). Interestingly, repeated transient ischaemia to the sinoatrial node artery is associated with an increase in locally released MEAP and increased vagal bradycardia (Deo et al. 2009) whilst δ1 receptor agonists significantly reduce infarct size in the rat (Schultz et al. 1998). Nevertheless, many questions still remain regarding the pathophysiological role of different neuropeptides.
Nitric oxide generated by nNOS has also been suggested to act as a co‐transmitter responsible for the cholinergic modulation of ventricular fibrillation threshold via NO acting independently of the muscarinic receptor (Brack et al. 2011). Early studies demonstrated that the anti‐fibrillatory effect of the vagus on the ventricle was abolished by the muscarinic receptor antagonist atropine (Corr & Gillis, 1974; Yoon et al. 1977; Vanoli et al. 1991). Muscarinic agonists such as oxotremorine and methacholine can also reduce ventricular arrhythmias following myocardial infarction (De Ferrari et al. 1992, 1993). However, in an isolated Langendorff perfused rabbit heart, recent work suggests the anti‐fibrillatory action of direct vagal stimulation appears to be preserved in the presence of atropine, abolished in the presence of hexamethonium (Brack et al. 2011), and dependent on the generation of nitric oxide by nNOS acting in a paracrine fashion (Ng et al. 2007; Brack et al. 2009). This seemed surprising given previous observations with atropine and muscarinic agonists in vivo, and the fact that NO generated by nNOS can facilitate acetylcholine release (Herring & Paterson, 2001). More recently, vagus nerve stimulation has been shown to be anti‐arrhythmic in the setting of ischaemia–reperfusion via effects on mitochondrial function with atropine abolishing this effect (Shinlapawittayatorn et al. 2014). Silencing vagal preganglionic neurons from the dorsal motor nucleus of the vagus using an optogenetic approach also limits infarct size during ischaemia–reperfusion via an atropine dependent pathway (Mastitskaya et al. 2012). Some beneficial changes in ventricular electrophysiology were still observed, suggesting there might be a paracrine action via NO, since this action was removed following NOS inhibition (Machhada et al. 2015). However, in this issue of The Journal of Physiology, Kalla et al. (2016) challenge the functional significance of the paracrine effect of NO. Whilst they demonstrate that the acetylcholine analogue carbamylcholine raises ventricular fibrillation threshold (VFT), they observed its effect was prevented by atropine, the nicotinic receptor antagonist mecamylamine, and inhibitors of neuronal nitric oxide synthase (nNOS) and soluble guanylyl cyclase (sGC). Moreover, the NO donor sodium nitroprusside could mimic the anti‐fibrillatory action of carbamylcholine, but its action was again ultimately dependent on muscarinic receptor stimulation, since all effects were blocked by atropine (Kalla et al. 2016).
Questions and controversies
What are the physiological roles of cardiac sympathetic and parasympathetic co‐transmitters?
Are they just neuromodulators or do they play a major role as co‐transmitters during cardiovascular disease?
Can these pathways be exploited therapeutically in patients receiving conventional pharmacological and interventional treatments?
Paracrine neuromodulators
Natriuretic peptides
The natriuretic peptide family is made up of at least three distinct peptides: atrial natriuretic peptide (ANP), brain derived natriuretic peptide (BNP), and C‐type natriuretic peptide (CNP) which share a disulphide linked 17 amino acid ring structure giving them their functionality. ANP is secreted from the atria in response to myocardial stretch producing natriuresis and vasorelaxation, whilst BNP is synthesized and secreted in the atria and ventricle, particularly during congestive heart failure where plasma levels are a prognostic indicator (Doust et al. 2005). CNP is the predominant natriuretic peptide of the nervous system (Sudoh et al. 1990) and is also found within vascular endothelium and the human ventricle (Wei et al. 1993). BNP is able to act directly on cardiac sympathetic neurons to reduce the intracellular calcium transient and release of NA during neuronal depolarization via a cGMP–PKG dependent pathway (Li et al. 2015). Conversely, exogenous BNP and CNP are also capable of directly augmenting vagal neurotransmission in a manner similar to NO via generation of cGMP (Herring et al. 2001 a). BNP may therefore be expected to result in a beneficial shift in cardiac autonomic balance towards sympathetic withdrawal and greater vagal tone during chronic heart failure, although cGMP dependent signalling may be limited by phosphodiesterase 2A (PDE2A) that is upregulated in this condition (Li et al. 2015). This may explain the lack of success of recombinant BNP (nesiritide), which has only been trialled in acute heart failure where results were disappointing (O'Connor et al. 2011).
Questions and controversies
Although a natriuretic peptide receptor agonist has been trialled unsuccessfully in acute decompensated heart failure, would their use as sympatholytic neuromodulating agents be beneficial in chronic stable heart failure if neuronal PDE2A levels can be controlled?
Ventricular myocytes as a source of acetylcholine
Rat ventricular myocytes express choline acetyltransferase (ChAT) and vesicular acetylcholine transporter, and the expression of these genes and the intracellular acetylcholine content can be modulated by muscarinic receptor inhibition (Kakinuma et al. 2009). This suggests that cardiomyocytes possess an acetylcholine synthesis system that is positively modulated by cholinergic stimuli and may therefore amplify the beneficial effects of vagal stimulation on the ventricles. In keeping with this, cardiac specific overexpression of ChAT in mice improves survival post myocardial infarction (Kakinuma et al. 2013). Whether such a system plays an important role in large mammals and humans is yet to be determined.
Angiotensin II
The renin–angiotensin system plays a key role in the regulation of blood pressure, circulating volume and neuronal function. In addition to the classical pathway via angiotensin converting enzyme (ACE) in the pulmonary vasculature, the presence of local cardiac ACE (Saito et al. 1987) raises the possibility of local AT II synthesis and influence on neurotransmission. Inhibitors of ACE and AT1 receptors facilitate the heart rate response to peripheral vagal nerve stimulation (Takata et al. 2004) whilst AT II inhibits myocardial acetylcholine release (Kawada et al. 2007) via the AT1 receptor, although the intracellular pathway potentially linking the AT1 receptor to transmitter release has not been elucidated. Moreover, cardiac AT II levels are higher in the SHR where it potentiates NA release reinforcing the sympathetic hyperactivity (Herring et al. 2011). The use of ACE inhibitors post myocardial infarction (e.g. AIRE trial) and in chronic heart failure (e.g. SOLVD and CONSENSUS trials) is well established where they confer a mortality benefit.
Neurohumoral impact on cardiac adaptations in disease
Role of sensory afferent nerves in the diseased heart
The cardiac autonomic nervous system is composed of efferent and afferent nerves. In contrast to sympathetic innervation, which has well‐defined roles in the progression of cardiac disease, sensory innervation and its alteration in diseased hearts have not been elucidated sufficiently. Sensory nerves within the heart express the peptides substance P and CGRP, and those peptides have differing effects in cardiovascular disease. Substance P induces adverse myocardial remodelling (Melendez et al. 2011), while CGRP has cardioprotective effects in pathophysiological conditions including cardiovascular disease and diabetes mellitus (Brain & Grant, 2004; Chai et al. 2006) (Fig. 2). CGRP is also an important angiogenic factor for microvascular formation in the diseased heart (Li et al. 2013 b). Experimentally, CGRP is cardioprotective by activating the PI3K/Akt and ERK1/2 mitogen‐activated protein kinase (MAPK) kinase pathways and restoring Bcl‐2/Bax to inhibit apoptosis in cultured rat cardiomyocytes (Schaeffer et al. 2003; Ma et al. 2013; Umoh et al. 2014). CGRP may also have a protective function in heart failure through decreased inflammation, cell death and fibrosis.
Figure 2. Diagram of the pathophysiological interactions between the cardiac sensory nerves and diseased state .

CGRP can cause activation of multiple signalling pathways and has cardioprotective effects. Cardiovascular disease and autonomic neuropathy can lead to downregulation of CGRP, which causes inflammation, vasoconstriction, impairing angiogenesis, fibrosis and apoptosis, and may ultimately lead to heart failure or sudden cardiac death. CGRP, calcitonin gene‐related peptide; ROS, reactive oxygen species; PI3K, phosphatidylinositol‐3 kinase; Akt, serine/threonine‐specific protein kinase; ERK, extracellular signal‐regulated kinases; Bcl‐2/Bax, B‐cell lymphoma 2/Bcl‐2‐associated X protein.
Role of sympathetic efferent nerves in the diseased heart
The contributions of sympathetic dysfunction to cardiovascular pathology have been the subject of many reviews (Esler et al. 1997; Lefkowitz et al. 2000; Gourine & Gourine, 2014; Shen & Zipes, 2014; Fukuda et al. 2015; Gardner et al. 2016). Under normal physiological conditions, NA released from sympathetic nerves impacts cardiac ion channels and Ca2+ handling to enact prototypical chronotropic and inotropic responses. Briefly, NA binds to β‐ARs on cardiac myocytes leading to phosphorylation of several intracellular targets, including L‐type Ca2+ channels, K+ channels, phospholamban and ryanodine receptors (RyRs) (Bers, 2002). Phosphorylation of the L‐type Ca2+ channel (Cav1.2) increases current, which by itself would be expected to increase the cardiac action potential duration (APD). However, this increase is counterbalanced by the effect of β‐AR on delayed rectifier K+ currents, most notably an increase in rapidly activating I Ks, but slowly activating I Kr may also be involved (Kagan et al. 2002; Marx et al. 2002). The net effect is typically a shortening of the APD, which is required to accommodate fast heart rates during sympathetic activity.
Phosphorylation of phospholamban leads to increased sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) activity and an increase in the Ca2+ content of the SR. Although increased SR Ca2+ content facilitates positive inotropy, SR Ca2+ overload can also occur, resulting in spontaneous opening of RyRs and Ca2+ release into the cytosol that is not in response to an action potential. This leads to Ca2+ extrusion from the cytosol via the Na+/Ca2+ exchanger (NCX), which produces a net inward current (Pogwizd et al. 2001). If the inward current is large enough, the cell membrane depolarizes and a triggered action potential occurs, also known as a delayed after‐depolarization (DAD).
Long term remodelling of these prototypical cardiomyocyte responses occurs as a result of chronic sympathetic hyperinnervation or denervation, and both of these conditions have been linked to increased risk of ventricular arrhythmias and sudden cardiac death (Cao et al. 2000 a,b; Liu et al. 2003; Li et al. 2004 b; Boogers et al. 2010; Nishisato et al. 2010; Vaseghi et al. 2012; Fallavollita et al. 2014). A loss of cardiomyocyte β‐AR responsiveness over time is a hallmark of sustained adrenergic stimulation and hyperinnervation. Changes in β‐AR sensitivity are probably due to altered expression of either β‐ARs or G‐protein receptor kinase 2 (GRK2, also known as βARK1) (Iaccarino et al. 1998; Yatani et al. 2006). In addition to decreased responsiveness to NA, APDs may be prolonged in hyperinnervated regions due to NGF‐induced downregulation of K+ currents (Heath et al. 1998). This increase in APD may also facilitate early after‐depolarizations (EADs), in which a secondary depolarization occurs before final repolarization. Conversely, chronic sympathetic denervation can lead to β‐AR supersensitivity, which results from upregulation of β‐ARs and increased responsiveness upon catecholamine binding (Vatner et al. 1985) due to downregulation of GRK2 (Yatani et al. 2006). Denervation also leads to a loss of I to, which is responsible for the initial repolarization in phase 1 of the action potential (Bru‐Mercier et al. 2003; Bai et al. 2008). Gardner et al. recently showed in a murine model that reperfused infarcts remain denervated and display β‐adrenergic supersensitivity, calcium mishandling, and decreased I to that are reversed by restoring the innervation (Gardner et al. 2015).
In addition to these cellular level responses, abnormally heterogeneous release of NA due to hyperinnervation or denervation leads to tissue level responses that also facilitate arrhythmia. Heterogeneous sympathetic nerve distribution may produce non‐uniform shortening of cardiac APD and increased dispersion of repolarization. Although non‐uniform APD shortening occurs even under normal sympathetic control (due to the normally heterogeneous sympathetic nerve distribution), Ajijola and colleagues found that following myocardial infarction, sympathetic stimulation further increased the dispersion of repolarization and altered activation and propagation patterns (Ajijola et al. 2013 b). These results were confirmed in human patients with myocardial infarction in which reflex sympathetic stimulation caused a 230% increase in dispersion compared to patients with structurally normal hearts (Vaseghi et al. 2012). Collectively, these increases in dispersion of repolarization increase the likelihood of conduction block and reentrant arrhythmias. Furthermore, localized release of adrenergic agonists (e.g. hyperinnervation) has been shown to induce propagating premature ventricular complexes (PVCs) and sustained focal ventricular tachycardia in intact hearts (Nash et al. 2001; Myles et al. 2012, 2015).
However, NA is not the only substance released by sympathetic nerves in the heart during pathological conditions. The impact of sympathetic NPY on coronary arteries was described previously, but the effect of ACh released from cardiac sympathetic nerves is not yet understood. Mice with heart failure and cholinergic transmission from cardiac sympathetic nerves had significantly improved survival and ventricular function compared with sympathetic nerve‐specific gp130‐deficient mice, which lacked sympathetic ACh (Kanazawa et al. 2010). This suggests that sympathetic cholinergic transdifferentiation might be an adaptive response that protects the heart. Consistent with this idea, cholinergic signalling via cardiac muscarinic acetylcholine receptors decreases the susceptibility of ventricular myocytes to chronic adrenergic stress, improving cardiac function (LaCroix et al. 2008). Furthermore, cardiomyocytes can also synthesize and release ACh, which is positively modulated by cholinergic stimuli. This suggests a positive feedback mechanism for ACh release, which may further any cardioprotective effects (Kakinuma et al. 2009). Similarly, augmenting parasympathetic nerve activity decreases mortality rates in heart failure, suggesting a beneficial effect of cholinergic signalling (Li et al. 2004 a). However, it remains controversial if release of ACh from sympathetic nerves, which are distributed differently from parasympathetic nerves within the myocardium, is a favourable or unfavourable event for cardiac performance and prognosis (DeMazumder et al. 2015).
Thus, the spatial and temporal innervation pattern and activity of sympathetic nerves directly impacts pathogenesis in the diseased heart (Fig. 3).
Figure 3. Crosstalk between cardiomyocyte and sympathetic nerve via humoral factors in diseased heart .

Failing cardiomyocytes induce NGF via an endothelin‐1 mediated pathway and LIF. NGF leads to hyperinnervation (anatomical modulation) and LIF leads to rejuvenation/cholinergic differentiation (functional modulation). This phenomenon shows the expression of catecholaminergic marker such as reduced TH and increased cholinergic (CHT, ChAT) and juvenile markers (PSA‐NCAM). PSA‐NCAM, polysialylated neural cell adhesion molecule; TrkA, tropomyosin‐related kinase A; NGF, nerve growth factor; LIF, leukemia inhibitory factor; LIFR, LIF receptor; TH, tyrosine hydroxylase.
Questions and controversies
Does sympathetic release of ACh protect the heart?
Does sympathetic ACh have the same effect in heart failure and acute myocardial infarction?
Neurohumoral regulation of calmodulin‐dependent protein kinase II in health and disease
The multifunctional Ca2+‐ and calmodulin‐dependent protein kinase II (CaMKII) is a serine/threonine kinase that is abundant in myocardium. CaMKII appears to augment myocardial performance in response to physiological stress, at least in part, by neurohumoral agonist stimulation. CaMKII catalyses phosphorylation events that activate many of the voltage‐gated sarcolemmal ion channels and Ca2+ homeostatic proteins orchestrating excitation–contraction coupling (Rokita & Anderson, 2012). The role of CaMKII‐catalysed phosphorylation of the type 2 intracellular Ca2+ release ryanodine receptor (RyR2) seems to be particularly important in augmenting release of myofilament activating Ca2+ from the SR during neurohumoral agonist stimulation (Grimm et al. 2015; Polakova et al. 2015) and during oxidant stress (Shirokova et al. 2014). In cardiac pacemaker cells, CaMKII activation contributes to fight or flight heart rate increases in response to physical activity and isoproterenol (isoprenaline) (Wu et al. 2009). Thus, CaMKII activation downstream to neurohumoral agonists acts to augment myocardial performance under physiological stress. However, a large body of evidence supports a view that CaMKII is also a nodal signal for promoting heart failure and arrhythmias (Swaminathan et al. 2012). Myocardial diseases are marked by sustained elevation of neurohumoral signalling and excessive CaMKII activity. Furthermore, CaMKII is activated by neurohumoral agonist pathways that are amongst the best clinically validated targets for heart failure therapeutics. Thus, it seems likely that high impact cardiovascular drugs, including β‐adrenergic receptor antagonists (β‐blockers), angiotensin II receptor blockers (ARBs), angiotensin converting enzyme (ACE) inhibitors and mineralocorticoid receptor (MR) antagonists are beneficial, at least in part, because they mitigate against CaMKII hyperactivation under conditions of sustained, pathological neurohumoral stress.
The CaMKII holoenzyme consists of two stacked hexamers and includes a total of 12 homologous or identical subunits (Chao et al. 2011). Each monomeric CaMKII subunit joins the holoenzyme by a C‐terminal association domain (Fig. 4). The kinase domain, containing the ATP binding pocket resides in the N‐terminus. The regulatory domain is interposed between the kinase and association domains and consists of an autoinhibitory region (N‐terminus) and a calmodulin (CaM) binding region (C‐terminus). Under resting conditions, CaMKII is largely inactive because the kinase domain engages the autoinhibitory region, which acts as a pseudosubstrate. However, when the concentration of intracellular Ca2+ ([Ca2+]i) rises and binds to CaM the calcified CaM (Ca2+/CaM) embraces the CaM binding region and induces a conformational change that distorts the relationship between the catalytic domain and the autoinhibitory region, leading to disinhibition and activation of CaMKII. Brief, low frequency [Ca2+]i elevations permit Ca2+/CaM unbinding and a return to autoinhibition. However, when [Ca2+]i elevations are rapid or sustained CaMKII transitions between Ca2+/CaM‐dependent and Ca2+/CaM‐autonomous conformations by an autophosphorylation process (Kuret & Schulman, 1985; Schworer et al. 1986). Autophosphorylation of a conserved threonine at position 286 or 287 (Thr 287, numbering varies slightly by isoform) in the regulatory domain enhances the avidity of Ca2+/CaM binding (so called CaM trapping) (Meyer et al. 1992), favouring sustained CaMKII activation, but also prevents effective autoinhibition even after Ca2+/CaM unbinding, allowing for Ca2+/CaM autonomous CaMKII activity. More recently, we discovered a mechanism for Ca2+/CaM independent CaMKII activity by oxidation of a methionine pair (281/282) (Erickson et al. 2008). Oxidized CaMKII (ox‐CaMKII) does not support CaM trapping and is reversible by a methionine reductase (MsrA, methionine sulfoxide reductase A). Interestingly, CaMKII activation can be sustained at very low [Ca2+]i in oxidative environments, suggesting that reactive oxygen species (ROS) sensitize the Ca2+/CaM dependence of CaMKII activation. Other post‐translational modifications to the regulatory domain shown to increase Ca2+/CaM autonomous CaMKII activity are O‐GlcNAcylation (at serine 280) (Erickson et al. 2013) and nitrosylation (at cysteine 290) (Gutierrez et al. 2013; Curran et al. 2014). Cysteine 290 may show more nitrosylation during catecholamine stimulation but other nitrosylation site(s) may lead to reduced CaMKII activity (Erickson et al. 2015). O‐GlcNAcylation is not presently known to be downstream to neurohumoral agonist stimulation. Thus, a rich variety of regulatory domain post‐translational modifications account for important aspects of CaMKII activation in response to neurohumoral agonist stimulation.
Figure 4. Neurohumoral activation of CaMKII .
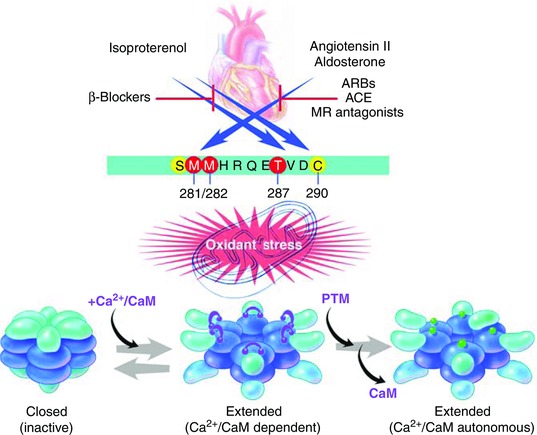
Catecholamines, angiotensin II and aldosterone are upstream activators of CaMKII by inducing post‐translational modifications (PTM) to the N‐terminus of the regulatory domain (blue horizontal bar, see text). Catecholamine agonists promote phosphorylation of threonine 287 and nitrosylation of cysteine 290. Angiotensin II and aldosterone increase oxidant stress leading to oxidation of methionines 281 and 282. Hyperglycaemia can also activate CaMKII directly (by O‐Glycnacylation of serine 280) and by increasing reactive oxygen species and oxidizing methionines 281 and 282. The inactive CaMKII holoenzyme exists in a compact configuration. Intracellular Ca2+ increases lead to calcification of calmodulin (Ca2+/CaM) that binds to the C‐terminus of the regulatory domain. Ca2+/CaM binding induces CaMKII into an extended, active conformation that is inactivated after Ca2+/CaM unbinding. Post‐translational modifications to the regulatory domain keep CaMKII in a persistantly extended, active state even after Ca2+/CaM unbinding. ARB, angiotensin II receptor blockers; ACE, angiotensin converting enzyme; MR, mineralocorticoid receptor.
The role of Ca2+/CaM independent CaMKII activation appears to be important for the role of CaMKII in neurohumoral agonist induced myocardial disease. At this time, neurohumoral agonist initiated pathways are best understood to activate Ca2+/CaM autonomous CaMKII activity by increasing Thr 287 autophosphorylation or Met 281/282 oxidation. Infusion of the mixed β1/2 receptor agonist isoproterenol is a model for catecholamine induced cardiomyopathy. In our hands, isoproterenol activates CaMKII and induces Ca2+/CaM independent CaMKII activity primarily by favouring Thr 287 autophosphorylation (Erickson et al. 2008). Features of isoproterenol induced myocardial toxicity include hypertrophy, cell death, fibrosis and depressed left ventricular contraction (Zhang et al. 2005; Yang et al. 2006). Mice with transgenic expression of a CaMKII inhibitory peptide (AC3‐I) (Zhang et al. 2005) or a protein kinase A (PKA) inhibitory peptide (PKI) (Zhang et al. 2013) are resistant to CaMKII activation and isoproterenol infusion‐induced myocardial injury. The precise nature of pathways for activating CaMKII in response to β‐adrenergic agonist stimulation remain controversial because some evidence supports exclusive, or near exclusive, CaMKII activation as a consequence of PKA‐induced intracellular Ca2+ mobilization (Zhang et al. 2013), while other studies support a role for the PKA‐independent, exchange proteins directly activated by cAMP (EPAC) pathway (Grimm et al. 2015).
Questions and controversies
Do β‐adrenergic receptors activate CaMKII solely through PKA, or does EPAC play a role?
Although detailed dose ranging studies are lacking, isoproterenol appears to be a more toxic myocardial agent compared to angiotensin II or aldosterone because these latter two agents do not induce substantial loss of left ventricular contraction in the absence of a ‘second hit’, such as myocardial infarction. We found that mice with myocardial targeted expression of a CaMKII inhibitory peptide, AC3‐I, were protected against isoproterenol infusion induced cardiomyocyte death (Yang et al. 2006), cardiomyopathy and myocardial infarction (Zhang et al. 2005), a condition where elevated neurohumoral signalling contributes to increased mortality. Mice with myocardial and mitochondrial CaMKII inhibition by transgenic expression of another CaMKII inhibitory peptide, CaMKIIN (Chang et al. 1998), are also protected against myocardial death after isoproterenol injection or myocardial infarction (Joiner et al. 2012), suggesting CaMKII actions in mitochondria contribute to catecholamine cardiotoxicity. CaMKIIδ knock‐out mice are also protected from pathological stress responses after aortic banding (Backs et al. 2009; Ling et al. 2009), a model of elevated afterload, where neurohumoral signalling probably contributes to adverse outcomes. Angiotensin II infusion, to approximate blood concentrations present in patients with heart failure, oxidizes CaMKII at Met 281/282 (ox‐CaMKII) leading to sinus node cell death (Swaminathan et al. 2011), inducing a heart rate disease phenotype resembling ‘sick sinus syndrome.’ Angiotensin II infusion increases susceptibility to rapid pacing induced atrial fibrillation and knock‐in mice lacking Met 281/282 on CaMKIIδ or knock‐in mice lacking a validated CaMKII phosphorylation site on RyR2 (serine 2814) are resistant to angiotensin II biasing of atrial fibrillation induction (Purohit et al. 2013). Aldosterone infusion, to achieve blood levels present in patients with heart failure after myocardial infarction, increases ox‐CaMKII and promotes myocardial rupture by enhancing ox‐CaMKII mediated expression of matrix metalloproteinase 9 in mice after myocardial infarction surgery (He et al. 2011). Taken together, these and other findings support a view that CaMKII is an important component of physiological and pathological pathways central to neurohumoral agonist signalling in myocardium.
Adapting novel molecular and cellular tools for neurocardiology research
Detailed studies of how whole‐heart function is altered by the activity of specific populations of neurons that innervate the heart provide insights into neurocardiac physiology as well as disease mechanisms and potential therapies. However, controlled measurements of the interplay between nerve activity and the myocardium can be challenging, particularly for in vivo animal studies and in excised perfused heart experiments. For example, in a hybrid perfused heart approach, parasympathetic neurons were activated by careful dissection and electrical stimulation of the left and right vagus nerve while sympathetic neurons were activated by electrical stimulation of the spinal canal at the second thoracic vertebra (Ng et al. 2001; Brack et al. 2004). This approach is powerful because it provides for controlled activation of the cardiac nerves in the absence of circulating compounds, although spinal cord stimulation activates both left and right sympathetic pathways, reducing left–right specificity (Brack et al. 2004). The approach has been primarily used in rabbits and could be difficult to implement in smaller species due to the additional steps required to prepare the heart for perfusion, isolate the nerve tissue, and position stimulation electrodes. For in vivo animal studies, electrodes are usually implanted around the vagus nerve for chronic parasympathetic stimulation (Li et al. 2004 a), but this has the limitation of potentially stimulating both efferent and afferent nerves. Exposing and electrically stimulating the stellate ganglia provides sympathetic stimulation in animals (Ajijola et al. 2013 a) while sympathetic cardiac neurons can be silenced by removing or ablating the stellate ganglia (Schwartz, 2014; Vaseghi et al. 2014).
In other studies, exogenous neurotransmitters, or their analogues, are administered to the coronary circulation to activate postsynaptic receptors to study the downstream effect of receptor activation on whole‐heart function (Liu et al. 1999; Lang et al. 2015). The neurotransmitters circulate through the myocardium to ubiquitously activate their receptors. Although this approach reveals, for example, important aspects of adrenergic receptor sensitivity and expression within the myocytes, it does not recapitulate neurotransmitter release and the physiological kinetics and spatially dependent concentration of transmitters released from cardiac neurons. This can be an important experimental limitation when the spatial arrangement of innervation (Freeman et al. 2014), disparate activity levels of nerve bundles (Ng et al. 2001), or altered autonomic tone (Thayer & Lane, 2007; Cauley et al. 2015) influences myocardial function.
Optogenetics is a powerful way to selectively target and activate cells in living tissue with unmatched spatiotemporal specificity using light‐actuated ion channels, such as channelrhodopsin‐2 (ChR2). Originally developed for neuroscience (Boyden et al. 2005), with ongoing developments in cardiac tissue (Ambrosi et al. 2014; Nussinovitch & Gepstein, 2015), optogenetics provides unparalleled specificity for cellular activation. This has created new opportunities for investigating the interaction between the cardiac nerves and the myocardium, overcoming previous approaches that required dissection and non‐specific electrical stimulation of bundles of heterogeneous nerve fibres. Precise cell‐type activation can be accomplished by regulating ChR2 expression using Cre‐Lox recombination and cell‐specific promoters. In a transgenic implementation of this system, a Cre responder animal would have a loxP‐flanked STOP cassette upstream of a ChR2‐enhanced yellow fluorescent protein (EYFP) fusion gene. This animal would then be mated with a Cre driver animal that expresses Cre recombinase under the control of a cell‐specific promoter such as α‐myosin heavy chain (cardiomyocytes) (Zaglia et al. 2015), tyrosine hydroxylase (TH, sympathetic neurons) (Wang et al. 2014; Wengrowski et al. 2015), or choline acetyltransferase (ChAT, parasympathetic neurons) (Kalmbach et al. 2012). In the progeny, ChR2‐EYFP would then have promoter‐specific expression only in targeted tissues.
This approach was recently used to study the neural control of parasympathetic tone in the brainstem of transgenic mice (Wang et al. 2014). ChR2 was expressed in noradrenergic cells using Cre‐Lox recombination and a TH promoter. Parasympathetic cardiac vagal neurons were labelled with the retrograde fluorescent tracer X‐rhodamine isothiocyanate (XRITC). Brains were then excised, sliced to reveal the locus coeruleus and cardiac vagal neurons, and noradrenergic (TH) neurons were photo‐stimulated to study their influence on the cardiac vagal neurons. Photo‐stimulated action potentials in the noradrenergic neurons increased the frequency of inhibitory postsynaptic currents in cardiac vagal neurons, providing a circuit substrate for the withdrawal of parasympathetic activity and increase in heart rate that occurs during stressful events. The selective activation of noradrenergic cells using optogenetics was a critical component in the success of these studies.
Questions and controversies
Could therapies that target the brainstem blunt autonomic imbalance that develops during heart failure?
In other studies, transgenic mice similar to those of Wang et al. (2014) were used for expression of ChR2 in noradrenergic (TH) neurons to elicit a cardiac β‐adrenergic response that originated from sympathetic nerve terminals within the heart (Wengrowski et al. 2015). Hearts were excised and perfused using a standard Langendorff approach. The epicardial surface was illuminated with blue light to photo‐stimulate noradrenergic neurons to generate a myocardial adrenergic response. Changes in contractile force, heart rate, and electrical activity were measured before and after photo‐stimulation. Results demonstrated pronounced increases in heart rate and contractile force during photo‐stimulation as well as recovery of prestimulation function after cessation of photo‐stimulation. Photo‐stimulation also shortened myocardial action potential duration, consistent with β‐adrenergic activation, and made hearts more susceptible to arrhythmia. Photo‐stimulation of sympathetic nerve terminals provided approximately half the increase in contractile force and heart rate that resulted from ubiquitous β‐adrenergic receptor activation using isoproterenol (5 μm) (Wengrowski et al. 2015).
Questions and controversies
Could a premature ventricular contraction be initiated by the simultaneous activation of a minimum number of sympathetic nerve terminals?
How would increased fibrosis and scar tissue during heart failure, or within an infarct, modulate that minimum number?
Optogenetics has also been instrumental in studies of cardiac ischaemia–reperfusion injury by providing for selective stimulation of specific populations of cardiac neurons. Using optogenetics to activate neurons in the brainstem, Mastitskaya et al. showed that cardioprotection provided by remote ischaemic preconditioning in rats is dependent upon the activity of brainstem vagal preganglionic neurons (Mastitskaya et al. 2012). Lenti‐ and adenoviral vectors were administered to specific locations within the dorsal motor nucleus of the vagus nerve (DVMN) for selective expression of a channelrhodopsin variant (ChIEF) (Lin et al. 2009) to activate neurons using light and a Drosophila allatostatin receptor (AlstR) to silence neurons using allatostatin (Tan et al. 2006). ChIEF and AlstR were selectively expressed in cholinergic vagal preganglionic neurons of the DVMN by driving expression with a PRSx8 promoter (Hwang et al. 2001), which is activated by Phox2, a transcription factor expressed in cholinergic vagal preganglionic neurons of the DVMN. Cardioprotection provided by remote preconditioning was abolished when the brainstem DVMN neurons were silenced with allatostatin. Interestingly, photo‐stimulation of the brainstem DVMN neurons before ischaemia and during reperfusion (in the absence of remote preconditioning) effectively mimicked the effect of remote preconditioning. This work has provided new insights into the mechanisms of preconditioning, which appear to be strongly mediated by vagal activity and cardiac muscarinic receptors.
Questions and controversies
How should optogenetic technologies evolve to enable new therapies for human heart disease?
Is the temporal control provided by optogenetics always necessary for cardiac therapies or could a drug delivered through the bloodstream also selectively activate an expressed neural actuator (DREADDS, for example) to achieve the same therapeutic result?
Selective activation as well as selective inhibition of neuron populations using new technologies, such as optogenetics and designer receptors exclusively activated by designer drugs (DREADDs), provides many exciting opportunities for investigating neurocardiac physiology. This includes the potential for unravelling fundamental functions in cardiac control that have largely eluded complete understanding, such as how parasympathetic neurons inhibit sympathetic activity at the level of the brainstem and cardiac ganglia. It is likely that technologies such as optogenetics will also facilitate new targets of opportunity for restoring autonomic balance to improve cardiac function in many diseases. For example, futuristic therapies for restoring autonomic balance may include implantable devices (Xu et al. 2014) that provide in vivo illumination using low power micro‐LEDs (Kim et al. 2010) with wireless control to activate neurons that express light‐activated ion channels.
Additional information
Competing interests
None declared.
Funding
This work was supported, in part, by National Institutes of Health (NIH) Grants HL079031, HL096652, HL070250 and HL071140 (M.E.A.), HL068231 and HL093056 (B.A.H.), HL111600 (C.M.R.) and GM107949 (D.B.H.), and by the British Heart Foundation (N.H., D.J.P.).
Acknowledgements
The authors thank Dr Rebecca Kreipke (Brandeis University) for contributions to the section on development, and thank Drs Matthew Kay and David Mendelowitz (George Washington University) for contributions to the section on novel molecular tools and critical reading of the manuscript. Mr Timothy Phelps contributed art work.
References
- Ahuja P, Perriard E, Pedrazzini T, Satoh S, Perriard JC & Ehler E (2007). Re‐expression of proteins involved in cytokinesis during cardiac hypertrophy. Exp Cell Res 313, 1270–1283. [DOI] [PubMed] [Google Scholar]
- Ajijola OA, Vaseghi M, Zhou W, Yamakawa K, Benharash P, Hadaya J, Lux RL, Mahajan A & Shivkumar K (2013. a). Functional differences between junctional and extrajunctional adrenergic receptor activation in mammalian ventricle. Am J Physiol Heart Circ Physiol 304, H579–H588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajijola OA, Yagishita D, Patel KJ, Vaseghi M, Zhou W, Yamakawa K, So E, Lux RL, Mahajan A & Shivkumar K (2013. b). Focal myocardial infarction induces global remodeling of cardiac sympathetic innervation: neural remodeling in a spatial context. Am J Physiol Heart Circ Physiol 305, H1031–H1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi CM, Klimas A, Yu J & Entcheva E (2014). Cardiac applications of optogenetics. Prog Biophys Mol Biol 115, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA & Liu DW (1991). Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 69, 1427–1449. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y & Sasayama S (2000). Augmented expression of cardiotrophin‐1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol 32, 1821–1830. [DOI] [PubMed] [Google Scholar]
- Ardell JL (2001). Neurohumoral control of cardiac function In Basic and Clinical Neurocardiology, 4th edn, ed. Armour JA. & Ardell JL, pp. 45–59. Academic Press, San Diego. [Google Scholar]
- Ardell JL, Rajendran PS, Nier HA, KenKnight BH & Armour JA (2015). Central‐peripheral neural network interactions evoked by vagus nerve stimulation: functional consequences on control of cardiac function. Am J Physiol Heart Circ Physiol 309, H1740–H1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Pfeufer A, Post W, Kao WH, Newton‐Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marban E, O'Donnell CJ, Hirschhorn JN, Kaab S, Spooner PM, Meitinger T & Chakravarti A (2006). A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet 38, 644–651. [DOI] [PubMed] [Google Scholar]
- Armour JA (2008). Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp Physiol 93, 165–176. [DOI] [PubMed] [Google Scholar]
- Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel‐Duby R, Maier LS & Olson EN (2009). The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA 106, 2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Ren C, Hao W, Wang R & Cao JM (2008). Chemical sympathetic denervation, suppression of myocardial transient outward potassium current, and ventricular fibrillation in the rat. Can J Physiol Pharmacol 86, 700–709. [DOI] [PubMed] [Google Scholar]
- Barber MJ, Mueller TM, Henry DP, Felten SY & Zipes DP (1983). Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation 67, 787–796. [DOI] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Berthoud HR & Neuhuber WL (2000). Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 85, 1–17. [DOI] [PubMed] [Google Scholar]
- Boogers MJ, Borleffs CJ, Henneman MM, van Bommel RJ, van Ramshorst J, Boersma E, Dibbets‐Schneider P, Stokkel MP, van der Wall EE, Schalij MJ & Bax JJ (2010). Cardiac sympathetic denervation assessed with 123‐iodine metaiodobenzylguanidine imaging predicts ventricular arrhythmias in implantable cardioverter‐defibrillator patients. J Am Coll Cardiol 55, 2769–2777. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K (2005). Millisecond‐timescale, genetically targeted optical control of neural activity. Nat Neurosci 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Brack KE, Coote JH & Ng GA (2004). Interaction between direct sympathetic and vagus nerve stimulation on heart rate in the isolated rabbit heart. Exp Physiol 89, 128–139. [DOI] [PubMed] [Google Scholar]
- Brack KE, Coote JH & Ng GA (2011). Vagus nerve stimulation protects against ventricular fibrillation independent of muscarinic receptor activation. Cardiovasc Res 91, 437–446. [DOI] [PubMed] [Google Scholar]
- Brack KE, Patel VH, Mantravardi R, Coote JH & Ng GA (2009). Direct evidence of nitric oxide release from neuronal nitric oxide synthase activation in the left ventricle as a result of cervical vagus nerve stimulation. J Physiol 587, 3045–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain SD & Grant AD (2004). Vascular actions of calcitonin gene‐related peptide and adrenomedullin. Physiol Rev 84, 903–934. [DOI] [PubMed] [Google Scholar]
- Bru‐Mercier G, Deroubaix E, Capuano V, Ruchon Y, Rucker‐Martin C, Coulombe A & Renaud JF (2003). Expression of heart K+ channels in adrenalectomized and catecholamine‐depleted reserpine‐treated rats. J Mol Cell Cardiol 35, 153–163. [DOI] [PubMed] [Google Scholar]
- Brunet S, Aimond F, Li H, Guo W, Eldstrom J, Fedida D, Yamada KA & Nerbonne JM (2004). Heterogeneous expression of repolarizing, voltage‐gated K+ currents in adult mouse ventricles. J Physiol 559, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, O'Meara CC, Ho NN, Gannon J, Cai L & Lee RT (2015). A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol 79, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G (2009). Autonomic neurotransmission: 60 years since Sir Henry Dale. Annu Rev Pharmacol Toxicol 49, 1–30. [DOI] [PubMed] [Google Scholar]
- Calupca MA, Locknar SA, Zhang L, Harrison TA, Hoover DB & Parsons RL (2001). Distribution of cocaine‐ and amphetamine‐regulated transcript peptide in the guinea pig intrinsic cardiac nervous system and colocalization with neuropeptides or transmitter synthetic enzymes. J Comp Neurol 439, 73–86. [DOI] [PubMed] [Google Scholar]
- Calupca MA, Vizzard MA & Parsons RL (2000). Origin of pituitary adenylate cyclase‐activating polypeptide (PACAP)‐immunoreactive fibers innervating guinea pig parasympathetic cardiac ganglia. J Comp Neurol 423, 26–39. [PubMed] [Google Scholar]
- Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, Lai WW, Karagueuzian HS, Wolf PL, Fishbein MC & Chen PS (2000. a). Nerve sprouting and sudden cardiac death. Circ Res 86, 816–821. [DOI] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS & Chen LS (2000. b). Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 101, 1960–1969. [DOI] [PubMed] [Google Scholar]
- Cauley E, Wang X, Dyavanapalli J, Sun K, Garrott K, Kuzmiak‐Glancy S, Kay MW & Mendelowitz D (2015). Neurotransmission to parasympathetic cardiac vagal neurons in the brain stem is altered with left ventricular hypertrophy‐induced heart failure. Am J Physiol Heart Circ Physiol 309, H1281–H1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai W, Mehrotra S, Jan Danser AH & Schoemaker RG (2006). The role of calcitonin gene‐related peptide (CGRP) in ischemic preconditioning in isolated rat hearts. Eur J Pharmacol 531, 246–253. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S & Soderling TR (1998). Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci USA 95, 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H & Kuriyan J (2011). A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin‐dependent kinase II holoenzyme. Cell 146, 732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun LL & Patterson PH (1977). Role of nerve growth factor in the development of rat sympathetic neurons in vitro. I. Survival, growth, and differentiation of catecholamine production. J Cell Biol 75, 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coote JH (2013). Myths and realities of the cardiac vagus. J Physiol 591, 4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corr PB & Gillis RA (1974). Role of the vagus nerves in the cardiovascular changes induced by coronary occlusion. Circulation 49, 86–97. [DOI] [PubMed] [Google Scholar]
- Cuculi F, Herring N, De Caterina AR, Banning AP, Prendergast BD, Forfar JC, Choudhury RP, Channon KM & Kharbanda RK (2013). Relationship of plasma neuropeptide Y with angiographic, electrocardiographic and coronary physiology indices of reperfusion during ST elevation myocardial infarction. Heart 99, 1198–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT & Shannon TR (2014). Nitric oxide‐dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One 9, e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler MJ, Jeyaraj D & Rosenbaum DS (2011). Cardiac electrical remodeling in health and disease. Trends Pharmacol Sci 32, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dae MW, O'Connell JW, Botvinick EH & Chin MC (1995). Acute and chronic effects of transient myocardial ischemia on sympathetic nerve activity, density, and norepinephrine content. Cardiovasc Res 30, 270–280. [PubMed] [Google Scholar]
- Dawson TA, Li D, Woodward T, Barber Z, Wang L & Paterson DJ (2008). Cardiac cholinergic NO‐cGMP signaling following acute myocardial infarction and nNOS gene transfer. Am J Physiol Heart Circ Physiol 295, H990–H998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GM, Salvati P, Grossoni M, Ukmar G, Vaga L, Patrono C & Schwartz PJ (1993). Pharmacologic modulation of the autonomic nervous system in the prevention of sudden cardiac death. A study with propranolol, methacholine and oxotremorine in conscious dogs with a healed myocardial infarction. J Am Coll Cardiol 22, 283–290. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Vanoli E, Curcuruto P, Tommasini G & Schwartz PJ (1992). Prevention of life‐threatening arrhythmias by pharmacologic stimulation of the muscarinic receptors with oxotremorine. Am Heart J 124, 883–890. [DOI] [PubMed] [Google Scholar]
- DeGeest H, Levy MN, Zieske H & Lipman RI (1965). Depression of ventricular contractility by stimulation of the vagus nerves. Circ Res 17, 222–235. [DOI] [PubMed] [Google Scholar]
- DeMazumder D, Kass DA, O'Rourke B & Tomaselli GF (2015). Cardiac resynchronization therapy restores sympathovagal balance in the failing heart by differential remodeling of cholinergic signaling. Circ Res 116, 1691–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo SH, Barlow MA, Gonzalez L, Yoshishige D & Caffrey JL (2009). Repeated arterial occlusion, delta‐opioid receptor (DOR) plasticity and vagal transmission within the sinoatrial node of the anesthetized dog. Exp Biol Med (Maywood) 234, 84–94. [DOI] [PubMed] [Google Scholar]
- Doust JA, Pietrzak E, Dobson A & Glasziou P (2005). How well does B‐type natriuretic peptide predict death and cardiac events in patients with heart failure: systematic review. BMJ 330, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ & Anderson ME (2008). A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J & Bers DM (2015). S‐Nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin‐dependent protein kinase II δ. J Biol Chem 290, 25646–25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM & Bers DM (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O‐linked glycosylation. Nature 502, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler M, Kaye D, Lambert G, Esler D & Jennings G (1997). Adrenergic nervous system in heart failure. Am J Cardiol 80, 7L–14L. [DOI] [PubMed] [Google Scholar]
- Fallavollita JA, Heavey BM, Luisi AJ Jr, Michalek SM, Baldwa S, Mashtare TL Jr, Hutson AD, Dekemp RA, Haka MS, Sajjad M, Cimato TR, Curtis AB, Cain ME & Canty JM Jr (2014). Regional myocardial sympathetic denervation predicts the risk of sudden cardiac arrest in ischemic cardiomyopathy. J Am Coll Cardiol 63, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias M 3rd, Jackson KE, Yoshishige D & Caffrey JL (2003). Cardiac enkephalins interrupt vagal bradycardia via δ2‐opioid receptors in sinoatrial node. Am J Physiol Heart Circ Physiol 284, H1693–H1701. [DOI] [PubMed] [Google Scholar]
- Farias M, Jackson K, Stanfill A & Caffrey JL (2001). Local opiate receptors in the sinoatrial node moderate vagal bradycardia. Auton Neurosci 87, 9–15. [DOI] [PubMed] [Google Scholar]
- Freeman K, Tao W, Sun H, Soonpaa MH & Rubart M (2014). In situ three‐dimensional reconstruction of mouse heart sympathetic innervation by two‐photon excitation fluorescence imaging. J Neurosci Methods 221, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregoso SP & Hoover DB (2012). Development of cardiac parasympathetic neurons, glial cells, and regional cholinergic innervation of the mouse heart. Neuroscience 221, 28–36. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Kanazawa H, Aizawa Y, Ardell JL & Shivkumar K (2015). Cardiac innervation and sudden cardiac death. Circ Res 116, 2005–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RT & Habecker BA (2013). Infarct‐derived chondroitin sulfate proteoglycans prevent sympathetic reinnervation after cardiac ischemia‐reperfusion injury. J Neurosci 33, 7175–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RT, Ripplinger CM, Myles RC & Habecker BA (2016). Molecular mechanisms of sympathetic remodeling and arrhythmias. Circulation 9, e001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RT, Wang L, Lang BT, Cregg JM, Dunbar CL, Woodward WR, Silver J, Ripplinger CM & Habecker BA (2015). Targeting protein tyrosine phosphatase sigma after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat Commun 6, 6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebova NO & Ginty DD (2005). Growth and survival signals controlling sympathetic nervous system development. Annu Rev Neurosci 28, 191–222. [DOI] [PubMed] [Google Scholar]
- Gourine A & Gourine AV (2014). Neural mechanisms of cardioprotection. Physiology (Bethesda) 29, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Ling H, Willeford A, Pereira L, Gray CB, Erickson JR, Sarma S, Respress JL, Wehrens XH, Bers DM & Brown JH (2015). CaMKIIδ mediates β‐adrenergic effects on RyR2 phosphorylation and SR Ca2+ leak and the pathophysiological response to chronic β‐adrenergic stimulation. J Mol Cell Cardiol 85, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritman K, Van Winkle DM, Lorentz CU, Pennica D & Habecker BA (2006). The lack of cardiotrophin‐1 alters expression of interleukin‐6 and leukemia inhibitory factor mRNA but does not impair cardiac injury response. Cytokine 36, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez DA, Fernandez‐Tenorio M, Ogrodnik J & Niggli E (2013). NO‐dependent CaMKII activation during β‐adrenergic stimulation of cardiac muscle. Cardiovasc Res 100, 392–401. [DOI] [PubMed] [Google Scholar]
- Habecker BA, Bilimoria P, Linick C, Gritman K, Lorentz CU, Woodward W & Birren SJ (2008). Regulation of cardiac innervation and function via the p75 neurotrophin receptor. Auton Neurosci 140, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth R (1991). Reflexes from the heart. Physiol Rev 71, 617–658. [DOI] [PubMed] [Google Scholar]
- Hansson M, Kjorell U & Forsgren S (2001). Ingrowth of sympathetic innervation occurs concomitantly with a decrease of ANP in the growing rat cardiac ventricles. Anat Embryol (Berl) 203, 35–44. [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al Hafez B, Bilgen M & Smith PG (2006). Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res 1124, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan W, Woodward WR & Habecker BA (2012). Altered atrial neurotransmitter release in transgenic p75−/− and gp130 KO mice. Neurosci Lett 529, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ & Anderson ME (2011). Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 17, 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath BM, Xia J, Dong E, An RH, Brooks A, Liang C, Federoff HJ & Kass RS (1998). Overexpression of nerve growth factor in the heart alters ion channel activity and β‐adrenergic signalling in an adult transgenic mouse. J Physiol 512, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton DA, Li D, Almond SC, Dawson TA, Wang L, Channon KM & Paterson DJ (2007). Gene transfer of neuronal nitric oxide synthase into intracardiac ganglia reverses vagal impairment in hypertensive rats. Hypertension 49, 380–388. [DOI] [PubMed] [Google Scholar]
- Hellweg R & Hartung HD (1990). Endogenous levels of nerve growth factor (NGF) are altered in experimental diabetes mellitus: a possible role for NGF in the pathogenesis of diabetic neuropathy. J Neurosci Res 26, 258–267. [DOI] [PubMed] [Google Scholar]
- Herring N (2015). Autonomic control of the heart: going beyond the classical neurotransmitters. Exp Physiol 100, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring N, Cranley J, Lokale MN, Li D, Shanks J, Alston EN, Girard BM, Carter E, Parsons RL, Habecker BA & Paterson DJ (2012). The cardiac sympathetic co‐transmitter galanin reduces acetylcholine release and vagal bradycardia: implications for neural control of cardiac excitability. J Mol Cell Cardiol 52, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring N, Golding S & Paterson DJ (2000). Pre‐synaptic NO‐cGMP pathway modulates vagal control of heart rate in isolated adult guinea pig atria. J Mol Cell Cardiol 32, 1795–1804. [DOI] [PubMed] [Google Scholar]
- Herring N, Lee CW, Sunderland N, Wright K & Paterson DJ (2011). Pravastatin normalises peripheral cardiac sympathetic hyperactivity in the spontaneously hypertensive rat. J Mol Cell Cardiol 50, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring N, Lokale MN, Danson EJ, Heaton DA & Paterson DJ (2008). Neuropeptide Y reduces acetylcholine release and vagal bradycardia via a Y2 receptor‐mediated, protein kinase C‐dependent pathway. J Mol Cell Cardiol 44, 477–485. [DOI] [PubMed] [Google Scholar]
- Herring N & Paterson DJ (2001). Nitric oxide‐cGMP pathway facilitates acetylcholine release and bradycardia during vagal nerve stimulation in the guinea‐pig in vitro . J Physiol 535, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring N, Rigg L, Terrar DA & Paterson DJ (2001. a). NO‐cGMP pathway increases the hyperpolarisation‐activated current, If, and heart rate during adrenergic stimulation. Cardiovasc Res 52, 446–453. [DOI] [PubMed] [Google Scholar]
- Herring N, Zaman JA & Paterson DJ (2001. b). Natriuretic peptides like NO facilitate cardiac vagal neurotransmission and bradycardia via a cGMP pathway. Am J Physiol Heart Circ Physiol 281, H2318–H2327. [DOI] [PubMed] [Google Scholar]
- Hildreth V, Anderson RH & Henderson DJ (2009). Autonomic innervation of the developing heart: origins and function. Clin Anat 22, 36–46. [DOI] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Airaksinen MS & Saarma M (2000). GDNF family receptors in the embryonic and postnatal rat heart and reduced cholinergic innervation in mice hearts lacking Ret or GFRα2. Dev Dyn 219, 28–39. [DOI] [PubMed] [Google Scholar]
- Hoard JL, Hoover DB, Mabe AM, Blakely RD, Feng N & Paolocci N (2008). Cholinergic neurons of mouse intrinsic cardiac ganglia contain noradrenergic enzymes, norepinephrine transporters, and the neurotrophin receptors tropomyosin‐related kinase A and p75. Neuroscience 156, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan K & Markos F (2006). Vasoactive intestinal polypeptide receptor antagonism enhances the vagally induced increase in cardiac interval of the rat atrium in vitro . Exp Physiol 91, 641–646. [DOI] [PubMed] [Google Scholar]
- Hoover DB, Ganote CE, Ferguson SM, Blakely RD & Parsons RL (2004). Localization of cholinergic innervation in guinea pig heart by immunohistochemistry for high‐affinity choline transporters. Cardiovasc Res 62, 112–121. [DOI] [PubMed] [Google Scholar]
- Hoover DB, Isaacs ER, Jacques F, Hoard JL, Page P & Armour JA (2009). Localization of multiple neurotransmitters in surgically derived specimens of human atrial ganglia. Neuroscience 164, 1170–1179. [DOI] [PubMed] [Google Scholar]
- Hoover DB, Shepherd AV, Southerland EM, Armour JA & Ardell JL (2008). Neurochemical diversity of afferent neurons that transduce sensory signals from dog ventricular myocardium. Auton Neurosci 141, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins DA, Macdonald SE, Murphy DA & Armour JA (2000). Pathology of intrinsic cardiac neurons from ischemic human hearts. Anat Rec 259, 424–436. [DOI] [PubMed] [Google Scholar]
- Hulting J, Sollevi A, Ullman B, Franco‐Cereceda A & Lundberg JM (1990). Plasma neuropeptide Y on admission to a coronary care unit: raised levels in patients with left heart failure. Cardiovasc Res 24, 102–108. [DOI] [PubMed] [Google Scholar]
- Hunt RA, Ciuffo GM, Saavedra JM & Tucker DC (1995). Sympathetic innervation modulates the expression of angiotensin II receptors in embryonic rat heart grafted in oculo . J Mol Cell Cardiol 27, 2445–2452. [DOI] [PubMed] [Google Scholar]
- Hwang DY, Carlezon WA Jr, Isacson O & Kim KS (2001). A high‐efficiency synthetic promoter that drives transgene expression selectively in noradrenergic neurons. Hum Gene Ther 12, 1731–1740. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Tomhave ED, Lefkowitz RJ & Koch WJ (1998). Reciprocal in vivo regulation of myocardial G protein‐coupled receptor kinase expression by β‐adrenergic receptor stimulation and blockade. Circulation 98, 1783–1789. [DOI] [PubMed] [Google Scholar]
- Ieda M & Fukuda K (2009). Cardiac innervation and sudden cardiac death. Curr Cardiol Rev 5, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fukuda K, Hisaka Y, Kimura K, Kawaguchi H, Fujita J, Shimoda K, Takeshita E, Okano H, Kurihara Y, Kurihara H, Ishida J, Fukamizu A, Federoff HJ & Ogawa S (2004). Endothelin‐1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J Clin Invest 113, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Kanazawa H, Ieda Y, Kimura K, Matsumura K, Tomita Y, Yagi T, Onizuka T, Shimoji K, Ogawa S, Makino S, Sano M & Fukuda K (2006). Nerve growth factor is critical for cardiac sensory innervation and rescues neuropathy in diabetic hearts. Circulation 114, 2351–2363. [DOI] [PubMed] [Google Scholar]
- Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Taniguchi M, Lee JK, Matsumura K, Tomita Y, Miyoshi S, Shimoda K, Makino S, Sano M, Kodama I, Ogawa S & Fukuda K (2007). Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nat Med 13, 604–612. [DOI] [PubMed] [Google Scholar]
- Jackson KE, Farias M, Stanfill A & Caffrey JL (2001). Delta opioid receptors inhibit vagal bradycardia in the sinoatrial node. J Cardiovasc Pharmacol Ther 6, 385–393. [DOI] [PubMed] [Google Scholar]
- Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS & Anderson ME (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan A, Melman YF, Krumerman A & McDonald TV (2002). 14‐3‐3 amplifies and prolongs adrenergic stimulation of HERG K+ channel activity. EMBO J 21, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma Y, Akiyama T & Sato T (2009). Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J 276, 5111–5125. [DOI] [PubMed] [Google Scholar]
- Kakinuma Y, Tsuda M, Okazaki K, Akiyama T, Arikawa M, Noguchi T & Sato T (2013). Heart‐specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia‐inducible factor‐1α‐related defense mechanisms. J Am Heart Assoc 2, e004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla M, Chotalia M, Coughlan C, Hao G, Crabtree MJ, Tomek J, Bub G, Paterson DJ & Herring N (2016). Protection against ventricular fibrillation via cholinergic receptor stimulation and the generation of nitric oxide. J Physiol 594, 3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach A, Hedrick T & Waters J (2012). Selective optogenetic stimulation of cholinergic axons in neocortex. J Neurophysiol 107, 2008–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi‐Manabe H, Matsuhashi T, Endo J, Sano M, Kawakami T, Kimura T, Monkawa T, Hayashi M, Iwanami A, Okano H, Okada Y, Ishibashi‐Ueda H, Ogawa S & Fukuda K (2010). Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130‐signaling cytokines in rodents. J Clin Invest 120, 408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marban E, Spooner PM, Burke GL & Chakravarti A (2009). Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community‐based populations. Circulation 119, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada T, Yamazaki T, Akiyama T, Li M, Zheng C, Shishido T, Mori H & Sugimachi M (2007). Angiotensin II attenuates myocardial interstitial acetylcholine release in response to vagal stimulation. Am J Physiol Heart Circ Physiol 293, H2516–H2522. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Vaddadi G, Gruskin SL, Du XJ & Esler MD (2000). Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res 86, E80–E84. [DOI] [PubMed] [Google Scholar]
- Kim RH, Kim DH, Xiao J, Kim BH, Park SI, Panilaitis B, Ghaffari R, Yao J, Li M, Liu Z, Malyarchuk V, Kim DG, Le AP, Nuzzo RG, Kaplan DL, Omenetto FG, Huang Y, Kang Z & Rogers JA (2010). Waterproof AlInGaP optoelectronics on stretchable substrates with applications in biomedicine and robotics. Nat Mater 9, 929–937. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ieda M & Fukuda K (2012). Development, maturation, and transdifferentiation of cardiac sympathetic nerves. Circ Res 110, 325–336. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ieda M, Kanazawa H, Yagi T, Tsunoda M, Ninomiya S, Kurosawa H, Yoshimi K, Mochizuki H, Yamazaki K, Ogawa S & Fukuda K (2007). Cardiac sympathetic rejuvenation: a link between nerve function and cardiac hypertrophy. Circ Res 100, 1755–1764. [DOI] [PubMed] [Google Scholar]
- Kimura K, Kanazawa H, Ieda M, Kawaguchi‐Manabe H, Miyake Y, Yagi T, Arai T, Sano M & Fukuda K (2010). Norepinephrine‐induced nerve growth factor depletion causes cardiac sympathetic denervation in severe heart failure. Auton Neurosci 156, 27–35. [DOI] [PubMed] [Google Scholar]
- Komuro I, Kurabayashi M, Takaku F & Yazaki Y (1988). Expression of cellular oncogenes in the myocardium during the developmental stage and pressure‐overloaded hypertrophy of the rat heart. Circ Res 62, 1075–1079. [DOI] [PubMed] [Google Scholar]
- Kostreva DR, Zuperku EJ, Cusick JF & Kampine JP (1977). Ventral root mapping of cardiac nerves in the canine using evoked potentials. Am J Physiol 232, H590–H595. [DOI] [PubMed] [Google Scholar]
- Kreipke RE & Birren SJ (2015). Innervating sympathetic neurons regulate heart size and the timing of cardiomyocyte cell cycle withdrawal. J Physiol 593, 5057–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristen AV, Kreusser MM, Lehmann L, Kinscherf R, Katus HA, Haass M & Backs J (2006). Preserved norepinephrine reuptake but reduced sympathetic nerve endings in hypertrophic volume‐overloaded rat hearts. J Card Fail 12, 577–583. [DOI] [PubMed] [Google Scholar]
- Kuehl M & Stevens MJ (2012). Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat Rev Endocrinol 8, 405–416. [DOI] [PubMed] [Google Scholar]
- Kuret J & Schulman H (1985). Mechanism of autophosphorylation of the multifunctional Ca2+/calmodulin‐dependent protein kinase. J Biol Chem 260, 6427–6433. [PubMed] [Google Scholar]
- LaCroix C, Freeling J, Giles A, Wess J & Li YF (2008). Deficiency of M2 muscarinic acetylcholine receptors increases susceptibility of ventricular function to chronic adrenergic stress. Am J Physiol Heart Circ Physiol 294, H810–H820. [DOI] [PubMed] [Google Scholar]
- Landis SC & Keefe D (1983). Evidence for neurotransmitter plasticity in vivo: developmental changes in properties of cholinergic sympathetic neurons. Dev Biol 98, 349–372. [DOI] [PubMed] [Google Scholar]
- Lang D, Holzem K, Kang C, Xiao M, Hwang HJ, Ewald GA, Yamada KA & Efimov IR (2015). Arrhythmogenic remodeling of β2 versus β1 adrenergic signaling in the human failing heart. Circ Arrhythm Electrophysiol 8, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rockman HA & Koch WJ (2000). Catecholamines, cardiac β‐adrenergic receptors, and heart failure. Circulation 101, 1634–1637. [DOI] [PubMed] [Google Scholar]
- Leger J, Croll RP & Smith FM (1999). Regional distribution and extrinsic innervation of intrinsic cardiac neurons in the guinea pig. J Comp Neurol 407, 303–317. [PubMed] [Google Scholar]
- Lewis ME, Al‐Khalidi AH, Bonser RS, Clutton‐Brock T, Morton D, Paterson D, Townend JN & Coote JH (2001). Vagus nerve stimulation decreases left ventricular contractility in vivo in the human and pig heart. J Physiol 534, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Lee CW, Buckler K, Parekh A, Herring N & Paterson DJ (2012). Abnormal intracellular calcium homeostasis in sympathetic neurons from young prehypertensive rats. Hypertension 59, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Lu CJ, Hao G, Wright H, Woodward L, Liu K, Vergari E, Surdo NC, Herring N, Zaccolo M & Paterson DJ (2015). Efficacy of B‐type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons. Hypertension 66, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Nikiforova N, Lu CJ, Wannop K, McMenamin M, Lee CW, Buckler KJ & Paterson DJ (2013. a). Targeted neuronal nitric oxide synthase transgene delivery into stellate neurons reverses impaired intracellular calcium transients in prehypertensive rats. Hypertension 61, 202–207. [DOI] [PubMed] [Google Scholar]
- Li D, Wang L, Lee CW, Dawson TA & Paterson DJ (2007). Noradrenergic cell specific gene transfer with neuronal nitric oxide synthase reduces cardiac sympathetic neurotransmission in hypertensive rats. Hypertension 50, 69–74. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM & Gerdes AM (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28, 1737–1746. [DOI] [PubMed] [Google Scholar]
- Li J, Levick SP, DiPette DJ, Janicki JS & Supowit SC (2013. b). Alpha‐calcitonin gene‐related peptide is protective against pressure overload‐induced heart failure. Regul Pept 185, 20–28. [DOI] [PubMed] [Google Scholar]
- Li M, Zheng C, Sato T, Kawada T, Sugimachi M & Sunagawa K (2004. a). Vagal nerve stimulation markedly improves long‐term survival after chronic heart failure in rats. Circulation 109, 120–124. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM & Habecker BA (2004. b). Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol 286, H2229–H2236. [DOI] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P & Tsien RY (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J 96, 1803–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D & Heller BJ (2009). Requirement for Ca2+/calmodulin‐dependent kinase II in the transition from pressure overload‐induced cardiac hypertrophy to heart failure in mice. J Clin Invest 119, 1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yasui K, Arai A, Kamiya K, Cheng J, Kodama I & Toyama J (1999). β‐Adrenergic modulation of L‐type Ca2+‐channel currents in early‐stage embryonic mouse heart. Am J Physiol 276, H608–H613. [DOI] [PubMed] [Google Scholar]
- Liu YB, Wu CC, Lu LS, Su MJ, Lin CW, Lin SF, Chen LS, Fishbein MC, Chen PS & Lee YT (2003). Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res 92, 1145–1152. [DOI] [PubMed] [Google Scholar]
- Lockhart ST, Mead JN, Pisano JM, Slonimsky JD & Birren SJ (2000). Nerve growth factor collaborates with myocyte‐derived factors to promote development of presynaptic sites in cultured sympathetic neurons. J Neurobiol 42, 460–476. [PubMed] [Google Scholar]
- Lockhart ST, Turrigiano GG & Birren SJ (1997). Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci 17, 9573–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhurst JC, Tjen‐A‐Looi SC & Fu LW (2001). Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann NY Acad Sci 940, 74–95. [DOI] [PubMed] [Google Scholar]
- Lorentz CU, Alston EN, Belcik T, Lindner JR, Giraud GD & Habecker BA (2010). Heterogeneous ventricular sympathetic innervation, altered β‐adrenergic receptor expression, and rhythm instability in mice lacking the p75 neurotrophin receptor. Am J Physiol Heart Circ Physiol 298, H1652–H1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CJ, Hao G, Nikiforova N, Larsen HE, Liu K, Crabtree MJ, Li D, Herring N & Paterson DJ (2015). CAPON modulates neuronal calcium handling and cardiac sympathetic neurotransmission during dysautonomia in hypertension. Hypertension 65, 1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Gao X, Gu J, Zhou L, Guo S, Hao W, Zhou Z & Cao JM (2012). Nerve sprouting contributes to increased severity of ventricular tachyarrhythmias by upregulating iGluRs in rats with healed myocardial necrotic injury. J Mol Neurosci 48, 448–455. [DOI] [PubMed] [Google Scholar]
- Luther JA & Birren SJ (2006). Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol 96, 946–958. [DOI] [PubMed] [Google Scholar]
- Luther JA & Birren SJ (2009). p75 and TrkA signaling regulates sympathetic neuronal firing patterns via differential modulation of voltage‐gated currents. J Neurosci 29, 5411–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YX, Guo Z & Sun T (2013). CGRP inhibits norepinephrine induced apoptosis with restoration of Bcl‐2/Bax in cultured cardiomyocytes of rat. Neurosci Lett 549, 130–134. [DOI] [PubMed] [Google Scholar]
- Mabe AM, Hoard JL, Duffourc MM & Hoover DB (2006). Localization of cholinergic innervation and neurturin receptors in adult mouse heart and expression of the neurturin gene. Cell Tissue Res 326, 57–67. [DOI] [PubMed] [Google Scholar]
- Mabe AM & Hoover DB (2009). Structural and functional cardiac cholinergic deficits in adult neurturin knockout mice. Cardiovasc Res 82, 93–99. [DOI] [PubMed] [Google Scholar]
- Mabe AM & Hoover DB (2011). Remodeling of cardiac cholinergic innervation and control of heart rate in mice with streptozotocin‐induced diabetes. Auton Neurosci 162, 24–31. [DOI] [PubMed] [Google Scholar]
- McAllen RM & Spyer KM (1976). The location of cardiac vagal preganglionic motoneurones in the medulla of the cat. J Physiol 258, 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machhada A, Ang R, Ackland GL, Ninkina N, Buchman VL, Lythgoe MF, Trapp S, Tinker A, Marina N & Gourine AV (2015). Control of ventricular excitability by neurons of the dorsal motor nucleus of the vagus nerve. Heart Rhythm 12, 2285–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud AI, O'Meara CC, Gemberling M, Zhao L, Bryant DM, Zheng R, Gannon JB, Cai L, Choi WY, Egnaczyk GF, Burns CE, Burns CG, MacRae CA, Poss KD & Lee RT (2015). Nerves regulate cardiomyocyte proliferation and heart regeneration. Dev Cell 34, 387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR & Kass RS (2002). Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N & Marks AR (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. [DOI] [PubMed] [Google Scholar]
- Mastitskaya S, Marina N, Gourine A, Gilbey MP, Spyer KM, Teschemacher AG, Kasparov S, Trapp S, Ackland GL & Gourine AV (2012). Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre‐ganglionic neurones. Cardiovasc Res 95, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez GC, Li J, Law BA, Janicki JS, Supowit SC & Levick SP (2011). Substance P induces adverse myocardial remodelling via a mechanism involving cardiac mast cells. Cardiovasc Res 92, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van LS, Spillmann F, Campesi I, Madeddu P, Quaini F & Emanueli C (2010). Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res 106, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L & Schulman H (1992). Calmodulin trapping by calcium‐calmodulin‐dependent protein kinase. Science 256, 1199–1202. [DOI] [PubMed] [Google Scholar]
- Moon JI & Birren SJ (2008). Target‐dependent inhibition of sympathetic neuron growth via modulation of a BMP signaling pathway. Dev Biol 315, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles RC, Wang L, Bers DM & Ripplinger CM (2015). Decreased inward rectifying K+ current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J Physiol 593, 1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles RC, Wang L, Kang C, Bers DM & Ripplinger CM (2012). Local β‐adrenergic stimulation overcomes source‐sink mismatch to generate focal arrhythmia. Circ Res 110, 1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabauer M, Beuckelmann DJ, Uberfuhr P & Steinbeck G (1996). Regional differences in current density and rate‐dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 93, 168–177. [DOI] [PubMed] [Google Scholar]
- Nalivaiko E, Antunes VR & Paton JF (2010). Control of cardiac contractility in the rat working heart–brainstem preparation. Exp Physiol 95, 107–119. [DOI] [PubMed] [Google Scholar]
- Nash MP, Thornton JM, Sears CE, Varghese A, O'Neill M & Paterson DJ (2001). Ventricular activation during sympathetic imbalance and its computational reconstruction. J Appl Physiol (1985) 90, 287–298. [DOI] [PubMed] [Google Scholar]
- Ng GA, Brack KE & Coote JH (2001). Effects of direct sympathetic and vagus nerve stimulation on the physiology of the whole heart – a novel model of isolated Langendorff perfused rabbit heart with intact dual autonomic innervation. Exp Physiol 86, 319–329. [DOI] [PubMed] [Google Scholar]
- Ng GA, Brack KE, Patel VH & Coote JH (2007). Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res 73, 750–760. [DOI] [PubMed] [Google Scholar]
- Nishisato K, Hashimoto A, Nakata T, Doi T, Yamamoto H, Nagahara D, Shimoshige S, Yuda S, Tsuchihashi K & Shimamoto K (2010). Impaired cardiac sympathetic innervation and myocardial perfusion are related to lethal arrhythmia: quantification of cardiac tracers in patients with ICDs. J Nucl Med 51, 1241–1249. [DOI] [PubMed] [Google Scholar]
- Norris JE, Foreman RD & Wurster RK (1974). Responses of the canine heart to stimulation of the first five ventral thoracic roots. Am J Physiol 227, 9–12. [DOI] [PubMed] [Google Scholar]
- Nussinovitch U & Gepstein L (2015). Optogenetics for in vivo cardiac pacing and resynchronization therapies. Nat Biotechnol 33, 750–754. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF Jr, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalan R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Mendez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F & Califf RM (2011). Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med 365, 32–43. [DOI] [PubMed] [Google Scholar]
- O'Shea JE & Evans BK (1985). Innervation of bat heart: cholinergic and adrenergic nerves innervate all chambers. Am J Physiol 249, H876–H882. [DOI] [PubMed] [Google Scholar]
- Olivas A, Gardner RT, Wang L, Ripplinger CM, Woodward WR & Habecker BA (2016). Myocardial infarction causes transient cholinergic transdifferentiation of cardiac sympathetic nerves via gp130. J Neurosci 36, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardini BJ, Lund DD & Schmid PG (1990). Innervation patterns of the middle cervical–stellate ganglion complex in the rat. Neurosci Lett 117, 300–306. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Nkadi P, Woodward WR, Schutz G & Habecker BA (2009). Infarction‐induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol 95, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauza DH, Skripka V, Pauziene N & Stropus R (2000). Morphology, distribution, and variability of the epicardiac neural ganglionated subplexuses in the human heart. Anat Rec 259, 353–382. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W & Bers DM (2001). Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium‐calcium exchange, inward rectifier potassium current, and residual β‐adrenergic responsiveness. Circ Res 88, 1159–1167. [DOI] [PubMed] [Google Scholar]
- Polakova E, Illaste A, Niggli E & Sobie EA (2015). Maximal acceleration of Ca2+ release refractoriness by β‐adrenergic stimulation requires dual activation of kinases PKA and CaMKII in mouse ventricular myocytes. J Physiol 593, 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN & Sadek HA (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El‐Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XH, Song LS, Dobrev D, Maier LS & Anderson ME (2013). Oxidized Ca2+/calmodulin‐dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Vulapalli RS, Stevens SY & Liang CS (2002). Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am J Physiol Heart Circ Physiol 282, H363–H371. [DOI] [PubMed] [Google Scholar]
- Qu J & Robinson RB (2004). Cardiac ion channel expression and regulation: the role of innervation. J Mol Cell Cardiol 37, 439–448. [DOI] [PubMed] [Google Scholar]
- Quan KJ, Lee JH, Geha AS, Biblo LA, Van Hare GF, Mackall JA & Carlson MD (1999). Characterization of sinoatrial parasympathetic innervation in humans. J Cardiovasc Electrophysiol 10, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Quan KJ, Lee JH, Van Hare GF, Biblo LA, Mackall JA & Carlson MD (2002). Identification and characterization of atrioventricular parasympathetic innervation in humans. J Cardiovasc Electrophysiol 13, 735–739. [DOI] [PubMed] [Google Scholar]
- Rana OR, Saygili E, Meyer C, Gemein C, Kruttgen A, Andrzejewski MG, Ludwig A, Schotten U, Schwinger RH, Weber C, Weis J, Mischke K, Rassaf T, Kelm M & Schauerte P (2009). Regulation of nerve growth factor in the heart: The role of the calcineurin‐NFAT pathway. J Mol Cell Cardiol 46, 568–578. [DOI] [PubMed] [Google Scholar]
- Rokita AG & Anderson ME (2012). New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin‐dependent kinase II (CaMKII). Circulation 126, 2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Gutkind JS & Saavedra JM (1987). Angiotensin II binding sites in the conduction system of rat hearts. Am J Physiol 253, H1618–H1622. [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Vandroux D, Thomassin L, Athias P, Rochette L & Connat JL (2003). Calcitonin gene‐related peptide partly protects cultured smooth muscle cells from apoptosis induced by an oxidative stress via activation of ERK1/2 MAPK. Biochim Biophys Acta 1643, 65–73. [DOI] [PubMed] [Google Scholar]
- Schmid H, Forman LA, Cao X, Sherman PS & Stevens MJ (1999). Heterogeneous cardiac sympathetic denervation and decreased myocardial nerve growth factor in streptozotocin‐induced diabetic rats: implications for cardiac sympathetic dysinnervation complicating diabetes. Diabetes 48, 603–608. [DOI] [PubMed] [Google Scholar]
- Schultz HD (2001). Cardiac vagal chemosensory afferents. Ann NY Acad Sci 940, 59–73. [PubMed] [Google Scholar]
- Schultz JE, Hsu AK & Gross GJ (1998). Ischemic preconditioning in the intact rat heart is mediated by δ1‐ but not μ‐ or κ‐opioid receptors. Circulation 97, 1282–1289. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ (2014). Cardiac sympathetic denervation to prevent life‐threatening arrhythmias. Nat Rev Cardiol 11, 346–353. [DOI] [PubMed] [Google Scholar]
- Schwarz P, Diem R, Dun NJ & Forstermann U (1995). Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ Res 77, 841–848. [DOI] [PubMed] [Google Scholar]
- Schworer CM, Colbran RJ & Soderling TR (1986). Reversible generation of a Ca2+‐independent form of Ca2+(calmodulin)‐dependent protein kinase II by an autophosphorylation mechanism. J Biol Chem 261, 8581–8584. [PubMed] [Google Scholar]
- Shanks J & Herring N (2013). Peripheral cardiac sympathetic hyperactivity in cardiovascular disease: role of neuropeptides. Am J Physiol Regul Integr Comp Physiol 305, R1411–R1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks J, Mane S, Ryan R & Paterson DJ (2013. a). Ganglion‐specific impairment of the norepinephrine transporter in the hypertensive rat. Hypertension 61, 187–193. [DOI] [PubMed] [Google Scholar]
- Shanks J, Manou‐Stathopoulou S, Lu CJ, Li D, Paterson DJ & Herring N (2013. b). Cardiac sympathetic dysfunction in the prehypertensive spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 305, H980–H986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS & Bers DM (2000). Reverse mode of the sarcoplasmic reticulum calcium pump and load‐dependent cytosolic calcium decline in voltage‐clamped cardiac ventricular myocytes. Biophys J 78, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MJ & Zipes DP (2014). Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res 114, 1004–1021. [DOI] [PubMed] [Google Scholar]
- Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Kumfu S, Kumphune S, Chattipakorn S, KenKnight BH & Chattipakorn N (2014). Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm 11, 2278–2287. [DOI] [PubMed] [Google Scholar]
- Shirokova N, Kang C, Fernandez‐Tenorio M, Wang W, Wang Q, Wehrens XH & Niggli E (2014). Oxidative stress and Ca2+ release events in mouse cardiomyocytes. Biophys J 107, 2815–2827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Singh S, Sayers S, Walter JS, Thomas D, Dieter RS, Nee LM & Wurster RD (2013). Hypertrophy of neurons within cardiac ganglia in human, canine, and rat heart failure: the potential role of nerve growth factor. J Am Heart Assoc 2, e000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonimsky JD, Yang B, Hinterneder JM, Nokes EB & Birren SJ (2003). BDNF and CNTF regulate cholinergic properties of sympathetic neurons through independent mechanisms. Mol Cell Neurosci 23, 648–660. [DOI] [PubMed] [Google Scholar]
- Stanton MS, Tuli MM, Radtke NL, Heger JJ, Miles WM, Mock BH, Burt RW, Wellman HN & Zipes DP (1989). Regional sympathetic denervation after myocardial infarction in humans detected noninvasively using I‐123‐metaiodobenzylguanidine. J Am Coll Cardiol 14, 1519–1526. [DOI] [PubMed] [Google Scholar]
- Steele PA & Choate JK (1994). Innervation of the pacemaker in guinea‐pig sinoatrial node. J Auton Nerv Syst 47, 177–187. [DOI] [PubMed] [Google Scholar]
- Sudhof TC & Rothman JE (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudoh T, Minamino N, Kangawa K & Matsuo H (1990). C‐type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem Biophys Res Commun 168, 863–870. [DOI] [PubMed] [Google Scholar]
- Swaminathan PD, Purohit A, Hund TJ & Anderson ME (2012). Calmodulin‐dependent protein kinase II: linking heart failure and arrhythmias. Circ Res 110, 1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ & Anderson ME (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest 121, 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentivanyi M, Pace JB, Wechsler JS & Randall WC (1967). Localized myocardial responses to stimulation of cardiac sympathetic nerves. Circ Res 21, 691–702. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Maehara K, Onuki N, Saito T & Maruyama Y (2003). Decreased contractility of the left ventricle is induced by the neurotransmitter acetylcholine, but not by vagal stimulation in rats. Jpn Heart J 44, 257–270. [DOI] [PubMed] [Google Scholar]
- Takata Y, Arai T, Suzuki S, Kurihara J, Uezono T, Okubo Y & Kato H (2004). Captopril enhances cardiac vagal but not sympathetic neurotransmission in pithed rats. J Pharmacol Sci 95, 390–393. [DOI] [PubMed] [Google Scholar]
- Tan EM, Yamaguchi Y, Horwitz GD, Gosgnach S, Lein ES, Goulding M, Albright TD & Callaway EM (2006). Selective and quickly reversible inactivation of mammalian neurons in vivo using the Drosophila allatostatin receptor. Neuron 51, 157–170. [DOI] [PubMed] [Google Scholar]
- Taquet H, Komajda M, Grenier O, Belas F, Landault C, Carayon A, Lechat P, Grosgogeat Y & Legrand JC (1992). Plasma calcitonin gene‐related peptide decreases in chronic congestive heart failure. Eur Heart J 13, 1473–1476. [DOI] [PubMed] [Google Scholar]
- Thayer JF & Lane RD (2007). The role of vagal function in the risk for cardiovascular disease and mortality. Biol Psychol 74, 224–242. [DOI] [PubMed] [Google Scholar]
- Thomas D, Kiehn J, Katus HA & Karle CA (2004). Adrenergic regulation of the rapid component of the cardiac delayed rectifier potassium current, IKr, and the underlying hERG ion channel. Basic Res Cardiol 99, 279–287. [DOI] [PubMed] [Google Scholar]
- Ullman B, Hulting J & Lundberg JM (1994). Prognostic value of plasma neuropeptide‐Y in coronary care unit patients with and without acute myocardial infarction. Eur Heart J 15, 454–461. [DOI] [PubMed] [Google Scholar]
- Umoh NA, Walker RK, Millis RM, Al‐Rubaiee M, Gangula PR & Haddad GE (2014). Calcitonin gene‐related peptide regulates cardiomyocyte survival through regulation of oxidative stress by PI3K/Akt and MAPK signaling pathways. Ann Clin Exp Hypertens 2, 1007. [PMC free article] [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis‐Davies GC & Lederer WJ (1995). Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science 267, 1997–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoli E, De Ferrari GM, Stramba‐Badiale M, Hull SS Jr, Foreman RD & Schwartz PJ (1991). Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res 68, 1471–1481. [DOI] [PubMed] [Google Scholar]
- Vaseghi M, Gima J, Kanaan C, Ajijola OA, Marmureanu A, Mahajan A & Shivkumar K (2014). Cardiac sympathetic denervation in patients with refractory ventricular arrhythmias or electrical storm: intermediate and long‐term follow‐up. Heart Rhythm 11, 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseghi M, Lux RL, Mahajan A & Shivkumar K (2012). Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Heart Circ Physiol 302, H1838–H1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatner DE, Lavallee M, Amano J, Finizola A, Homcy CJ & Vatner SF (1985). Mechanisms of supersensitivity to sympathomimetic amines in the chronically denervated heart of the conscious dog. Circ Res 57, 55–64. [DOI] [PubMed] [Google Scholar]
- Wang L, Henrich M, Buckler KJ, McMenamin M, Mee CJ, Sattelle DB & Paterson DJ (2007). Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J Mol Cell Cardiol 43, 717–725. [DOI] [PubMed] [Google Scholar]
- Wang LH, Zhou SX, Li RC, Zheng LR, Zhu JH, Hu SJ & Sun YL (2012). Serum levels of calcitonin gene‐related peptide and substance P are decreased in patients with diabetes mellitus and coronary artery disease. J Int Med Res 40, 134–140. [DOI] [PubMed] [Google Scholar]
- Wang X, Pinol RA, Byrne P & Mendelowitz D (2014). Optogenetic stimulation of locus ceruleus neurons augments inhibitory transmission to parasympathetic cardiac vagal neurons via activation of brainstem α1 and β1 receptors. J Neurosci 34, 6182–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Heublein DM, Perrella MA, Lerman A, Rodeheffer RJ, McGregor CG, Edwards WD, Schaff HV & Burnett JC Jr (1993). Natriuretic peptide system in human heart failure. Circulation 88, 1004–1009. [DOI] [PubMed] [Google Scholar]
- Wengrowski AM, Wang X, Tapa S, Posnack NG, Mendelowitz D & Kay MW (2015). Optogenetic release of norepinephrine from cardiac sympathetic neurons alters mechanical and electrical function. Cardiovasc Res 105, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White IA, Gordon J, Balkan W & Hare JM (2015). Sympathetic reinnervation is required for mammalian cardiac regeneration. Circ Res 117, 990–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XH, Mohler PJ, Song LS & Anderson ME (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci USA 106, 5972–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenopoulos NP & Applegate RJ (1994). The effect of vagal stimulation on left ventricular systolic and diastolic performance. Am J Physiol 266, H2167–H2173. [DOI] [PubMed] [Google Scholar]
- Xu L, Gutbrod SR, Bonifas AP, Su Y, Sulkin MS, Lu N, Chung HJ, Jang KI, Liu Z, Ying M, Lu C, Webb RC, Kim JS, Laughner JI, Cheng H, Liu Y, Ameen A, Jeong JW, Kim GT, Huang Y, Efimov IR & Rogers JA (2014). 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat Commun 5, 3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamori T, Fukada K, Aebersold R, Korsching S, Fann MJ & Patterson PH (1989). The cholinergic neuronal differentiation factor from heart cells is identical to leukemia inhibitory factor. Science 246, 1412–1416. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP & Anderson ME (2006). Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol 291, H3065–H3075. [DOI] [PubMed] [Google Scholar]
- Yatani A, Shen YT, Yan L, Chen W, Kim SJ, Sano K, Irie K, Vatner SF & Vatner DE (2006). Down regulation of the L‐type Ca2+ channel, GRK2, and phosphorylated phospholamban: protective mechanisms for the denervated failing heart. J Mol Cell Cardiol 40, 619–628. [DOI] [PubMed] [Google Scholar]
- Yoon MS, Han J, Tse WW & Rogers R (1977). Effects of vagal stimulation, atropine, and propranolol on fibrillation threshold of normal and ischemic ventricles. Am Heart J 93, 60–65. [DOI] [PubMed] [Google Scholar]
- Younes A, Pepe S, Barron BA, Spurgeon HA, Lakatta EG & Caffrey JL (2000). Cardiac synthesis, processing, and coronary release of enkephalin‐related peptides. Am J Physiol Heart Circ Physiol 279, H1989–H1998. [DOI] [PubMed] [Google Scholar]
- Zaglia T, Pianca N, Borile G, Da Broi F, Richter C, Campione M, Lehnart SE, Luther S, Corrado D, Miquerol L & Mongillo M (2015). Optogenetic determination of the myocardial requirements for extrasystoles by cell type‐specific targeting of ChannelRhodopsin‐2. Proc Natl Acad Sci USA 112, E4495–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak R (1973). Cell proliferation during cardiac growth. Am J Cardiol 31, 211–219. [DOI] [PubMed] [Google Scholar]
- Zarzoso M, Rysevaite K, Milstein ML, Calvo CJ, Kean AC, Atienza F, Pauza DH, Jalife J & Noujaim SF (2013). Nerves projecting from the intrinsic cardiac ganglia of the pulmonary veins modulate sinoatrial node pacemaker function. Cardiovasc Res 99, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ & Anderson ME (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11, 409–417. [DOI] [PubMed] [Google Scholar]
- Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY & Chen X (2013). Cardiotoxic and cardioprotective features of chronic β‐adrenergic signaling. Circ Res 112, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B & Chen PS (2004). Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res 95, 76–83. [DOI] [PubMed] [Google Scholar]