

Abstract
Background and Purpose
Ageing is an important risk factor of non‐alcoholic fatty liver disease (NAFLD). Here, we investigated whether the deficiency of nicotinamide adenine dinucleotide (NAD+), a ubiquitous coenzyme, links ageing with NAFLD.
Experimental Approach
Hepatic concentrations of NAD+, protein levels of nicotinamide phosphoribosyltransferase (NAMPT) and several other critical enzymes regulating NAD+ biosynthesis, were compared in middle‐aged and aged mice or patients. The influences of NAD+ decline on the steatosis and steatohepatitis were evaluated in wild‐type and H247A dominant‐negative, enzymically‐inactive NAMPT transgenic mice (DN‐NAMPT) given normal or high‐fat diet (HFD).
Key Results
Hepatic NAD+ level decreased in aged mice and humans. NAMPT‐controlled NAD+ salvage, but not de novo biosynthesis pathway, was compromised in liver of elderly mice and humans. Given normal chow, middle‐age DN‐NAMPT mice displayed systemic NAD+ reduction and had moderate NAFLD phenotypes, including lipid accumulation, enhanced oxidative stress, triggered inflammation and impaired insulin sensitivity in liver. All these NAFLD phenotypes, especially release of pro‐inflammatory factors, Kupffer cell accumulation, monocytes infiltration, NLRP3 inflammasome pathway and hepatic fibrosis (Masson's staining and α‐SMA staining), deteriorated further under HFD challenge. Oral administration of nicotinamide riboside, a natural NAD+ precursor, completely corrected these NAFLD phenotypes induced by NAD+ deficiency alone or HFD, whereas adenovirus‐mediated SIRT1 overexpression only partially rescued these phenotypes.
Conclusions and Implications
These results provide the first evidence that ageing‐associated NAD+ deficiency is a critical risk factor for NAFLD, and suggest that supplementation with NAD+ substrates may be a promising therapeutic strategy to prevent and treat NAFLD.
Abbreviations
- ABCG
ATP‐binding cassette sub‐family G
- Ad
adenovirus
- ALT
alanine transaminase
- a‐SMA
a‐smooth muscle actin
- AST
aspartate transaminase
- DN
dominant negative
- HE
haematoxylin and eosin
- HFD
high‐fat diet
- IDO
indolamine 2,3‐dioxygenase
- LXR
liver X receptor
- MDA
malondialdehyde
- MTTP
microsomal triglyceride transfer protein
- NADS
NAD synthetase
- NAFLD
non‐alcoholic fatty liver disease
- NAMPT
nicotinamide phosphoribosyltransferase
- NASH
non‐alcoholic steatohepatitis
- NEFA
non‐esterified fatty acids
- NLRP3
NLR family, pyrin domain‐containing 3
- NMNAT
nicotinamide mononucleotide adenylyl transferase
- NR
nicotinamide riboside
- PKCε
protein kinase Cε
- SIRT
sirtuin
- WT
wild type
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | Nuclear hormone receptors c |
| IDO, indolamine 2,3‐dioxygenase | LXR |
| SIRT1, sirtuin 1 | Transporters d |
| PKCε, protein kinase Cε | ABCG1 |
| Catalytic receptors b | ABCG5 |
| NLRP3 | ABCG8 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a b c dAlexander et al., 2015a, 2015b, 2015c, 2015d).
Introduction
Non‐alcoholic fatty liver disease (NAFLD) encompasses a continuum, from simple hepatic steatosis with moderate fatty infiltration to non‐alcoholic steatohepatitis (NASH) with focal inflammation. A small portion of NAFLD may progress to advanced fibrosis, cirrhosis and eventually hepatocellular carcinoma (Cohen et al., 2011). At least 40% and 10% of amiddle‐aged population were diagnosed as NAFLD and NASH respectively (Williams et al., 2011). NAFLD is widely considered as a hepatic manifestation of the excessive fat/energy intake and subsequent metabolic dysfunction and is therefore linked to the development of obesity and insulin resistance (Cohen et al., 2011). Hepatic inflammation and injury or death of hepatocytes play crucial roles in the progression from NAFLD to NASH (Cohen et al., 2011). Age is an important risk factor of NAFLD by facilitating steatohepatitis (Fontana et al., 2013) and the prevalence of NAFLD in humans increases with age (Ford et al., 2002). However, although a number of therapeutic agents for NAFLD have been developed (Pathil et al., 2014; Liang et al., 2015; Tziomalos et al., 2015), the molecular mechanisms underlying the relationship between ageing and NAFLD are not fully understood.
NAD+ is a ubiquitous pyridine nucleotide that functions as an essential cofactor in mitochondrial oxidative phosphorylation. Recently, an entirely different role of NAD+ as a critical signalling regulator has been revealed with the identification of a group of NAD+‐consuming proteins such as the sirtuins (SIRTs) and PARPs (Canto et al., 2015). Liver is the major site of de novo biosynthesis of NAD+ from tryptophan (Heyes et al., 1997) but the predominant mechanism for maintaining intracellular NAD+ levels in mammals is the salvage pathway. The rate‐limiting enzyme in this pathway, nicotinamide phosphoribosyltransferase (NAMPT), converts nicotinamide to nicotinamide mononucleotide (Rongvaux et al., 2002). The pool of NAD+ appears to be limited as it is regulated by time (Nakahata et al., 2009; Ramsey et al., 2009) and nutrient availability (Yang et al., 2007; Canto et al., 2009). Importantly, recent evidence in animal models showed that a decrease in the NAD+ pool is a significant feature of ageing (Braidy et al., 2011; Yoshino et al., 2011; Gomes et al., 2013; Mouchiroud et al., 2013). Moreover, this decline of NAD+ plays crucial role in ageing by increasing oxidative stress and disrupting nuclear‐mitochondrial communication (Braidy et al., 2011; Gomes et al., 2013; Mouchiroud et al., 2013). Increasing the pool of NAD+ by dietary supplementation also has been shown to ameliorate age‐induced obesity and diabetes in mice (Yoshino et al., 2011). However, the causal relationship between the age‐related decrease in NAD+ and NAFLD development has not been established yet.
In this study, we tried to explore the potential role of the decrease in NAD+ with age in the onset and development of NAFLD. We confirmed the decrease of NAD+ content in livers from elderly patients compared with those from middle‐aged patients and found that the NAMPT‐mediated NAD+ salvage pathway, but not indolamine 2,3‐dioxygenase (IDO)‐mediated NAD+ de novo pathway, was impaired in livers from elderly humans compared with those from middle‐aged patients. We also demonstrated that deficiency of NAMPT‐mediated NAD+ biosynthesis alone was able to initiate hepatic steatosis and potetiate diet‐induced steatohepatitis even in middle‐aged mice. These findings suggest that the age‐related decrease in the NAD+ pool would render the aged liver more susceptible to NAFLD pathogenesis.
Methods
Animals
All animal care and experimental procedures were approved by the Scientific Investigation Board of the Second Military Medical University. The animal studies are reported as recommended by the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Male C57BL/6J mice were purchased from Sino‐British SIPPR/BK Lab Animal Ltd. (Shanghai, China). The enzyme‐dead H247A dominant negative NAMPT transgenic mouse model (referred as DN‐NAMPT mice) has been described in our previous report (Wang et al., 2014b; Zhao et al., 2015). Mice were housed separately and had ad libitum access to water and standard normal chow (Sino‐British SIPPR/BK Lab Animal Ltd.) and were kept under a 12 h dark–light cycle.
Experimental protocols
Mice were divided into different groups and selected for all experiments randomly by laboratory technicians. During the experiment and data analysis, single‐blind study design was applied. To induce diet‐induced NAFLD, 8‐week‐old mice were fed with a high‐fat diet (HFD) containing 60 kcal% fat, (D12492; Research Diets Inc., New Brunswick, NJ, USA) for 16 weeks. For dietary supplementation with nicotinamide riboside (NR), mice were fed a diet of pellets made from HFD powder mixed with NR (200 mg−1·kg−1·d−1), according to the daily intake of food. This HFD+NR diet was given for 4 weeks (Canto et al., 2012).
Human samples
The use of human tissue was approved by the Research Ethics Committee of Second Military Medical University, China. Written informed consent was obtained from all the patients. All specimens were handled and made anonymous according to the ethical and legal standards. Twelve patients (age >60 years, n = 6; age <45 years, n = 6) with hepatocellular carcinoma or hepatolithiasis were included in this study. There was no diabetes in these patients and none of the patients had received chemotherapy or radiotherapy prior to surgery. They underwent hepatectomy in the Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai, China. A small piece of normal liver tissue around the lesions was obtained during the hepatectomy and then rapidly frozen in liquid nitrogen for further biological experiments. The clinical information for these patients was shown in Supporting Information Table S1.
Biochemical assays
Mice were deeply anaesthetized with phenobarbital sodium (i.p.; 35 mg·kg−1), the thoracic cavity was opened and blood (1.0~1.5 ml) was obtained from the inferior vena cava. The blood sample was kept in an anticoagulation tube at ‐80°C. The plasma sample (~300 μl) was used to determine the plasma triglyceride, total cholesterol, non‐esterified fatty acids (NEFA), alanine transaminase (ALT) and aspartate transaminase (AST) levels, with an automatic biochemistry analyzer (Beckman) according to the manufacturer's instructions. Blood NAMPT concentration was measured using a commercial NAMPT ELISA Kit (AG‐45A‐0007Y, AdipoGen, Epalinges, Switzerland). Liver triglyceride and cholesterol contents were determined using commercial kits (#10010303 and #10007640) from Cayman. Liver DAG level was measured by a commercial EIA kit (LS‐F10645, LifeSpan BioSciences, Seattle, WA, USA). Malondialdehyde (MDA) and total‐SOD activity in liver tissue were determined by commercial kits from Cell Biolabs (San Diego, CA, USA).
Primary cultures of mouse macrophages
Primary macrophages were isolated and cultured as described previously (Liu et al., 2009). Briefly, mice were injected i.p. with 10% thioglycollate (2 mL per mouse) once a day for 3 days. Then the elicited peritoneal macrophages were collected from the peritoneal cavity by flushing with 5 mL of ice‐cold Hanks' balanced salt solution containing 10 U·mL−1 heparin. Macrophages were plated at a density of 5 × 104 cells mL−1 in DMEM supplemented with 10% FBS and were left to adhere for 6 h in a humidified atmosphere at 37°C with 5% CO2. Macrophages were washed twice and incubated with LPS (20 ng·mL−1) for 24h, in DMEM medium supplemented with 10% FBS. The medium was then collected for TNF‐α and IL‐6 determination with ELISA kits (Thermo Fisher Scientific, Waltham, MA, USA).
Histology and Oil Red O staining
Mice were deeply anaesthetized with i.p. phenobarbital sodium (35 mg·kg−1) and killed by cervical dislocation. The animals were then perfused with PBS solution (20mL), followed by 4% paraformaldehyde (20mL). Livers were then isolated and cut into small pieces which were fixed in 4% paraformaldehyde for an additional 8 hours. Then, the samples were embedded in paraffin and cut into sections (8 mm) and stained with haematoxylin and eosin (H&E), and Masson's trichrome as described previously (Zhong et al., 2009; Wang et al., 2014b). For Oil Red O staining, livers were embedded in JUNG tissue freezing medium (Leica, Wetzlar, Germany), and then cut to sections in freezing microtome (Leica). The frozen sections were stained with Oil Red O as described previously (Wang et al., 2014a).
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
The GTT and ITT experiments were performed as described previously (Wang et al., 2012). Mice were fasted overnight prior to GTT and for 4 h prior to ITT. For GTT, mice received an i.p. injection of glucose (2 g·kg−1 body weight). Blood samples (a few drops) were collected from the tail vein at 0, 15, 30, 60 and 120 min to measure blood glucose. For ITT, the mice received one i.p. injection of insulin (2.0 U·kg−1). Blood samples were taken at 0, 15, 30, 60, 90 and 120 min to measure blood glucose. Blood glucose values were determined in whole blood using an automatic glucose monitor (One Touch® Ultra, Lifescan, Johnson & Johnson, Milpitas, CA, USA).
Real‐time PCR
Total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. DNase I (Thermo Fisher Scientific) was used to remove DNA contamination. Real‐time PCR was performed with a SYBR green PCR kit (Applied Biosystems) and ABI7500 PCR system (Applied Biosystems) as described previously (Zhang et al., 2011). Each reaction was performed in triplicate, and analysis was performed by the 2−ΔΔCt method. The primers are listed in Supporting Information Table S2.
Immunoblotting
Immunoblotting was performed in Odyssey Infrared Fluorescence Imaging System (Li‐Cor, Lincoln, NE, USA) as described previously (Wang et al., 2012). Tissues were washed in PBS (0.1 mmol·L−1) and homogenized with the buffer (Tris–HCl pH 7.5, 20 mmol·L−1; EDTA, 2 mmol·L−1; NP‐40, 1%; Triton‐100, 1%; PMSF, 2 mmol·L−1; leupeptin, 50 μg·mL−1; aprotinin, 25 μg·mL−1; pesptatin A, 10 μg·mL−1; and DTT, 2 mmol·L−1). The cultured cells were lysed with Cell Protein Extraction Reagent (Pierce) supplemented with a protease inhibitor cocktail (Pierce). The total protein concentration was determined by the Bradford assay. Samples (about 30 μg ) were run on 10% SDS‐PAGE. The proteins were electrotransferred to nitrocellulose membranes, probed with primary antibody overnight and then incubated with Infrared‐Dyes‐conjugated secondary antibodies (Li‐Cor). The images were obtained through Odyssey Infrared Fluorescence Imaging System. All immunoblotting experiments were repeated at least three times.
The following antibodies were used: NAMPT (C20 clone, sc‐46440, Santa Cruz Biotechnology, Dallas, TX, USA, 1:1000 dilution), NAMPT (#11776‐1‐AP, sc‐67020, PTG Lab, Rosemont, IL, USA 1:1000 dilution for detecting endogenous NAMPT), α‐SMA (ab7817, Abcam, Cambridge, UK, 1:1500 dilution), NLRP3 (ab17267, Abcam, 1:800 dilution), apoptosis‐associated speck‐like protein (PAB6098, Abnova, Taiwan, 1:1000 dilution), IL‐1β (#12242, Cell Signaling Technology, Danvers, MA, USA, 1:800), IL‐18 (sc‐6179, Santa Cruz Biotechnology, 1:1000 dilution), Casp‐1 p20 (AG‐20B‐0042‐C100, AdipoGen, 1:1000 dilution), p‐Akt (#4060, Cell Signaling Technology, 1:1000 dilution), t‐Akt (#4691, Cell Signaling Technology, 1:1000 dilution), PKCε (#4691, Cell Signaling Technology, 1:1000 dilution), sirtuin‐1(SIRT1; S5196, Sigma‐Aldrich, 1:1000 dilution), acetylated lysines (4G12, Millipore, Billerica, MA, USA, 1:800 dilution), liver X receptor (LXR) (sc‐1000, Santa Cruz Biotechnology, 1:500 dilution), IDO (ab134197, Abcam, 1:800 dilution), NAD synthetase (NADS; ab139561, Abcam, 1:500 dilution) and nicotinamide mononucleotide adenylyl transferase (NMNAT; sc‐30841, Santa Cruz Biotechnology, 1:500 dilution).
Immunohistochemistry
For immunochemistry experiments, frozen sections (8μm) were fixed in 4% paraformaldehyde, blocked by 8% normal goat serum and incubated in specific primary antibodies. After being washed three times by PBS, the sections and cells were incubated with horseradish peroxidase‐conjugated secondary antibodies. Staining is visualized using substrate DAB. Images were obtained by digital microscope (Leica). The following antibodies were used: NAMPT (C20 clone, sc‐46440, Santa Cruz Biotechnology, 1:300 dilution), IL‐6 (ab6672, Abcam, 1: 600 dilution), α‐SMA (ab7817, Abcam, 1: 600 dilution), TNF‐α (ab6671, Abcam, 1: 600 dilution), F4/80 (LS‐C96373‐100, Lifespan, 1:1000 dilution), (ab134197, Abcam, 1:300 dilution), NADS (ab139561, Abcam, 1:300 dilution) and NMNAT (sc‐30841, Santa Cruz Biotechnology, 1:300 dilution).
SIRT1 overexpression
Adenovirus overexpressing mouse SIRT1 was produced using RAPAd® Adenoviral Expression Systems (Cell Biolabs, Inc.) and purified to 1011 PFU. DN‐NAMPT mice were injected with the Ad‐LacZ (control, 50 μL) and Ad‐SIRT1 (50 μL) twice (once every week) to overexpress SIRT1. At 5 days after the first injection, the overexpression of SIRT1 in liver was determined by immunoblotting. The effects of SIRT1 overexpression on liver accumulation, hepatic fibrosis and steatohepatitis were examined at 5 days after the second injection.
NAD+ determination and SIRT1 activity
NAD+ levels in liver samples were determined using a commercial kit (Catalogue: K337‐100, BioVision, Milpitas, CA, USA) as previously described (Wang et al., 2014b). SIRT1 activity was evaluated by two methods based on a commercial kit and immunoblotting of LXR deacetylation, respectively (Wang et al., 2014b). For the former, liver extracts were obtained using a RAPI lysis buffer plus protease inhibitor mix. Samples were incubated for 10 min at 30°C to allow for NAD+ degradation and incubated for 10 additional minutes with dithiothreitol 2 mM. SIRT1 in the tissues lysed by RIPA buffer was enriched by immunoprecipitation using a monoclonal anti‐SIRT1 mAb and then subjected to a deacetylation assay using the Fluorimetric Drug Discovery Kit (AK‐555, Biomol, Hamburg, Germany). For immunoblotting of LXR deacetylation, the LXR was immunoprecipitated, using a specific anti‐LXR antibody, and the deacetylation of LXR was determined by immunoblotting with an antibody against acetylated lysine.
Data and statistical analysis
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All values are presented as the mean ± SEM and analyzed by Student's t‐test or ANOVA followed by Tukey post hoc test. P < 0.05 was considered statistically significant. Data were analyzed with graphpad prism‐5 statistic software (GraphPad Software, Inc., La Jolla, CA, USA).
Materials
All chemicals and reagents were purchased from Sigma‐Aldrich (St. Louis, MO, USA) unless stated otherwise. Nicotinamide riboside (NR; CAS No. 1341‐23‐7) was synthesized and provided by Biochempartner Co., Ltd. (Shanghai, China). The purity of NR is >98% according to the quality inspection.
Results
Age‐related depletion of the NAD+ pool and dysfunction of NAMPT‐mediated NAD+ salvage pathway in livers from mice and human patients
In line with previous animal studies showing systemic NAD+ levels decrease with age (Braidy et al., 2011; Yoshino et al., 2011; Mouchiroud et al., 2013), we detected a significant reduction of hepatic NAD+ level in old (20‐month‐old) mice compared with middle‐aged (12‐month‐old) or young adult (4‐month‐old) mice (Figure 1A). The increased activity of NAD+‐consuming proteins was reported to play a substantial role in the gradual depletion of NAD+ pool with age by reducing available NAD+ (Imai and Guarente, 2014). Apart from the increase in NAD+‐consuming processes, we speculated the dysfunction of NAD+ biosynthesis might also contribute to the NAD+ pool depletion with age. To this end, we examined the protein expression of several major enzymes governing NAD biosynthesis in mammal: NAMPT, IDO, NADS and NMNAT (Fig. S1). NAMPT and IDO are rate‐limiting enzymes for NAD+ salvage and de novo biosynthesis pathways, respectively (Ruggieri et al., 2015). Interestingly, we found that NAMPT was down‐regulated in aged mice liver, whereas IDO, NADS and NMNAT were up‐regulated (Figure 1B).
Figure 1.
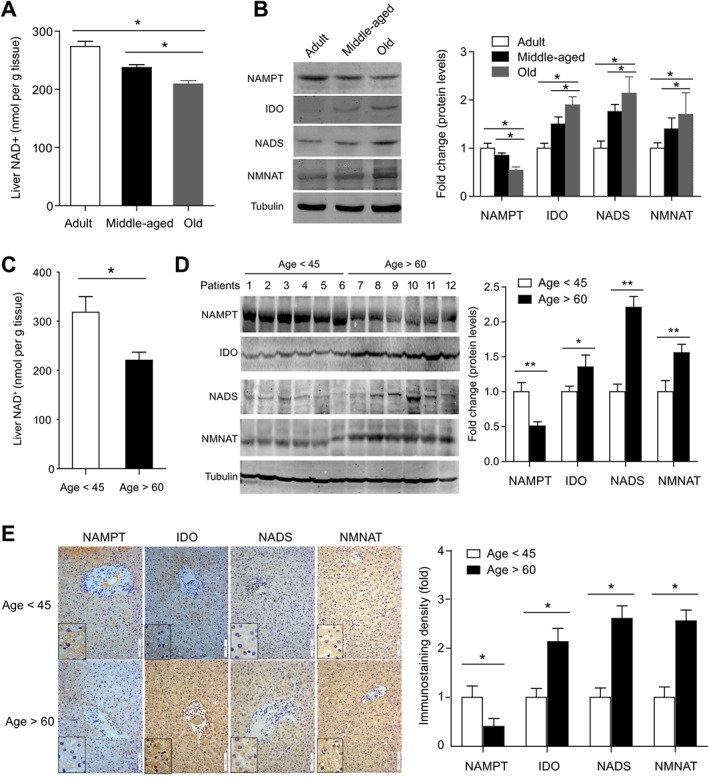
Age‐related decrease in the hepatic NAD+ pool and impaired NAD+ salvage biosynthesis pathway in mice and humans. (A) NAD+ level in liver tissues obtained from adult (4‐month‐old), middle‐aged (12‐month‐old) and old (20‐month‐old) mice. *P < 0.05. n = 8 for each group. (B) Immunoblotting analysis of the protein levels of NAMPT, IDO, NADS and NMNAT in liver tissues from adult, middle‐aged and old mice. *P < 0.05. n = 6 for each group. (C) NAD+ level in liver tissues obtained by hepatectomy from patients. *P < 0.05. n = 6 for each group. (D) Immunoblotting analysis of the protein levels of NAMPT, IDO, NADS and NMNAT in liver tissues obtained by hepatectomy from patients. *P < 0.05. n = 6 for each group. (E) Immunohistochemistry analysis of the protein levels of NAMPT, IDO, NADS and NMNAT in liver tissues obtained by hepatectomy from patients. *P < 0.05. n = 6 for each group.
To confirm the decrease of the hepatic NAD+ pool in mice, we measured the NAD+ level in non‐pathological liver tissues, isolated during hepatectomy from elderly patients (age >60 years) and middle‐aged patients (age <45 years) (see information about these patients in Supporting Information Table S1). The hepatic NAD+ levels in elderly patients were about ~70% (Figure 1C) of those in middle‐aged patients. Immunoblotting analysis demonstrated a significant down‐regulation of NAMPT in liver tissues from elderly patients compared with those from middle‐aged patients (Figure 1D). In contrast, NADS and NMNAT in liver tissues from elderly patients were up‐regulated (Figure 1D). Immunohistochemistry staining also demonstrated that all these three enzymes were mainly localized in hepatocyte cytoplasm, and confirmed that NAMPT was down‐regulated, but NMNAT and NADS were up‐regulated in aged human livers (Figure 1E). All these animal and human data suggest that the decline of NAMPT‐controlled NAD+ salvage biosynthesis was a causal factor in the age‐related NAD+ pool depletion, while the increased NAD+ de novo biosynthesis may be adaptive responses.
Deficiency of NAMPT‐mediated NAD+ pool impairs lipid homeostasis and favours hepatic steatosis in middle‐aged mice
To answer whether the lower level of NAMPT protein would lead to depletion of NAD+ in liver and thereby affect the progress to NAFLD, we used H247A DN‐NAMPT mice, which have been described in detail previously (Wang et al., 2014b) to mimic the systemic reduction of NAMPT and NAD+. These mice have lower endogenous NAMPT expression (Supporting Information Figure S2A and B) and a partly depleted NAD+ pool (Supporting Information Figure S2C) in liver. There were no differences of body weight or liver weight between adult wild‐type (WT) and DN‐NAMPT mice, although the liver to body weight ratio in DN‐NAMPT mice was slightly but significantly higher (Supporting Information Figure S2D).
As middle‐aged (12‐month‐old) DN‐NAMPT mice have already displayed impaired hepatic NAMPT expression and a lower NAD+ pool, as are present in elderly patients, we examined whether these mice also exhibited greater susceptibility to development of NAFLD. The hepatic triglyceride and cholesterol contents were increased in 12‐month‐old DN‐NAMPT mice compared with WT mice (Figure 2A). Plasma NEFA level was unaffected (Figure 2B). GTT and ITT revealed a slight insulin resistance in DN‐NAMPT mice (Figure 2C and D). Quantitative real‐time PCR assay showed that hepatic mRNA levels of triglyceride and cholesterol efflux genes, such as microsomal triglyceride transfer protein (MTTP), ATP‐binding cassette sub‐family G member 1 (ABCG1), ABCG5, ABCG8 and LDL receptor (LDL‐R), were reduced in DN‐NAMPT mice (Figure 2E and F). H&E staining showed small vacuoles (black arrows) and irregular arrangement of hepatocyes in DN‐NAMPT mice liver (Figure 2G). Furthermore, DN‐NAMPT mice displayed extensive hepatic steatosis revealed by a more prominent staining with Oil Red‐O even with a normal chow diet (Figure 2H). Taken together, these data suggest that, systemic deficiency of NAMPT‐NAD+ could impair lipid homeostasis and induce moderate fat accumulation in liver even in middle‐aged mice.
Figure 2.
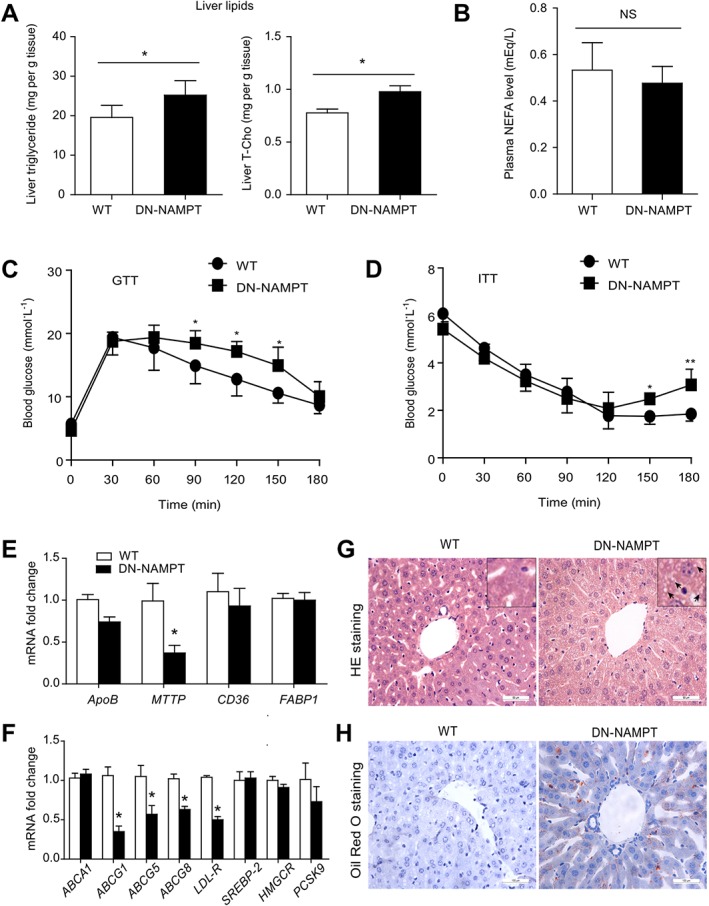
Impaired systemic lipid homeostasis and moderate hepatic steatosis in DN‐NAMPT mice given normal chow. (A) The levels of triglyceride and total‐cholesterol in liver tissue of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group. (B) Plasma NEFA levels of WT and DN‐NAMPT mice. (C and D) GTT and ITT showing insulin resistance in DN‐NAMPT mice. *P < 0.05. n = 8 for each group. (E and F) Relative mRNA levels of the genes regulating transport of triglyceride (E) and cholesterol (F) in liver tissues of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group. (G) Representative images for H&E staining of liver tissues of WT and DN‐NAMPT mice. (H) Representative images for Oil Red O staining of liver tissues of WT and DN‐NAMPT mice.
Deficiency of the NAD+ pool, mediated by NAMPT, induces moderate hepatic inflammation and injury under normal diet condition
We further investigated the hepatic inflammation in our system. In macrophages isolated from DN‐NAMPT mice, the NAMPT expression and NAD+ level (Supplementary Fig. 3A‐C) were reduced as expected. As observed earlier (Van Gool et al., 2009), the release of the proinflammatory TNF‐α from DN‐NAMPT macrophages upon acute LPS challenge was slightly lower than those from WT macrophages, whereas the release of IL‐6 was not affected (Supplementary Fig. S3D). Interestingly, enhanced TNF‐α protein and mRNA expression was observed in livers from middle‐aged DN‐NAMPT mice, whereas IL‐6 protein and mRNA expression was not different in these two strains of mice (Figure 3A and B). Immunohistochemical staining for F4/80 and CD‐11b revealed that the numbers of accumulated Kupffer cells and infiltrated monocytes were higher in middle‐aged DN‐NAMPT mice (Figure 3C). The inflammasome NLRP3 promoted steatohepatitis and fibrosis (Wree et al., 2014) but we did not find any difference in the activation of NLRP3 and concentrations of IL‐1β and IL‐18, two pro‐inflammatory cytokines evoked by NLRP3 inflammasome, between WT and DN‐NAMPT mice, given normal chow (Supplementary Fig. 4A‐B). DN‐NAMPT mice had higher levels of MDA, a widely accepted lipid peroxidation marker (Figure 3D, left panel). There was no significant difference of anti‐oxidant activity in liver between middle‐aged WT and DN‐NAMPT mice (Figure 3D, right panel). Next, we assessed hepatic fibrosis. Three hepatic fibrosis genes, including pro‐collagen α1 (Pro‐Col1α1), TGF‐β and tissue inhibitor of metalloproteinases‐1 were analyzed. Only Pro‐Col1α1 mRNA expression was increased in DN‐NAMPT mice liver (Figure 3E). Protein level of α‐smooth muscle actin (α‐SMA), another fibrosis marker, was unchanged (Figure 3F). Masson's staining also demonstrated that NAMPT‐NAD+ deficiency alone did not induce significant collagen deposition in liver (Figure 3G). Plasma levels of ALT and AST in DN‐NAMPT were higher than those in WT mice (Figure 3H). Collectively, these results suggest that deficiency of the NAD+ pool, mediated by NAMPT, did induce moderate liver injury in middle‐aged mice, given a normal diet.
Figure 3.
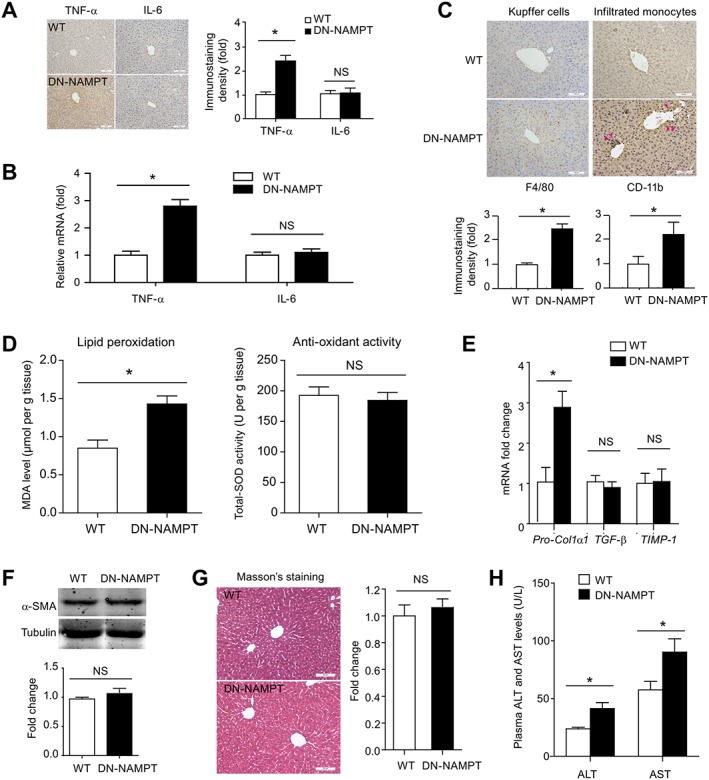
Hepatic inflammation and injury in DN‐NAMPT mice given normal chow. Immunohistochemistry (A) and real‐time PCR (B) analyses of the pro‐inflammatory factors TNF‐α and IL‐6 in liver tissues of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group. Scale bar, 100 μM. (C) Representative images and quantitative analysis of Kupffer cell numbers and infiltrated monocytes by F4/80 (left panel) and CD‐11b (right panel) immunohistochemistry staining in liver tissues of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group. Scale bar, 100 μM. (D) Lipid peroxidation and antioxidant activity were evaluated by MDA level and total SOD activity respectively. *P < 0.05. n = 8 for each group. (E) The mRNA levels of Pro‐Col1α1, TGF‐β and TIMP‐1 in WT and DN‐NAMPT mice livers. *P < 0.05. NS, no significance. n = 8 for each group. (F) The protein level of α‐SMA in WT and DN‐NAMPT mice livers. NS, no significance. n = 8 for each group. (G) Masson's staining in WT and DN‐NAMPT mice livers. NS, no significance. n = 8 for each group. (H) Plasma ALT and AST levels of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group.
Deficiency of the NAD+ pool, mediated by NAMPT, promotes steatohepatitis in diet‐induced NAFLD model
We determined the influence of HFD on hepatic NAD+ level and NAMPT expression in mice. The hepatic NAD+ level in middle‐aged mice with NAFLD, induced by HFD for 16 weeks, was significantly reduced (Supporting Information Figure S5A). The reduction of NAD+ content was also observed in other organs such as skeletal muscle (data not shown). Plasma NAMPT levels in NAFLD mice were found to be reduced by ~25% (Supporting Information Figure S5B). NAMPT protein was also reduced by ~40% in mice with NAFLD induced by HFD (Supporting Information Figure S5C). These results indicate a systemic reduction of the NAD+ pool, mediated by NAMPT, during development of NAFLD, which is consistent with previous findings in NAFLD patients (Dahl et al., 2010) and rodents (Yoshino et al., 2011; Zhang et al., 2014).
Next, the middle‐aged WT and DN‐NAMPT mice were fed with HFD for additional 16 weeks to further investigate whether this intervention would produce more pronounced NAFLD phenotypes. Both WT and DN‐NAMPT mice displayed NAFLD phenotypes after HFD administration, while DN‐NAMPT mice exhibited greater accumulation of triglyceride and total cholesterol (Figure 4A) in liver. The changes in lipid metabolism‐related genes are shown in Supporting Information Figure S5D and E. H&E staining showed that hepatocellular ballooning was present throughout the livers of DN‐NAMPT mice and to a lesser degree in WT mice (Figure 4B). Oil Red O staining also demonstrated greater hepatic lipid accumulation in livers from DN‐NAMPT mice (Figure 4C). Analysis of apoptosis by TUNEL assays showed more apoptotic cells in livers of DN‐NAMPT mice (Figure 4D). DN‐NAMPT mice also exhibited significantly higher levels of TNF‐α protein and mRNA in liver tissue after HFD treatment (Figure 4E and F) and a greater activation of the NLRP3 inflammasome pathway (Figure 4G).
Figure 4.
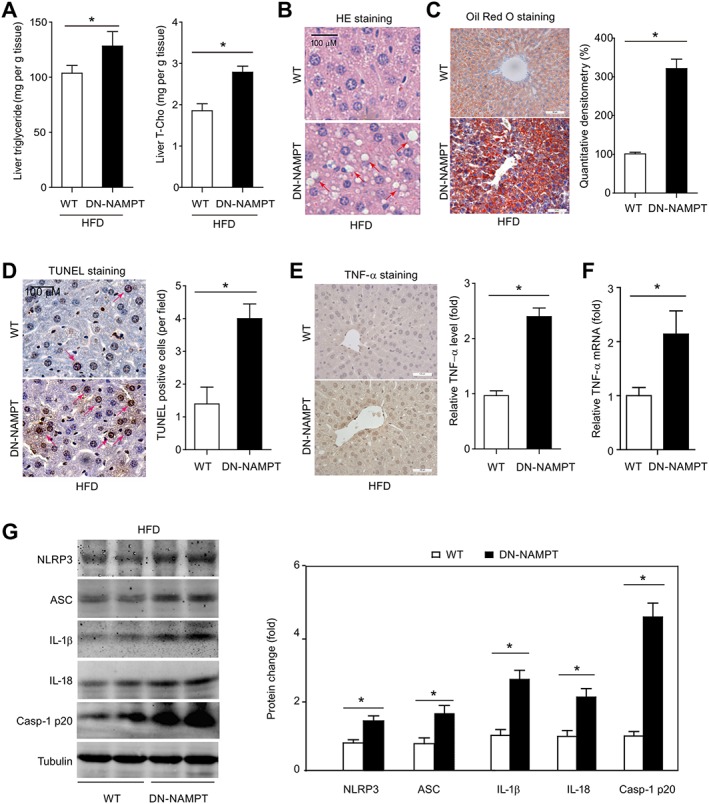
NAD+ pool deficiency promotes steatohepatitis in diet‐induced NAFLD model. (A) Lipid profile in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 8 for each group. (B) HE staining in livers of WT and DN‐NAMPT mice with NAFLD. (C) Oil Red O staining in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 8 for each group. (D) Apoptosis in livers of WT and DN‐NAMPT mice with NAFLD was determined by TUNEL staining. *P < 0.05. n = 8 for each group. TNF‐α protein (E) and mRNA (F) expression in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 8 for each group. (G) Activation of NLRP‐3 inflammasome pathway and production of IL‐1β and IL‐18 in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 4 for each group. ASC, apoptosis‐associated speck‐like protein.
Deficiency of the NAD+ pool, mediated by NAMPT, increases liver fibrosis and insulin resistance in diet‐induced NAFLD model
We next compared liver fibrosis between WT and DN‐NAMPT mice given the HFD. Higher mRNA levels of pro‐fibrotic genes, including α‐SMA, TGF‐β, TIMP‐2, Pro‐Col1α1 and Pro‐Col1α2 were observed in livers from HFD‐fed DN‐NAMPT mice, compared with WT mice (Figure 5A). This enhancement of hepatic fibrosis in DN‐NAMPT livers by HFD was confirmed by α‐SMA immunoblotting (Figure 5B), α‐SMA immunohistochemistry (Figure 5C) and Masson's staining (Figure 5D). As shown in Figure 2, NAMPT‐NAD+ deficiency alone was sufficient to induce whole‐body insulin resistance with normal diets. We therefore assessed hepatic insulin resistance after HFD, by measuring phosphorylation of Akt in liver tissue. After a bolus injection of insulin, Akt was phosphorylated rapidly in livers from WT mice but to a much lesser degree, in livers from DN‐NAMPT mice (Figure 5E). We also examined the hepatic DAG‐protein kinase Cε (PKCε) signalling pathway (Fabbro et al., 2015), which is a common mechanism for the development of insulin resistance in fatty liver disease (Jornayvaz and Shulman, 2012). Hepatic DAG content (Figure 5F) and PKCε expression (Figure 5G) in DN‐NAMPT mice were significantly higher, indicating that the NAMPT‐NAD+ deficiency increased the insulin resistance in this model of diet‐induced NAFLD.
Figure 5.
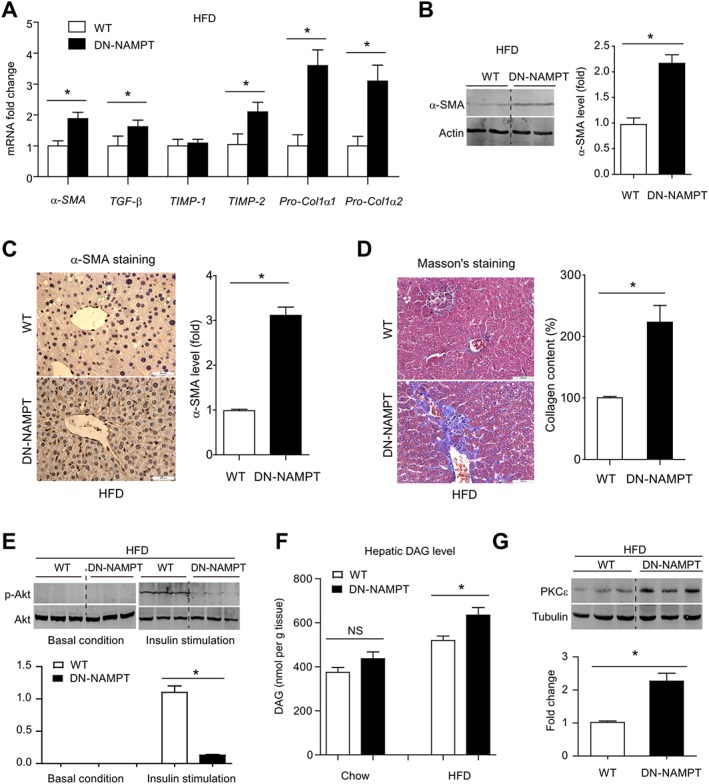
NAD+ pool deficiency increases fibrosis and insulin resistance in diet‐induced NAFLD model. (A) The mRNA levels of pro‐fibrosis genes in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 8 for each group. Protein levels of α‐SMA in livers of WT and DN‐NAMPT mice with NAFLD were detected by immunoblotting (B) and immunohistochemistry (C). *P < 0.05. n = 8 for each group. (D) Masson's staining in livers of WT and DN‐NAMPT mice with NAFLD. *P < 0.05. n = 8 for each group. (E) Phosphorylation of Akt in liver tissues of WT and DN‐NAMPT mice with NAFLD before and after bolus injection of insulin. *P < 0.05. n = 8 for each group. (F) DAG levels in liver tissues of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group. (G) Protein level of PKCε in liver tissues of WT and DN‐NAMPT mice. *P < 0.05. n = 8 for each group.
Replenishment with the natural NAD+ precursor NR, but not SIRT1 overexpression, completely corrects the development of NAFLD, induced by NAMPT‐NAD+ deficiency
As SIRT1 is the most intensively studied NAD+‐dependent effector (Canto and Auwerx, 2012), we measured SIRT1 expression and activity in livers from middle‐aged WT and DN‐NAMPT mice. Hepatic SIRT1 mRNA and protein expression remained unchanged in DN‐NAMPT mice given normal chow or HFD (Supporting Information Figure S6). However, the SIRT1 activity in DN‐NAMPT livers was lower than that in WT livers, after either normal chow or HFD (Supporting Information Figure S7A). The deacetylation of LXR in DN‐NAMPT livers was also lower than that in WT livers, after either normal chow or HFD (Supporting Information Figure S7B), consistent with the deacetylation of LXR being controlled by SIRT1 (Li et al., 2007).
We next asked whether the SIRT1 deficiency induced by depletion of the NAD+ pool could account totally for the susceptibility to hepatic steatosis and steatohepatitis in DN‐NAMPT mice. In DN‐NAMPT mice, we compared the effects of SIRT1 overexpression and replenishment with NR, a NAD+ precursor, widely used for increasing NAD+ content (Canto et al., 2012; Mouchiroud et al., 2013). Injection of adenovirus carrying SIRT1 (Ad‐SIRT1) into the tail vein significantly increased SIRT1 protein in livers from DN‐NAMPT mice (Supporting Information Figure S8). SIRT1 overexpression reversed the decrease in ABCG1, ABCG5, ABCG8 and LDL‐R mRNA but had no effect on the decrease of MTTP mRNA in DN‐NAMPT livers (Figure 6A). In contrast, feeding the mice on a diet supplemented with NR for 4 weeks reversed the down‐regulation of all these genes (Figure 6A). In addition, Oil Red O staining demonstrated that NR supplementation markedly inhibited the accumulation of lipids in DN‐NAMPT livers, whereas the effect of SIRT1 overexpression was weaker (Figure 6B). Dietary supplementation with NR also abolished the accumulation of triglyceride and cholesterol in DN‐NAMPT livers, while SIRT1 overexpression only partly prevented this hepatic lipid accumulation (Figure 6C).
Figure 6.
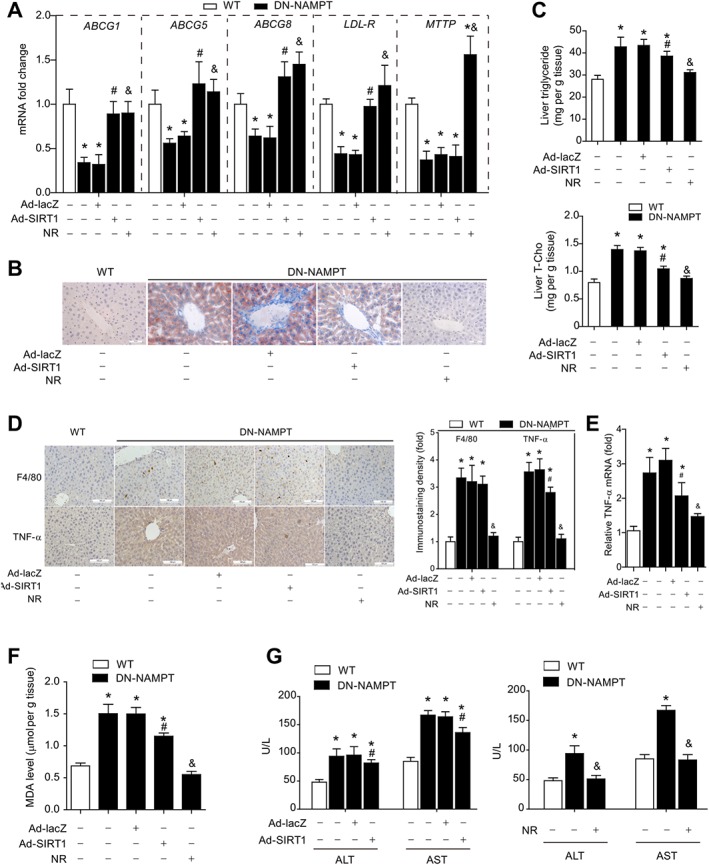
Dietary supplementation with NR, but not SIRT1 overexpression, completely corrects hepatic steatosis and steatohepatitis in DN‐NAMPT mice under normal chow. (A) Effect of SIRT1 overexpression or NR on expression of lipid regulatory genes in livers of WT and DN‐NAMPT mice. *P < 0.05 versus WT, # P < 0.05 DN‐NAMPT + Ad‐SIRT1 versus DN‐NAMPT + Ad‐lacZ, & P < 0.05 DN‐NAMPT + NR versus DN‐NAMPT by ANOVA analysis. n = 8 for each group. (B) Representative images of Oil Red O staining for the livers of WT and DN‐NAMPT mice. (C) Effect of SIRT1 overexpression or NR on lipid triglyceride and cholesterol in livers of WT and DN‐NAMPT mice. T‐Cho, total cholesterol. *P < 0.05 versus WT, # P < 0.05 DN‐NAMPT + Ad‐SIRT1 versus DN‐NAMPT + Ad‐lacZ, & P < 0.05 DN‐NAMPT + NR versus DN‐NAMPT by ANOVA analysis. n = 8 for each group. (D–E) Immunohistochemistry staining showing the effects of NR and SIRT1 overexpression on Kupffer cell accumulation (D) and TNF‐α protein (D) and mRNA (E) expression. *P < 0.05 versus WT, # P < 0.05 DN‐NAMPT + Ad‐SIRT1 versus DN‐NAMPT + Ad‐lacZ, & P < 0.05 DN‐NAMPT + NR versus DN‐NAMPT by ANOVA analysis. n = 8 for each group. Effects of NR and SIRT1 overexpression on hepatic MDA level (F) and blood ALT and AST levels (G). *P < 0.05 versus WT, # P < 0.05 DN‐NAMPT + Ad‐SIRT1 versus DN‐NAMPT + Ad‐lacZ, & P < 0.05 DN‐NAMPT + NR versus DN‐NAMPT by ANOVA analysis. n = 8 for each group.
We further evaluated the influence of SIRT1 overexpression and NR supplementation on steatohepatitis. Immunohistochemistry analyses showed that SIRT1 overexpression failed to block the Kupffer cell accumulation although it appeared to slightly reduce TNF‐α level in livers from DN‐NAMPT mice (Figure 6D). Unlike SIRT1 overexpression, NR supplementation successfully decreased both Kupffer cell accumulation and TNF‐α levels in DN‐NAMPT livers (Figure 6D and E). In addition, NR supplementation, but not SIRT1 overexpression, totally suppressed the lipid peroxidation in DN‐NAMPT livers (Figure 6F). Accordingly, the enhancement of blood ALT and AST levels was completely prevented by dietary supplementation with NR, while SIRT1 overexpression only partially corrected the increases of ALT/AST (Figure 6G).
Dietary supplementation with NR prevents the progress of NAFLD induced by HFD in WT mice
As NR supplementation completely corrected the hepatic steatosis and steatohepatitis induced by NAD+‐deficiency and HFD resulted in a decrease in the hepatic NAD+ pool, we further investigated whether NR supplementation could prevent diet‐induced NAFLD in WT mice. Dietary supplementation with NR for 4 weeks increased NAD level in liver tissues in HFD‐fed mice (Supporting Information Figure S9), potently blocked the HFD‐induced increases of body weight (Figure 7A) and reduced the marked increases of hepatic triglyceride and cholesterol levels (Figure 7B). NR supplementation also reduced liver weight and the ratio of liver weight to body weight (Figure 7C) and Oil Red O staining confirmed the decline of hepatic lipid content (Figure 7D). In addition, α‐SMA staining showed NR supplementation lowered hepatic fibrosis (Figure 7E) and the enhanced production of TNF‐α and IL‐1β in liver tissue (Figure 7F). These results demonstrated that dietary supplementation with NR prevented the development of diet‐induced NAFLD in WT mice.
Figure 7.
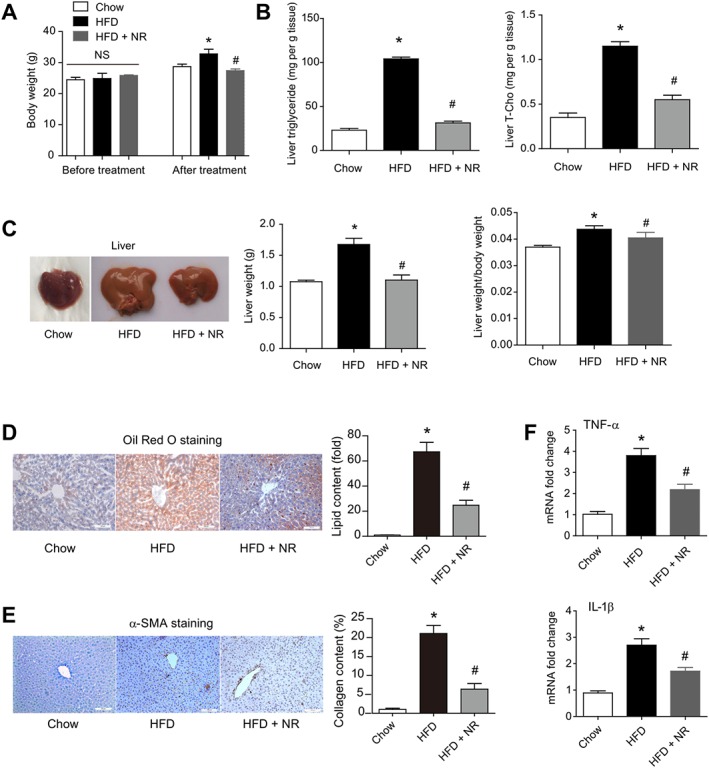
Dietary supplementation with NR corrects HFD‐induced hepatic steatosis in WT mice. (A) Changes of body weight in WT mice fed chow, HFD and HFD + NR. *P < 0.05. n = 8 for each group. HFD was fed for 16 weeks and NR was administrated for 4 weeks. *P < 0.05 versus chow, # P < 0.05 versus HFD by ANOVA analysis. n = 8 for each group. (B) Lipid triglyceride and cholesterol levels in livers of mice fed chow, HFD and HFD + NR. T‐Cho, total cholesterol. *P < 0.05 versus chow, # P < 0.05 versus HFD by ANOVA analysis. n = 8 for each group. (C) Representative images of liver, liver weight and liver weight/body weight ratio in mice fed HFD or HFD plus NR. *P < 0.05 versus chow, # P < 0.05 versus HFD by ANOVA analysis. n = 8 for each group. Oil Red O staining (D) and immunohistochemistry staining of α‐SMA (E) in livers of mice fed chow, HFD and HFD + NR. *P < 0.05 versus chow, # P < 0.05 versus HFD by ANOVA analysis. n = 8 for each group. (F) Pro‐inflammatory factors production in livers of mice fed chow, HFD and HFD + NR. *P < 0.05 versus chow, # P < 0.05 versus HFD by ANOVA analysis. n = 8 for each group.
Discussion and conclusions
Our data provide the first evidence that a deficiency of NAD+ links ageing to NAFLD. Age is a well‐known risk factor for NAFLD as clinical data demonstrate that, although NAFLD can be found in all age groups, its prevalence increases with age (Ford et al., 2002). Although previous work indicated that DNA damage, oxidative stress and inflammatory activation may be crucial causes for NAFLD pathogenesis in ageing (Brenner et al., 2013; Sheedfar et al., 2013), the molecular mechanism for ageing‐induced fatty liver remains poorly understood. Following the original description of NAD+ over 100 years ago, it has become evident that this compound holds a key position in the control of fundamental cellular processes (Canto et al., 2015). The NAD+ concentration in the organism appears to be variable, changing with space, time, nutrition availability and age. This phenomenon results in a limitation of the NAD+ available for a range of biological activities, under certain conditions. Several groups have demonstrated that the NAD+ content in various organs or tissues declines with age in animal models (Braidy et al., 2011; Yoshino et al., 2011; Gomes et al., 2013; Mouchiroud et al., 2013). Moreover, this decrease in the NAD+ pool appears to be a deleterious factor, exhibiting harmful actions because increasing NAD+ content ameliorates aged‐induced diabetes in mice (Yoshino et al., 2011) and increasing NAD+ biosynthesis extends lifespan in yeast (Belenky et al., 2007). Our results confirmed earlier experiments (Braidy et al., 2011; Yoshino et al., 2011; Mouchiroud et al., 2013) showing that the hepatic NAD+ level in old mice was significantly lower. Moreover, we found that the hepatic NAD+ level in elderly patients with age >60 was about ~65% of that in middle‐aged patients with age <45. Notably, IDO, NMNAT and NADS, three important enzymes in the de novo NAD+ biosynthesis pathway, were up‐regulated in older patients. This suggests the de novo NAD+ biosynthesis pathway may be enhanced with ageing. By contrast, the expression of NAMPT was significantly lower in livers from elderly patients, implying that the NAD+ salvage biosynthesis is compromised with increasing age. This deficiency in NAD+ salvage biosynthesis, together with the increased level of NAD+‐consuming biological processes (Imai and Guarente, 2014), cause an overall depletion of the NAD+ pool, and thus increase susceptibility of aged liver to NAFLD development and even to hepatocellular carcinoma, as loss of NAD+ is known to induce liver tumorigenesis (Tummala et al., 2014). In support of this speculation, middle‐aged DN‐NAMPT mice with decreased hepatic NAD+, exhibited fatty liver phenotypes, even when fed normal chow. By contrast, middle‐aged WT mice did not have fatty livers. As expected, these fatty liver phenotypes were further strengthened by HFD. All these results suggest that age‐related NAD+ deficiency increased susceptibility to hepatic steatosis and promoted diet‐induced steatohepatitis.
In our experiments, NAD+ levels not only decreased with age but also with the HFD. Hepatic NAD+ content or NAMPT expression is known to decrease in HFD‐fed animal (Tao et al., 2011; Yoshino et al., 2011; Zhang et al., 2014; Gariani et al., 2016). Hepatic or blood NAMPT levels were also reduced in patients with NAFLD (Dahl et al., 2010) or liver cirrhosis (de Boer et al., 2009). In line with these studies, our data demonstrate that hepatic NAD+ content, hepatic NAMPT expression and blood NAMPT concentration were reduced in mice with NAFLD induced by HFD, suggesting a systemic NAD+ deficit in NAFLD. There is however a report of enhanced NAD+ content and NAMPT expression in HFD‐fed mice (Penke et al., 2015). This difference in outcome might be explained by the composition and duration of the HFD and differences in the strain of mice used. Several critical factors involved in the development of NAFLD, such as HFD, inflammation, insulin resistance and ageing, are associated with the NAD+ pool (Canto et al., 2015). Thus, increasing the NAD+ pool should, theoretically, tend to prevent NAFLD, as it reduced hepatic triglycerides (Tao et al., 2011), reduced obesity (Canto et al., 2012) and improved insulin secretion (Revollo et al., 2007). However, increasing the NAD+ pool may also facilitate the inflammatory reaction and promote macrophage survival (Jia et al., 2004; Van Gool et al., 2009; Moschen et al., 2011), effects that would be detrimental for NAFLD. Intriguingly, we found that the DN‐NAMPT mice displayed hepatic lipid accumulation and steatosis on normal chow diet. The down‐regulation of triglyceride/cholesterol efflux genes including MTTP, ABCG1, ABCG5, ABCG8 and LDL‐R could account for the disturbed lipid regulation in these mice. Among these genes, ABCG1, ABCG5 and ABCG8 are targets for LXR, which was regulated by SIRT1 (Li et al., 2007). So the down‐regulation of ABCG1, ABCG5 and ABCG8 in livers of DN‐NAMPT mice may reflect a defect in the NAD+‐SIRT1 cascade. However, the MTTP and LDL‐R down‐regulation cannot be explained by such a defect in AD+–SIRT1 activity, suggesting the involvement of other NAD+‐regulated signalling pathways in the lipid accumulation. Recently, Bowlby et al . showed that NAMPT‐mediated NAD biosynthesis is required for de novo lipogenesis in tumour cells (Bowlby et al., 2012). As there is a connection between NAMPT, NAD+ metabolism and lipogenesis, the altered lipogenesis in DN‐NAMPT mice may also be involved in the lipid accumulation in this strain. Although the secretion of TNF‐α by macrophages from DN‐NAMPT mice seemed to be impaired, we found enhanced inflammation in livers of DN‐NAMPT mice, compared with those of WT mice, shown by the increases of TNF‐α production with normal chow diets, and the strengthened IL‐1β/IL‐18 release and NLRP3 inflammasome activation with HFD. The discrepancy of TNF‐α secretion between in vitro and in vivo experiments can be explained by the fact that the excessive lipid accumulation in DN‐NAMPT mice liver attracts more macrophages into liver and thus increased the total secretion of TNF‐α. Overall, these results suggest that blockade of NAD+ biosynthesis may not inhibit, but rather enhance, steatohepatitis during NAFLD.
We did not expect the effects of dietary supplementation with NR on steatosis and steatohepatitis would be greater than those of SIRT1 overexpression, in our rescue experiments. Among the NAD+‐consuming sirtuin deacetylases, SIRT1 is the best characterized regulator in metabolism, inflammation and ageing (Imai and Guarente, 2014). However, we found that although SIRT1 overexpression partly prevented the hepatic lipid accumulation, Kupffer cell activation and TNF‐α production in livers from DN‐NAMPT mice, it failed to correct all the pathogenic phenotypes. By contrast, dietary supplementation with NR almost totally corrected the steatosis and steatohepatitis in DN‐NAMPT livers, after either normal chow or HFD treatment. These results imply that, apart from SIRT1, other NAD+‐consuming factors may also participate in the development of NAFLD. For example, SIRT3, but not SIRT1, contributes to the protective effects of NAD+ on hearing loss (Brown et al., 2014). Also, a very recent study reported that increasing NAD+ levels prevented and reversed NAFLD by inducing a SIRT1‐dependent and SIRT3‐dependent mitochondrial unfolded protein response (Gariani et al., 2016). PARP‐1 also mediates liver cell death, inflammation and fibrosis (Mukhopadhyay et al., 2014).
Our study has a significant clinical relevance as NR was reported to decrease obesity (Canto et al., 2012; Mouchiroud et al., 2013), rescue hearing loss (Brown et al., 2014) and prevent age‐associated decline (Canto et al., 2012; Mouchiroud et al., 2013). Our work highlights the therapeutic value of dietary supplementation with NR in NAFLD. In fact, other NAD+ substrates such as nicotinamide can also prevent fatty liver and fibrosis (Varela‐Rey et al., 2010). Thus, replenishing the NAD+ pool by supplementation with NR appears to provide a master control improving the lipid regulation, inflammation and fibrosis, and thereby decrease the development of NAFLD in the elderly. The potential clinical evaluation of dietary supplementation with NR or other NAD substrates in NAFLD is warranted. However, there is a limitation in our study that the sample size of our study in humans was relatively small. As our primary aim in this work was only to confirm the phenotypes observed in mice rather than conduct a full clinical investigation, we did not include more patients. This limitation might cause bias in our study. Nonetheless, as a ‘proof‐of‐concept’ translational study, our data on the decrease in the NAD pool in elderly humans are intriguing. Obviously, these results need to be confirmed in large number of patients in the future.
In conclusion, our results demonstrate that the age‐related NAD+ deficiency is a risk factor for NAFLD pathogenesis, and suggest that supplementation with NAD+ precursors may be a promising therapeutic strategy to prevent and treat NAFLD in the elderly.
Author contributions
P.W. and C.Y.M. were responsible for the study concept and design; P.W., C.C.Z., X.Y., X.H., J.L., M.B.F., G.Q.L. and J.S. acquired the data; P.W., C.C.Z., X.Y., X.H., Y.T.X., Z.Y.L. and Y.F.G. analysed and interpreted the data; P.W. and C.Y.M. wrote the manuscript; P.W., C.C.Z. and X.Y. carried out the statistical analysis; P.W. and C.Y.M. obtained funding.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Illustration of NAD+ de novo and salvage biosynthesis pathway. NAMPT is the step‐limiting enzyme for salvage biosynthesis, whereas NADS and NMNAT are two important enzymes for NAD+ de novo biosynthesis.
Figure S2 Decrease in the NAD+ pool in DN‐NAMPT mice. (A) Endogenous NAMPT protein expression in WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (B) Detecting His‐tag in WT, NAMPT and DN‐NAMPT transgenic mice. (C) Liver NAD+ levels in liver tissues of WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (D) Body weight, liver weight and liver/body weight ratio in WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group.
Figure S3 Macrophages isolated from WT and DN‐NAMPT mice. (A) Representative images of isolated and cultured primary macrophages from WT and DN‐NAMPT mice. (B) Endogenous NAMPT detection using a specific antibody against full‐length NAMPT. *P < 0.05 by Student's t‐test. n = 6 for each group. (C) Intracellular NAD+ levels in macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. (D) Determination of TNF‐ α and IL‐6 release from macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. NS, no significance.
Figure S4 NLRP3 inflammasome pathway in livers of WT and DN‐NAMPT mice under normal chow. (A) Representative images of isolated and cultured primary macrophages from WT and DN‐NAMPT mice. (B) Endogenous NAMPT detection using a specific antibody against full‐length NAMPT. *P < 0.05 by Student's t‐test. n = 6 for each group. (C) Intracellular NAD+ levels in macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. (D) Determination of TNF‐α and IL‐6 release from macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. NS, no significance.
Figure S5 Decrease in the NAD+ pool in HFD‐induced NAFLD mice and DN‐NAMPT mice and lipid profiles in HFD‐fed WT and DN‐NAMPT mice. (A) Liver NAD+ levels in liver tissues of HFD‐induced NAFLD mice. The mice were fed with HFD for 16 weeks. *P < 0.05 by Student's t‐test. n = 8 for each group. (B–C) Decline of NAMPT protein in plasma (B) and liver (C) of NAFLD mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (D–E) Expression of triglyceride and cholesterol efflux genes in HFD‐fed WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group.
Figure S6 SIRT1 mRNA and protein levels in WT and DN‐NAMPT mice under normal chow or HFD. (A) SIRT1 mRNA level in livers of WT and DN‐NAMPT mice. NS, no significance. n = 6 for each group. (B) SIRT1 protein level in livers of WT and DN‐NAMPT mice. NS, no significance. n = 6 for each group.
Figure S7 SIRT1 activity in WT and DN‐NAMPT mice under normal chow or HFD. (A) SIRT1 activity in liver tissues of WT and DN‐NAMPT mice under control and NAFLD conditions. *P < 0.05 by Student's t‐test. n = 8 for each group. (B) Acetylation of LXR in liver tissues of WT and DN‐NAMPT mice under control and NAFLD conditions. *P < 0.05 by Student's t‐test. n = 6 for each group.
Figure S8 Adenovirus‐mediated SIRT1 overexpression in liver tissue of DN‐NAMPT mice. Representative image and quantitative analysis of adenovirus‐mediated SIRT1 overexpression in liver tissue of DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 4 for each group.
Figure S9 NR treatment enhances hepatic NAD+ level in HFD‐fed mice. *P < 0.05 by Student's t‐test. n = 6 for each group.
Table S1 Clinical information for the patients with hepatectomy.
Table S2 Sequences of primers for PCR analysis.
Supporting Info Item
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (no. 81373414, no. 81130061, no. 81473208 and no. 81422049) and the National 863 Plan Young Scientist Program of China (no. 2015AA020943).
Zhou, C. ‐C. , Yang, X. , Hua, X. , Liu, J. , Fan, M. ‐B. , Li, G. ‐Q. , Song, J. , Xu, T. ‐Y. , Li, Z. ‐Y. , Guan, Y. ‐F. , Wang, P. , and Miao, C. ‐Y. (2016) Hepatic NAD+ deficiency as a therapeutic target for non‐alcoholic fatty liver disease in ageing. British Journal of Pharmacology, 173: 2352–2368. doi: 10.1111/bph.13513.
References
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015d). The concise guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C (2007). Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129: 473–484. [DOI] [PubMed] [Google Scholar]
- Bowlby SC, Thomas MJ, D'Agostino RB Jr, Kridel SJ (2012). Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One 7: e40195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan‐Ling T, Poljak A, Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brenner C, Galluzzi L, Kepp O, Kroemer G (2013). Decoding cell death signals in liver inflammation. J Hepatol 59: 583–594. [DOI] [PubMed] [Google Scholar]
- Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W et al. (2014). Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise‐induced hearing loss. Cell Metab 20: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J (2012). Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev 64: 166–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y et al. (2012). The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high‐fat diet‐induced obesity. Cell Metab 15: 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J (2015). NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH (2011). Human fatty liver disease: old questions and new insights. Science 332: 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl TB, Haukeland JW, Yndestad A, Ranheim T, Gladhaug IP, Damas JK et al. (2010). Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down‐regulated in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 95: 3039–3047. [DOI] [PubMed] [Google Scholar]
- De Boer JF, Bahr MJ, Boker KH, Manns MP, Tietge UJ (2009). Plasma levels of PBEF/Nampt/visfatin are decreased in patients with liver cirrhosis. Am J Physiol Gastrointest Liver Physiol 296: G196–G201. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Cowan‐Jacob SW, Moebitz H (2015). Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol 172: 2675–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ (2013). Aging promotes the development of diet‐induced murine steatohepatitis but not steatosis. Hepatology 57: 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ES, Giles WH, Dietz WH (2002). Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287: 356–359. [DOI] [PubMed] [Google Scholar]
- Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER et al. (2016). Eliciting the mitochondrial unfolded protein response via NAD repletion reverses fatty liver disease. Hepatology 63: 1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 155: 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Chen CY, Major EO, Saito K (1997). Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem J 326 (Pt 2): 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD et al. (2004). Pre‐B cell colony‐enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest 113: 1318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI (2012). Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab 15: 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L (2007). SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28: 91–106. [DOI] [PubMed] [Google Scholar]
- Liang W, Verschuren L, Mulder P, van der Hoorn JW, Verheij J, van Dam AD et al. (2015). Salsalate attenuates diet induced non‐alcoholic steatohepatitis in mice by decreasing lipogenic and inflammatory processes. Br J Pharmacol 172: 5293–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Shen FM, Le YY, Kong Y, Liu X, Cai GJ et al. (2009). Antishock effect of anisodamine involves a novel pathway for activating alpha7 nicotinic acetylcholine receptor. Crit Care Med 37: 634–641. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschen AR, Gerner R, Schroll A, Fritz T, Kaser A, Tilg H (2011). A key role for Pre‐B cell colony‐enhancing factor in experimental hepatitis. Hepatology 54: 675–686. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C et al. (2013). The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Cao Z, Horvath B, Park O, Wang H et al. (2014). Poly (ADP‐ribose) polymerase‐1 is a key mediator of liver inflammation and fibrosis. Hepatology 59: 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone‐Corsi P (2009). Circadian control of the NAD+ salvage pathway by CLOCK‐SIRT1. Science 324: 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathil A, Mueller J, Ludwig JM, Wang J, Warth A, Chamulitrat W et al. (2014). Ursodeoxycholyl lysophosphatidylethanolamide attenuates hepatofibrogenesis by impairment of TGF‐beta1/Smad2/3 signalling. Br J Pharmacol 171: 5113–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penke M, Larsen PS, Schuster S, Dall M, Jensen BA, Gorski T et al. (2015). Hepatic NAD salvage pathway is enhanced in mice on a high‐fat diet. Mol Cell Endocrinol 412: 65–72. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B et al. (2009). Circadian clock feedback cycle through NAMPT‐mediated NAD+ biosynthesis. Science 324: 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A et al. (2007). Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 6: 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O et al. (2002). Pre‐B‐cell colony‐enhancing factor, whose expression is up‐regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol 32: 3225–3234. [DOI] [PubMed] [Google Scholar]
- Ruggieri S, Orsomando G, Sorci L, Raffaelli N (2015). Regulation of NAD biosynthetic enzymes modulates NAD‐sensing processes to shape mammalian cell physiology under varying biological cues. Biochim Biophys Acta 1854: 1138–1149. [DOI] [PubMed] [Google Scholar]
- Sheedfar F, Di Biase S, Koonen D, Vinciguerra M (2013). Liver diseases and agingageing: friends or foes? AgingAgeing Cell 12: 950–954. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Wei D, Gao H, Liu Y, DePinho RA, Dong XC (2011). Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem 286: 14681–14690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala KS, Gomes AL, Yilmaz M, Grana O, Bakiri L, Ruppen I et al. (2014). Inhibition of de novo NAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell 26: 826–839. [DOI] [PubMed] [Google Scholar]
- Tziomalos K, Athyros VG, Paschos P, Karagiannis A (2015). Nonalcoholic fatty liver disease and statins. Metabolism 64: 1215–1223. [DOI] [PubMed] [Google Scholar]
- Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A et al. (2009). Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin‐dependent manner. Nat Med 15: 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela‐Rey M, Martinez‐Lopez N, Fernandez‐Ramos D, Embade N, Calvisi DF, Woodhoo A et al. (2010). Fatty liver and fibrosis in glycine N‐methyltransferase knockout mice is prevented by nicotinamide. Hepatology 52: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Zhao Y, Li ZY, Lan XH et al. (2014a). Vascular smooth muscle cell apoptosis is an early trigger for hypothyroid atherosclerosis. Cardiovasc Res 102: 448–459. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu H et al. (2014b). ARRB1/beta‐arrestin‐1 mediates neuroprotection through coordination of BECN1‐dependent autophagy in cerebral ischemia. Autophagy 10: 1535–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhang RY, Song J, Guan YF, Xu TY, Du H et al. (2012). Loss of AMP‐activated protein kinase‐alpha2 impairs the insulin‐sensitizing effect of calorie restriction in skeletal muscle. Diabetes 61: 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M et al. (2011). Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle‐aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 140: 124–131. [DOI] [PubMed] [Google Scholar]
- Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME et al. (2014). NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 92: 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ et al. (2007). Nutrient‐sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130: 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 14: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZF, Fan SH, Zheng YL, Lu J, Wu DM, Shan Q et al. (2014). Troxerutin improves hepatic lipid homeostasis by restoring NAD(+)‐depletion‐mediated dysfunction of lipin 1 signaling in high‐fat diet‐treated mice. Biochem Pharmacol 91: 74–86. [DOI] [PubMed] [Google Scholar]
- Zhang RY, Qin Y, Lv XQ, Wang P, Xu TY, Zhang L et al. (2011). A fluorometric assay for high‐throughput screening targeting nicotinamide phosphoribosyltransferase. Anal Biochem 412: 18–25. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC et al. (2015). Regenerative Neurogenesis After Ischemic Stroke Promoted by Nicotinamide Phosphoribosyltransferase‐Nicotinamide Adenine Dinucleotide Cascade. Stroke 46: 1966–1974. [DOI] [PubMed] [Google Scholar]
- Zhong W, Shen WF, Ning BF, Hu PF, Lin Y, Yue HY et al. (2009). Inhibition of extracellular signal‐regulated kinase 1 by adenovirus mediated small interfering RNA attenuates hepatic fibrosis in rats. Hepatology 50: 1524–1536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Illustration of NAD+ de novo and salvage biosynthesis pathway. NAMPT is the step‐limiting enzyme for salvage biosynthesis, whereas NADS and NMNAT are two important enzymes for NAD+ de novo biosynthesis.
Figure S2 Decrease in the NAD+ pool in DN‐NAMPT mice. (A) Endogenous NAMPT protein expression in WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (B) Detecting His‐tag in WT, NAMPT and DN‐NAMPT transgenic mice. (C) Liver NAD+ levels in liver tissues of WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (D) Body weight, liver weight and liver/body weight ratio in WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group.
Figure S3 Macrophages isolated from WT and DN‐NAMPT mice. (A) Representative images of isolated and cultured primary macrophages from WT and DN‐NAMPT mice. (B) Endogenous NAMPT detection using a specific antibody against full‐length NAMPT. *P < 0.05 by Student's t‐test. n = 6 for each group. (C) Intracellular NAD+ levels in macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. (D) Determination of TNF‐ α and IL‐6 release from macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. NS, no significance.
Figure S4 NLRP3 inflammasome pathway in livers of WT and DN‐NAMPT mice under normal chow. (A) Representative images of isolated and cultured primary macrophages from WT and DN‐NAMPT mice. (B) Endogenous NAMPT detection using a specific antibody against full‐length NAMPT. *P < 0.05 by Student's t‐test. n = 6 for each group. (C) Intracellular NAD+ levels in macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. (D) Determination of TNF‐α and IL‐6 release from macrophages isolated from WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 6 for each group. NS, no significance.
Figure S5 Decrease in the NAD+ pool in HFD‐induced NAFLD mice and DN‐NAMPT mice and lipid profiles in HFD‐fed WT and DN‐NAMPT mice. (A) Liver NAD+ levels in liver tissues of HFD‐induced NAFLD mice. The mice were fed with HFD for 16 weeks. *P < 0.05 by Student's t‐test. n = 8 for each group. (B–C) Decline of NAMPT protein in plasma (B) and liver (C) of NAFLD mice. *P < 0.05 by Student's t‐test. n = 8 for each group. (D–E) Expression of triglyceride and cholesterol efflux genes in HFD‐fed WT and DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 8 for each group.
Figure S6 SIRT1 mRNA and protein levels in WT and DN‐NAMPT mice under normal chow or HFD. (A) SIRT1 mRNA level in livers of WT and DN‐NAMPT mice. NS, no significance. n = 6 for each group. (B) SIRT1 protein level in livers of WT and DN‐NAMPT mice. NS, no significance. n = 6 for each group.
Figure S7 SIRT1 activity in WT and DN‐NAMPT mice under normal chow or HFD. (A) SIRT1 activity in liver tissues of WT and DN‐NAMPT mice under control and NAFLD conditions. *P < 0.05 by Student's t‐test. n = 8 for each group. (B) Acetylation of LXR in liver tissues of WT and DN‐NAMPT mice under control and NAFLD conditions. *P < 0.05 by Student's t‐test. n = 6 for each group.
Figure S8 Adenovirus‐mediated SIRT1 overexpression in liver tissue of DN‐NAMPT mice. Representative image and quantitative analysis of adenovirus‐mediated SIRT1 overexpression in liver tissue of DN‐NAMPT mice. *P < 0.05 by Student's t‐test. n = 4 for each group.
Figure S9 NR treatment enhances hepatic NAD+ level in HFD‐fed mice. *P < 0.05 by Student's t‐test. n = 6 for each group.
Table S1 Clinical information for the patients with hepatectomy.
Table S2 Sequences of primers for PCR analysis.
Supporting Info Item