

Abstract
Background and Purpose
Endoplasmic reticulum (ER) stress is increasingly recognized as an important causal factor of many diseases. Targeting ER stress has now emerged as a new therapeutic strategy for treating cardiovascular diseases. Here, we investigated the effects and underlying mechanism of ginkgolide K (1,10‐dihydroxy‐3,14‐didehydroginkgolide, GK) on cardiac ER stress.
Experimental Approach
Cell death, apoptosis and ER stress‐related signalling pathways were measured in cultured neonatal rat cardiomyocytes, treated with the ER stress inducers tunicamycin, hydrogen peroxide and thapsigargin. Acute myocardial infarction was established using left coronary artery occlusion in mice, and infarct size was measured by triphenyltetrazolium chloride staining. Echocardiography was used to assess heart function and transmission electron microscopy for evaluating ER expansion.
Key Results
Ginkgolide K (GK) significantly decreased ER stress‐induced cell death in both in vitro and in vivo models. In ischaemic injured mice, GK treatment reduced infarct size, rescued heart dysfunction and ameliorated ER dilation. Mechanistic studies revealed that the beneficial effects of GK occurred through enhancement of inositol‐requiring enzyme 1α (IRE1α)/X box‐binding protein‐1 (XBP1) activity, which in turn led to increased ER‐associated degradation‐mediated clearance of misfolded proteins and autophagy. In addition, GK was also able to partly repress the pro‐apoptotic action of regulated IRE1‐dependent decay and JNK pathway.
Conclusions and Implications
In conclusion, GK acts through selective activation of the IRE1α/XBP1 pathway to limit ER stress injury. GK is revealed as a promising therapeutic agent to ameliorate ER stress for treating cardiovascular diseases.
Abbreviations
- ASK1
apoptotic signalling kinase‐1
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- CHOP
C/EBP homologous protein
- ER
endoplasmic reticulum
- ERAD
ER‐associated degradation
- eIF2α
eukaryotic translation initiation factor 2α
- GB
ginkgolide B
- GK
ginkgolide K
- GRP78
glucose‐regulated protein 78 kD
- IRE1α
inositol‐requiring enzyme 1α
- NRCMs
neonatal rat cardiomyocytes
- PERK
pancreatic eIF2α kinase
- RIDD
regulated IRE1‐dependent decay
- TRAF2
TNF receptor‐associated factor 2
- UPR
unfolded protein response
- XBP1
X box‐binding protein‐1
Tables of Links
| LIGANDS |
|---|
| Thapsigargin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
The endoplasmic reticulum (ER) is a membranous organelle that plays an important role in the maintenance of cellular processes such as protein processing, calcium homeostasis and lipid biosynthesis. Effective ER function is essential for the folding of secretory and membrane proteins (Wei and Hendershot, 1996). A number of cellular stresses disturb the balance between the folding capacity of the ER and the burden of incoming proteins, including oxidative stress, ischaemic insult, abnormal ER calcium content, and enhanced expression of normal and/or folding‐defective proteins. These events lead to the accumulation of unfolded proteins, a condition referred to as ER stress. The ER is capable of sensing a wide variety of perturbations, which triggers an adaptive process, known as the unfolded protein response (UPR), to maintain ER homeostasis and mitigate or eliminate the stress. This UPR consists of three characteristic components: (i) translational attenuation to reduce the translocation of nascent proteins into the ER; (ii) transcriptional activation of genes encoding ER chaperones and enzymes to improve protein folding capacity; and (iii) transcriptional activation of genes for components of the ER‐associated degradation (ERAD) system and autophagy to degrade the misfolded protein. ER stress engages the ER‐located molecular chaperone, glucose‐regulated protein 78 kD (GRP78), and three ER transmembrane proteins – inositol‐requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6) and pancreatic eukaryotic translation initiation factor 2α (eIF2α) kinase (PERK) – to mobilize UPR. PERK stimulates phosphorylation of eIF2α as an immediate response. IRE1α is a dual protein kinase/endoribonuclease (RNase) and processes the mRNA encoding unspliced X box‐binding protein 1 (XBP1u) to spliced XBP1 (XBP1s) through its RNase activity. XBP1s encodes an active transcription factor that controls the genes encoding proteins involved in protein folding, ERAD, protein quality control and phospholipid synthesis. Meanwhile, ATF6 cooperates with IRE1α for the induction of XBP1 transcription (Sozen et al., 2015). These proteins work in concert to balance the unfolded protein/chaperone system to provide ER homeostasis. If the cell fails to recover from ER stress, the UPR represses the adaptive response and triggers apoptosis (Chen and Brandizzi, 2013).
Malfunction of the UPR has been implicated in a wide range of diseases including cancer, diabetes, neurodegenerative diseases and age‐related disorders. Numerous lines of evidence also indicate that the UPR and ER stress play an important role in the development of cardiovascular diseases such as ischaemia/reperfusion injury (Vekich et al., 2012; Wang et al., 2014), atherosclerosis (McAlpine and Werstuck, 2013; Chistiakov et al., 2014) and heart failure (Dickhout et al., 2011; Groenendyk et al., 2013). Although prolonged UPR activation is deleterious, leading to cell death, the UPR acts as a protective response at an early stage during cell injury. For UPR action, three ER sensors work synergistically through distinct sets of target genes. Thus, strategies that are able to differentially regulate individual components of UPR to alleviate ER stress may present promising avenues to develop novel therapeutic interventions for cardiovascular diseases.
Ginkgolide K (1,10‐dihydroxy‐3,14‐didehydroginkgolide, GK) is a diterpene lactone isolated from the leaves of Ginkgo biloba, a natural product with a long history of therapeutic application for cardiovascular diseases in humans (Liu et al., 2014). G. biloba extract EGb761 is a widely used and well‐defined extract of G. biloba leaves (Montes et al., 2015). Many studies have shown that ginkgolides are the most important constituents of G. biloba , which play a vital role in prevention and treatment of many diseases, including cardiovascular diseases (Tosaki et al., 1996; Liu et al., 2012) and neurological disorders (Maclennan et al., 2002). Previous studies have reported that ginkgolide B (GB) is a natural specific antagonist of the PAF receptor that exerts protective effects against ischaemic injury (Reinstein et al., 2013), arrhythmia (Koltai et al., 1989; Zhao et al., 2013) and inflammation (Liu et al., 2012; Liu et al., 2014). Recently, GB has been reported to attenuate apoptosis as well as ER stress through its antioxidant properties (Zhang et al., 2011). As a derivative of GB, GK has been shown to possess protective effects on cerebral ischaemia/reperfusion injury (Ma et al., 2012b) and markedly protect PC12 cells against H2O2‐ or glutamate‐induced cytotoxicity by ameliorating mitochondrial dysfunction, oxidative stress and Ca2 + overload (Ma et al., 2012a; Ma et al., 2014). A pharmacokinetic study of GK in rats provided some important information that would promote further study of GK in human diseases (Fan et al., 2015). However, the effects of GK on ER stress remain undefined.
In the present study, we demonstrate that GK enhanced IRE1α phosphorylation and XBP1 mRNA splicing and partly repressed regulated IRE1‐dependent decay (RIDD) and the JNK pathway. The relief of ER stress by GK was further confirmed by in vivo studies in mice subjected to acute myocardial infarction (AMI), which is a clinically relevant model to mimic the ER stress‐caused cardiac injury. Our findings suggest that the IRE1α/XBP1 pathway plays a pivotal role in GK‐mediated cardioprotection.
Methods
Animals
All animal care and in vitro experimental procedures were reviewed and approved by the Ethics Committee, University of Manchester. All animal care and experimental procedures for the in vivo experiments were approved by the Institutional Animal Care and Use Committee and the Ethics Committee of Chinese Academy of Medical Sciences and Peking Union Medical College. Male adult ICR mice (8–10 weeks old) were obtained from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The experiments were performed in the Experimental Animal Center of Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College. The animals were housed under controlled conditions at a room temperature of 25°C in a 12 h light/dark cycle with ad libitum access to water and food. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2015).
Primary culture of NRCMs
Neonatal rat cardiomyocytes (NRCMs) were isolated from 1‐ to 2‐day‐old Sprague Dawley rats (Charles River UK, Ltd. Margate, Kent, UK) using the standard enzymic method described previously (Kimura et al., 2010).
Assessment of cell viability using LDH assays
To assess cell viability, ER stress was elicited by addition of fresh media containing 2.5 μg·mL−1 tunicamycin, 300 nM thapsigargin or 100 μM H2O2. NRCMs were pretreated with GK at the indicated concentrations for 12 h followed by incubation with tunicamycin, thapsigargin or vehicles (0.05% DMSO in culture media) for 24 h or H2O2 for 4 h. Cell death was evaluated by measuring released LDH in the media from dead cells using CytoTox 96 Non‐Radioactive Cytotoxicity Assay (G1780; Promega, Madison, WI, USA) according to the manufacturer's protocol.
Apoptosis assay
For cultured NRCMs, the fluorescent in situ TUNEL cell death detection kit was used (Roche, Indianapolis, IN, USA). Briefly, cells were fixed with 4% paraformaldehyde and then permeabilized. DNA nicks were labelled with terminal deoxynucleotidyltransferase and nucleotide mixture containing fluoresceinisothiocyanate‐conjugated dUTP. Nuclei were counter stained with DAPI, and images were collected on a fluorescence microscope (BX51; Olympus, Tokyo, Japan) and metavue Software (Molecular Devices, Sunnyvale, CA, USA). The percentage of TUNEL positive was calculated using imagej software. The operator and data analysis were blinded to the treatment groups.
For paraffin‐embedded heart sections, the colorimetric in situ cell death detection kit (Roche Applied Science) was used following the manufacturer's instructions. Individual nuclei were visualized at a magnification of 400‐fold, and the percentage of apoptotic nuclei was calculated from six randomly chosen fields per slide and averaged from four slides per heart for statistical analysis. The operator and data analysis were blinded to the treatment groups.
PCR and primers
Total RNA was extracted using Trizol reagent. A total of 10 μg of total RNA was subjected to RT‐PCR using Superscript III First Strand (Invitrogen, Carlsbad, CA, USA). Then 20–50 ng of cDNA was amplified by standard PCR with the HotStarTaq Master Mix Kit (Qiagen, Hilden, Germany) or real‐time quantitative PCR (qPCR) using SYBR® Select Master Mix. For standard PCR, the PCR products were analysed in 2% agarose gel, and images were collected with the Bio‐Rad ChemiDoc MP imaging system and image lab software (Bio‐Rad, Hercules, CA, USA). qPCR was performed on StepOnePlus Real‐Time PCR System by comparative 2−ΔΔCt method. The used primers were synthesized by Sigma as below: for XBP1, 5′‐TTACGAGAGAAAACTCATGGGC‐3′ and 5′‐GGGTCCAACTTGTCCAGAATGC‐3′; for EDEM1, 5′‐AAAGCCCTCTGGAACCTTCG‐3′ and 5′‐AAGGGATTCTTGGTCGCCTG‐3′; for HRD1, 5′‐CCGGTGCTAAGAGATTGCCT‐3′ and 5′‐TCTTCTGCAGTGCTCACAGG‐3′; for Gapdh, 5′‐GTTACCAGGGCTGCCTTCTC‐3′ and 5′‐CTCGTGGTTCACACCCATCA‐3′; for LC3b, 5′‐TGTCCGACTTATTCGAGAGCAGCA‐3′ and 5′‐TTCACCAACAGGAAGAAGGCCTGA‐3′; and for Beclin1, 5′‐AGGTTGAGAAAGGCGAGACA‐3′ and 5′‐TTTTGATGGAATAGGAGCCG‐3′. The primers of Blos1, Hgnat, Scara3, Pmp22, Pdgfrb and Col6 were obtained from Qiagen (Cat. Nos: QT00102060, QT00175938, QT00194866, QT01298696, QT01063069 and QT01599430).
Electrophoretic analysis of XBP1 mRNA splicing
To examine XBP1 mRNA splicing, the restriction endonuclease Pst I was used to detect XBP1s by only digesting the XBP1u isoform as described before (Margariti et al., 2013). Briefly, half of the RT‐PCR product was digested with Pst I and resolved on 2% agarose gels and visualized with ethidium bromide.
RNA interference
Specific short interfering RNA for IRE1α (siIRE1, Cat. No. SI02937914) and XBP1 (siXBP1, Cat. No. SI01973482) was purchased from Qiagen. Negative control siRNA (scramble, Cat. No. SR‐CL000–005) was obtained from Eurogenetec, Liège, Belgium. NRCMs were transfected with siRNA (15 nM) using Lipofectamine LTX and Plus reagents according to the manufacturer's instructions (Invitrogen). To assess the specific silencing effect of siIRE1 and siXBP1, total IRE1α and XBP1 protein were detected by immunoblot analysis 72 h post‐transfection.
Assessment of autophagic flux
Autophagic flux was detected by microtubule‐associated protein 1 light chain 3 type 2 (LC3‐II) and p62 turnover in the presence and absence of a cysteine protease inhibitor E64d (10 μg·mL−1; Calbiochem, Darmstadt, Germany) and an aspartic protease inhibitor pepstatin A (10 μg·mL−1; Sigma) (Mizushima and Yoshimori, 2007). NRCMs were pretreated with E64d and pepstatin A for 2 h to inhibit lysosomal proteases followed by incubation with or without 10 μM GK for 12 h and then with or without tunicamycin for additional 24 h incubation. At the end point, LC3‐II and p62 levels were analysed by Western blot.
Acute myocardial infarction (AMI) model
Mice (male ICR) were anaesthetized with 2% isoflurane inhalation using an isoflurane delivery system. AMI was performed by permanent occlusion of the left descending coronary artery (LCA) as previously described (Gao et al., 2010). All of the animals were randomly assigned to three experimental groups (n = 12 per group): (i) Sham group (undergoing the same surgical procedure except for occlusion); (ii) AMI + Vehicle group; and (iii) AMI + GK group. All animals were monitored and received one dose of buprenorphine (0.3 mg·kg−1) within 6 h post‐surgery. GK was dissolved in Tween 80 (<20%; Sigma) before administration. Mice in AMI + GK10 group were injected intraperitoneally with GK (10 mg·kg−1 body weight) immediately after LCA occlusion. The mice in Sham and AMI + Vehicle groups were administered with a corresponding dose of the vehicle. Another dose was administered at 12 h after surgery. Mice were subjected to 24 h of myocardial ischaemia followed by echocardiographic and infarct size analysis. At the end of the study, animals were killed by cervical dislocation. The tissue samples for transmission electron microscopy (TEM), TUNEL and Western blot analysis were prepared from the non‐infarcted region adjacent to the border zone of myocardial infarcts at the anterior wall of left ventricle. The operator and data analysis were blinded to the treatment groups.
Assessment of infarct size
Myocardial infarct size was determined by triphenyltetrazolium chloride (TTC) staining. Briefly, the hearts were frozen rapidly and sliced into five 2 mm transverse sections. The sections were incubated at 37°C with 1% TTC in phosphate buffer (pH 7.4) for 15 min, fixed in 10% formaldehyde solution, photographed with a digital camera (Canon E450; Canon Inc., Tokyo, Japan) and calculated in a blinded manner using imagej software (National Institutes of Health, Bethesda, MD, USA). The infarct size was expressed as a percentage of left ventricle circumference.
Echocardiography
All mice were examined by transthoracic echocardiography at 24 h after surgery (prior to killing) using the Vevo 770 High‐Resolution In Vivo Micro‐Imaging System (FujiFilm VisualSonics Inc., Toronto, Canada). Left ventricular internal diameter end diastole (LVIDd) and end systole (LVIDs) were measured perpendicularly to the long axis of the ventricle. Ejection fraction (EF) was calculated automatically by the echocardiography software according to LVIDd and LVIDs. Pulse wave velocity was used to measure aorta blood flow velocity (AV). The operator and data analysis were blinded.
Transmission electron microscopy (TEM)
Small heart samples, about 1 mm3, were fixed immediately by immersion in 4% ice‐cold glutaraldehyde, postfixed for 1 h in 1% OsO4 in 0.1 M cacodylate buffer, dehydrated and embedded in Epon 812 at 60°C for 48 h. Routine 60 nm ultrathin sections were cut and mounted on coated grids and stained with 2% uranyl acetate and 0.2% lead acetate. The sections were assessed with a electron microscope (H‐7650; Hitachi, Tokyo, Japan). The operator and data analysis were blinded to the treatment groups.
Western blot
After treatment, proteins were extracted from NRCMs or heart tissue in Triton lysis buffer (20 mM Tris HCl, pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X‐100, 25 mM β‐glycerophosphate, 10% glycerol, 1 mM orthovanadate, 1 mM phenylsulphfonylfluoride, 10 μg·mL−1 leupeptin and 10 μg·mL−1 aprotinin). Cytochrome c release was measured by a mitochondria isolation kit (Pierce, Appleton, WI, USA). Equal protein extracts (20 μg) were analysed by Western blotting, with antibodies against C/EBP homologous protein (CHOP), cleaved caspase 3, cleaved PARP, Beclin1, LC3A/B, p62, phospho‐PERK, PERK, Bcl‐2, ATF6, activating transcription factor 4 (ATF4), eIF2α, phospho‐eIF2α, apoptotic signalling kinase‐1 (ASK1), phospho‐ASK1, JNK, phospho‐JNK and ubiquitin (Cell Signalling, San Diego, CA, USA); cytochrome c, COX4, GRP78, ER degradation‐enhancing α‐mannosidase‐like protein 1 (EDEM1), IRE1α, phospho‐IRE1α (Ser724) and GAPDH (Abcam, Cambridge, UK); Bax (Santa Cruz, Dallas, TX, USA); and XBP1s (BioLegend, San Diego, CA, USA). Immunoblots were detected by enhanced chemiluminescence with anti‐mouse or anti‐rabbit immunoglobulin G coupled to horseradish peroxidase as the secondary antibody (Amersham‐Pharmacia, Amersham, UK).
Data analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as the means ± SEM and assessed with a two‐tailed Student's t‐test or a one‐way ANOVA followed by Tukey's post hoc test using graphpad prism software version 6.0 (GraphPad Software, La Jolla, CA, USA). The post hoc tests for multiple comparisons were performed only if F achieved P < 0.05 and there was no significant variance in homogeneity. A value of P < 0.05 was considered statistically significant.
Materials
GK (purity >98%) was obtained from Jiangsu Kanion Pharmaceutical Co., Ltd., Lianyungang, China, and freshly prepared before experiments. Tunicamycin, hydrogen peroxide and thapsigargin were purchased from Sigma (St. Louis, MO, USA). Tunicamycin and thapsigargin were dissolved in DMSO at a concentration of 5 mg·mL−1 and 600 μM respectively.
Other reagents and solvents were of analytical grade.
Results
GK alleviated ER stress‐induced injury in NRCMs
To evaluate the effects of GK on ER stress, we used tunicamycin to mimic ER stress injury in NRCMs, and degree of cell death was measured using the LDH method. The results showed that tunicamycin treatment at 2.5 μg·mL−1 for 24 h induced considerable cell toxicity, which was significantly decreased by GK pretreatment (10 or 20 μM) (Figure 1A). Next, we examined whether GK could prevent cytotoxicity exerted by the other ER stressors, such as thapsigargin and H2O2, which markedly increased cell death in NRCMs, and here also pretreatment with GK (10 μM or 20 μM) concentration‐dependently reduced the cytotoxicity (Figure 1B, C). TUNEL staining was performed to assess the apoptosis in cultures of NRCMs. TUNEL‐positive cells after GK pretreatment (10 μM or 20 μM) were significantly fewer than in the group treated with tunicamycin alone (Figure 1D, E). Western blot analysis showed that pretreatment with GK reduced the level of the ER stress‐induced apoptosis mediator CHOP in a concentration‐dependent manner (Figure 1F, G) and the levels of cleaved caspase 3 and cleaved PARP were also reduced after pretreatment with GK (10 μM or20 μM), compared with tunicamycin control (Figure 1F, H, I). Moreover, 10 μM GK increased the expression of anti‐apoptotic protein Bcl‐2, thus decreased the ratio of Bax/Bcl‐2 following tunicamycin‐induced ER stress. GK also blocked tunicamycin‐induced cytochrome c release from mitochondria, as indicated by a decrease in the ratio of cytosolic cytochrome c to mitochondrial cytochrome c (Figure 1J–L). In addition, we found that treatment with GK alone at the indicated concentrations for up to 36 h did not affect the viability of NRCMs or the levels of Bcl‐2, Bax, cytochrome c and cleaved caspase 3 (Supporting Information Fig. S1), suggesting that GK per se has no cytotoxicity.
Figure 1.
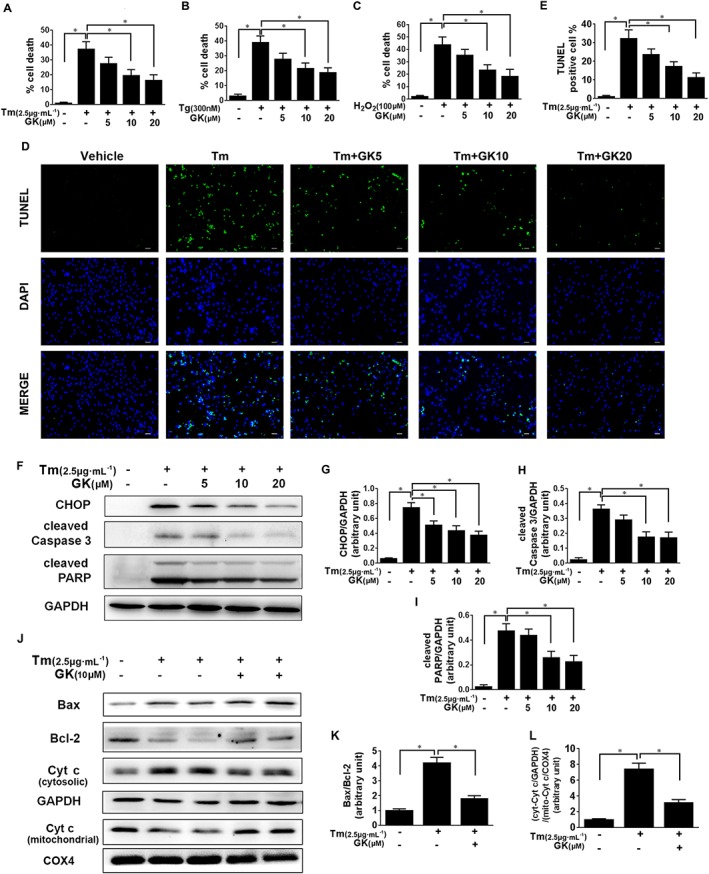
GK decreased ER stress‐induced cell death and apoptosis in NRCMs. NRCMs were pretreated with or without GK at the indicated concentrations (5, 10 and 20 μM respectively) for 12 h followed by treatment with ER stressors (tunicamycin, Tm, 2.5 μg·mL−1, 24 h; thapsigargin, Tg, 300 nM, 24 h; H2O2, 100 μM, 4 h). (A–C) ER stressor‐induced cell death was measured by the LDH method. (D, E) Apoptosis was detected by the TUNEL assay (scale bar, 40 μm), and the TUNEL‐positive cells were quantified. (F–L) Western blot analysis showed that tunicamycin‐induced CHOP, cleaved caspase 3, cleaved PARP and cytosolic cytochrome c were suppressed by pretreatment with GK. However, tunicamycin‐suppressed Bcl‐2 and mitochondrial cytochrome c were increased by pretreatment with GK. COX4 and GAPDH, which are exclusively expressed in the mitochondria and cytosol, respectively, were used as controls for loading and fractionation. Data are presented as means ± SEM (n = 5); *P < 0.05. Cyt c, cytochrome c; mito, mitochondrial.
GK promoted IRE1α activation and XBP1 splicing in tunicamycin‐treated NRCMs
To investigate the mechanism whereby GK prevents cell death in response to ER stress, we next examined the effects of GK on the UPR signal in stressed cardiomyocytes. Western blot analysis showed that tunicamycin treatment for 24 h increased GRP78 level and activated three arms of the UPR compared with non‐treated vehicle controls (Figure 2A–H). In comparison with tunicamycin control, 12 h pretreatment with 10 μM GK further enhanced GRP78 expression and increased phosphorylation at Ser724 of IRE1α, whilst there were no significant effects on the active form of ATF6, the phosphorylation of PERK and eIF2α as well as ATF4, suggesting that GK could selectively activate the IRE1α branch in the UPR. To verify the activation of the IRE1α branch, we assessed the mRNA level of XBP1s, which is the readout of the RNase activity of activated IRE1α. The XBP1 mRNA splicing assay was performed as shown in Figure 2I, and the mRNA level of XBP1s in NRCMs treated with 10 μM GK was nearly twofold higher than that in the tunicamycin control, indicating increased IRE1α RNase activity (Figure 2I). Consistent with these findings, Western blot analysis confirmed that 10 μM GK treatment increased the protein expression of XBP1s (Figure 2J). These results prompted us to explore whether GK alone can modulate the ER stress signalling pathways. The results in Supporting Information Fig. S2 showed that GK alone had no effects on ER stress marker proteins (Supporting Information Fig. S2a). Taken together, these data suggest that GK promotes activation of the IRE1α/XBP1 pathway to protect NRCMs from sustained ER stress.
Figure 2.
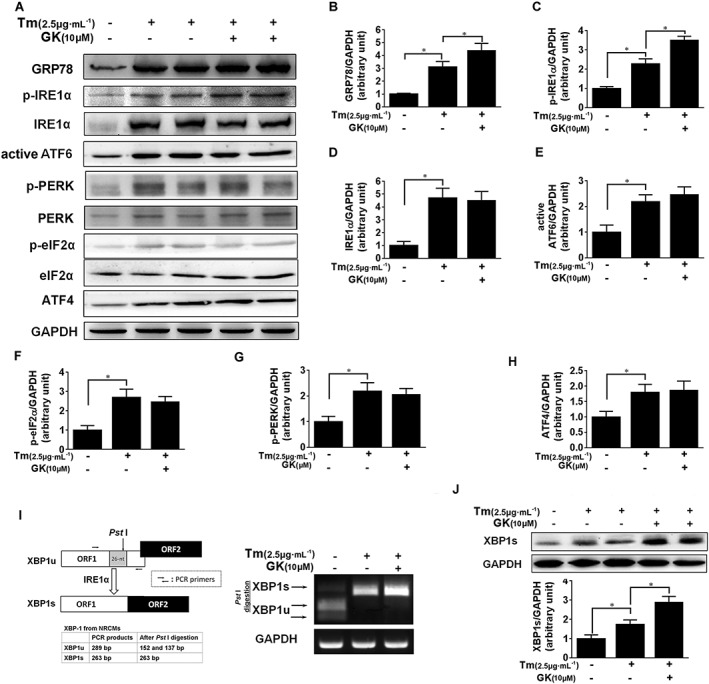
GK promoted IRE1α activation and XBP1 mRNA splicing in tunicamycin‐treated NRCMs. NRCMs were pretreated with or without 10 μM GK for 12 h followed by treatment with 2.5 μg·mL−1 tunicamycin (Tm) for 24 h. (A–H) Western blot analysis was performed on cell lysates. Tunicamycin‐induced p‐IRE1α, but not ATF6, p‐PERK, p‐eIF2α and ATF4, was increased by GK. (I) mRNA level of XBP1 was amplified by PCR followed by splicing assay. The digestion products of XBP1u and XBP1s by Pst I were analysed by agarose gel. (J) XBP1s protein level was analysed by Western blot. Data are presented as means ± SEM (n = 5); *P < 0.05.
IRE1α/XBP1 plays a pivotal role in GK‐mediated cytoprotection
To further confirm the involvement of the IRE1α/XBP1 pathway in GK‐mediated cytoprotection in cardiomyocytes, we used siRNA to silence IRE1α or XBP1 in NRCMs. Western blot analysis demonstrated a significant knockdown of IRE1α or XBP1 in NRCMs (Figure 3A). Cell death assayed by LDH release showed that IRE1α or XBP1 knockdown nearly abolished the protective effects of GK in tunicamycin‐challenged NRCMs (Figure 3B). The same results were also revealed in the TUNEL assay. As showed in Figure 3C–D and Supporting Information Fig. S3, the beneficial effects of 10 μM GK were blunted in the presence of siIRE1 or siXBP1. Taken together, these results support a critical role for the IRE1α/XBP1 pathway in GK‐mediated cytoprotection.
Figure 3.
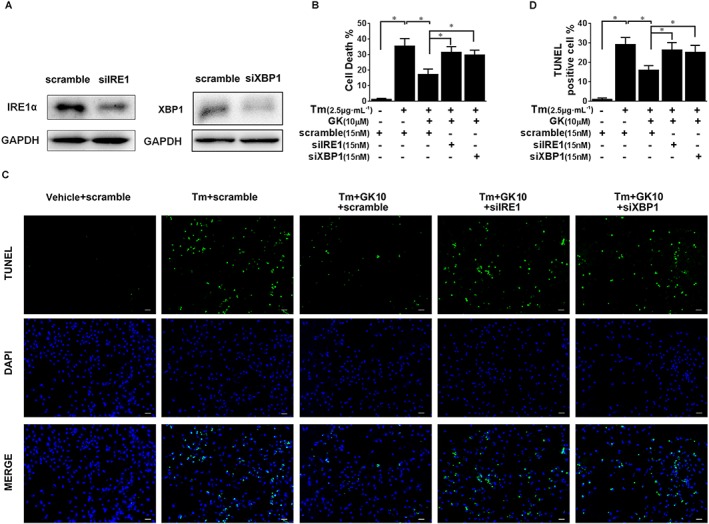
Knockdown of IRE1α or XBP1 abolished GK‐mediated cytoprotection. NRCMs were transfected with negative control siRNA (scramble) and specific siRNA of IRE1α (siIRE1) and XBP1 (siXBP1), respectively, for 72 h, and then stimulated with tunicamycin (Tm) or 24 h with or without 10 μM GK pretreatment. (A) Western blot analysis demonstrated a significant knockdown of IRE1α and XBP1 in NRCMs after treatment with siIRE1 or siXBP1. (B) LDH assay showed that IRE1α or XBP1 knockdown abolished the protective effect of GK in tunicamycin‐challenged NRCMs. (C, D) Apoptosis was detected by the TUNEL assay (scale bar, 40 μm) and quantitatively analysed using TUNEL‐positive cardiomyocytes percentage per field. The beneficial effect of GK was blunted in the presence of siIRE1 or siXBP1. Data are presented as means ± SEM (n = 5); *P < 0.05.
GK partially repressed IRE1α‐induced RIDD activity and ASK1/JNK activation
Prolonged ER stress allows IRE1α to exert its RNase activity on many ER‐localized mRNAs and some anti‐apoptotic miRNAs. This phenomenon is referred to as RIDD, which is commonly regarded as a mechanism of IRE1α‐dependent apoptosis (Byrd and Brewer, 2013; Ghosh et al., 2014). In this study, we examined RIDD activity by qPCR analysis using mRNA expression of six RIDD‐specific substrates. We found that the ER stressor tunicamycin notably reduced Blos1, Hgnat, Scara3, Pmp22 and Col6 in NRCMs (Figure 4A); and the other ER stressor, thapsigargin, reduced Blos1, Hgnat, Pdgfrb and Pmp22 (Supporting Information Fig. S4b), which was in line with previous studies (Hollien et al., 2009; Mendez et al., 2013; Maurel et al., 2013). In contrast, GK (10 μM) elevated Blos1, Hgnat and Scara3 in tunicamycin‐treated NRCMs and 4μ8C, a known IRE1α inhibitor, was also found to significantly elevate the levels of Blos1, Hgnat, Scara3, Pmp22 and Col6 in tunicamycin‐treated NRCMs (Figure 4A). In addition, GK alone had no effect on mRNA expression of RIDD substrates (Supporting Information Fig. S4a). Collectively, these data demonstrate that GK partly repressed the IRE1α‐induced RIDD activity in tunicamycin‐treated NRCMs.
Figure 4.
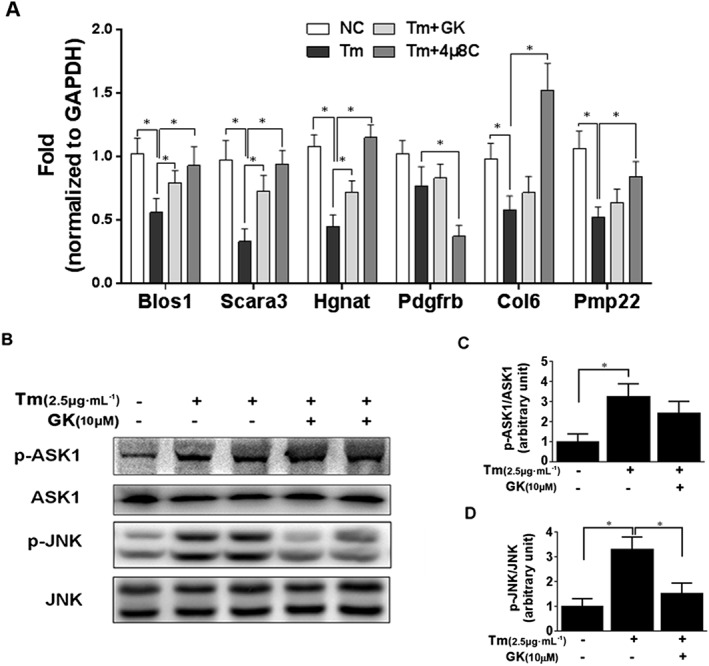
GK partly repressed IRE1α‐induced RIDD activity and JNK activation. (A) RIDD mRNA substrates Blos1, Hgnat, Scara3, Pmp22, Pdgfrb and Col6 were assayed by qPCR. The ER stressor tunicamycin (Tm ) notably reduced Blos1, Hgnat, Scara3, Pmp22 and Col6 in NRCMs. 4μ8C, a known IRE1α inhibitor, significantly elevated Blos1, Hgnat, Scara3, Pmp22 and Col6 in tunicamycin‐treated NRCMs; 10 μM GK elevated Blos1, Hgnat and Scara3 in tunicamycin‐treated NRCMs. (B–D) Western blot analysis showed that GK treatment repressed the increase in phospho‐JNK, but not phospho‐ASK1. Data are presented as means ± SEM (n = 5); *P < 0.05.
Activated IRE1α can form a hetero‐oligomeric complex with tumour necrosis factor receptor‐associated factor 2 (TRAF2), which stimulates activation of ASK1 and downstream JNK to promote apoptosis (Chen and Brandizzi, 2013). Thus, we investigated the effects of GK on the ASK1/JNK pathway. As shown in Figure 4B‐D, tunicamycin treatment increased the phosphorylation level of ASK1 and JNK compared with the non‐treated vehicle control. However, compared with the tunicamycin control, GK treatment significantly decreased the level of phospho‐JNK but not the level of phospho‐ASK (Figure 4B–D).
GK stimulated ERAD in tunicamycin‐treated NRCMs
Proteins that fail to correctly fold or assemble in the ER are retro‐translocated into the cytosol for poly‐ubiquitination and thereafter degraded by the proteasome system to preserve protein homeostasis, via a process known as ERAD (Amm et al., 2014; Christianson and Ye, 2014). It is generally agreed that ubiquitin conjugation to substrates drives the retro‐translocation process and transfers the ubiquitinated substrates to the cytosolic 26S‐proteasome for digestion. Thus, levels of ubiquitin‐conjugated proteins can reflect the activity of degradation (Su and Wang, 2010; Zhang et al., 2014). As shown in Figure 5A and Supporting Information Fig. S2b, Western blot analysis using ubiquitin antibody for detecting ubiquitinated proteins demonstrated that pretreatment with 10 μM GK profoundly reduced the level of ubiquitin‐conjugated proteins compared with the tunicamycin control, suggesting a positive effect of GK to remove ubiquitinated proteins following tunicamycin‐induced ER stress. In order to exclude the possibility due to a decline in the onset of ubiquitination, two important components of the ERAD machinery were examined; an ubiquitin ligase HMG‐CoA reductase degradation protein 1 homologue (HRD1) and EDEM1. PCR results showed that 10 μM GK treatment increased EDEM1 and HRD1 mRNA compared with the tunicamycin control (Figure 5B, C). Western blot analysis showed that tunicamycin (N‐linked glycosylation blocker) considerably elevated the level of non‐glycosylated EDEM1 protein, and treatment with 10 μM GK further enhanced the level of EDEM1 protein, regardless of glycosylated and non‐glycosylated status (Figure 5D, E). These results clearly demonstrated that GK stimulated ERAD to eliminate misfolded protein in ER‐stressed NRCMs.
Figure 5.
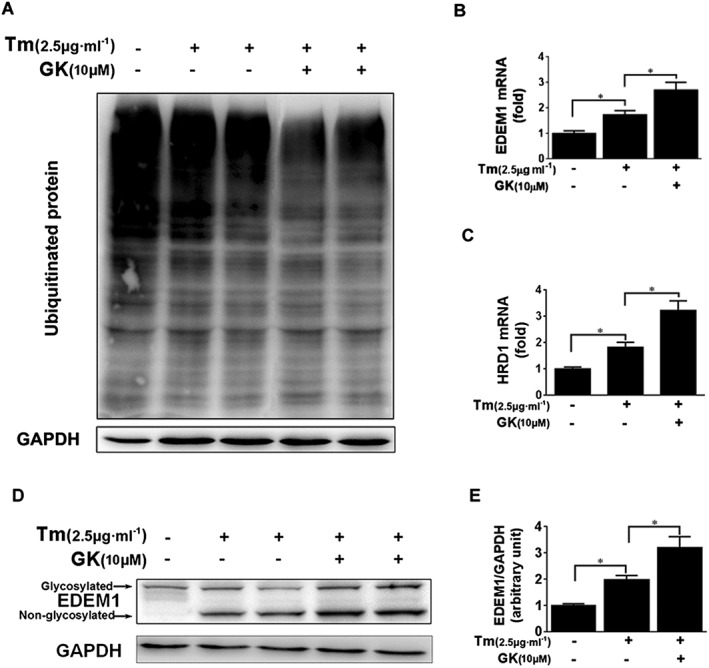
GK stimulated ERAD in tunicamycin (Tm )‐treated NRCMs. (A) Western blot analysis to detect ubiquitination showed that pretreatment with 10 μM GK profoundly reduced the level of ubiquitin‐conjugated proteins. (B, C) qPCR analysis showed that GK increased the mRNA levels of EDEM1 and HRD1. (D, E) Western blot analysis showed that GK further enhanced the expression of non‐glycosylated EDEM1 protein. Data are presented as means ± SEM (n = 5); *P < 0.05.
GK prompted autophagic flux in tunicamycin‐treated NRCMs
Although ERAD has been regarded as a primary pathway for the degradation of misfolded proteins, recent reports have shown that autophagy is an alternative cellular pathway to eliminate unfolded proteins (Groenendyk et al., 2013; Margariti et al., 2013; Amm et al., 2014). We showed by quantitative PCR that 10 μM GK treatment increased the transcriptional level of autophagy markers Beclin1 and LC3B when compared with tunicamycin control (Figure 6A, B). In addition, Western blot analysis demonstrated induced protein expression of Beclin1 after 10 μM GK treatment, indicating activation of the autophagy process (Figure 6C, D). Furthermore, detection of the autophagosome‐specific isoform of LC3 (known as LC3‐II) and p62 was performed to assess the autophagic flux in the presence of lysosome‐mediated proteolysis inhibitors, pepstatin A and E64d. In the presence of tunicamycin, 10 μM GK induced a notable elevation in LC3‐II (Figure 6E, F). Meanwhile, 10 μM GK induced additional accumulation of LC3‐II protein in the presence of pepstatin A (10 μg·mL−1) and E64d (10 μg·mL−1) (Figure 6E, F), suggesting a real increase in LC3‐II. The observation of decreased p62 after 10 μM GK treatment confirmed that GK improved activation of autophagy in NRCMs (Figure 6E, G). Of note, 10 μM GK alone had no effect on expression of LC3 and p62, with or without treatment with pepstatin A and E64d (Supporting Information Fig. S2c–f). These results indicate that GK increases an autophagic response in stressed NRCMs.
Figure 6.
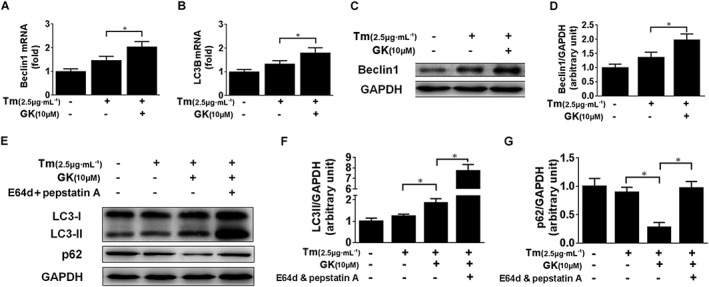
GK prompted autophagic flux in tunicamycin‐treated NRCMs. NRCMs were pretreated with or without 10 μM GK for 12 h and then treated with or without lysosome inhibitors pepstatin A and E64d for 2 h followed by treatment with 2.5 μg·mL−1 tunicamycin (Tm ) for a further 24 h. (A, B) qPCR analysis demonstrated that GK increased the transcriptional level of autophagy markers Beclin1 and LC3B. (C, D) Western blot analysis demonstrated that GK increased the protein expression of Beclin1. (E, F) LC3‐II was notably elevated by GK and was further elevated in the presence of pepstatin A and E64d, suggesting a genuine increase in LC3‐II. (E, G) Decreased p62 after GK treatment confirmed that GK improved activation of autophagy in NRCMs. Data are presented as means ± SEM (n = 5); *P < 0.05.
GK exerted cardioprotection and attenuated ER stress‐induced apoptosis in the mouse model of AMI
Numerous studies provide important evidence for the critical role of ER stress in cardiomyocytes apoptosis after myocardial infarction (Mitra et al., 2013; Luo et al., 2015). Thus, the AMI model is an ideal tool to study ER stress in the context of heart disease. In the present study, the AMI model was induced by coronary ligation in 8‐ to 10‐week‐old male ICR mice to evaluate the effects of GK on ER stress‐mediated injury in vivo. After 24 h of coronary ligation, a myocardial infarct size of up to 33% of the left ventricular circumference was induced in AMI + Vehicle mice. Intraperitoneal injection of GK with two doses of 10 mg·kg−1, immediately after and 12 h after coronary ligation, significantly diminished the infarction size (Figure 7A). As shown in Figure 7B–F, echocardiography analysis showed that heart function in AMI + Vehicle mice was compromised ‐ LVIDd and LVIDs were significantly increased and EF and AV were lowered. However, 10 mg·kg−1 GK treatment considerably improved heart function with decreased LVIDd and LVIDs as well as increased EF and AV. TUNEL assay on heart sections displayed that the percentage of TUNEL‐positive cells in AMI + GK10 group was significantly lower than that in AMI + Vehicle group, indicating decreased apoptosis following GK treatment (Figure 7G, H).
Figure 7.
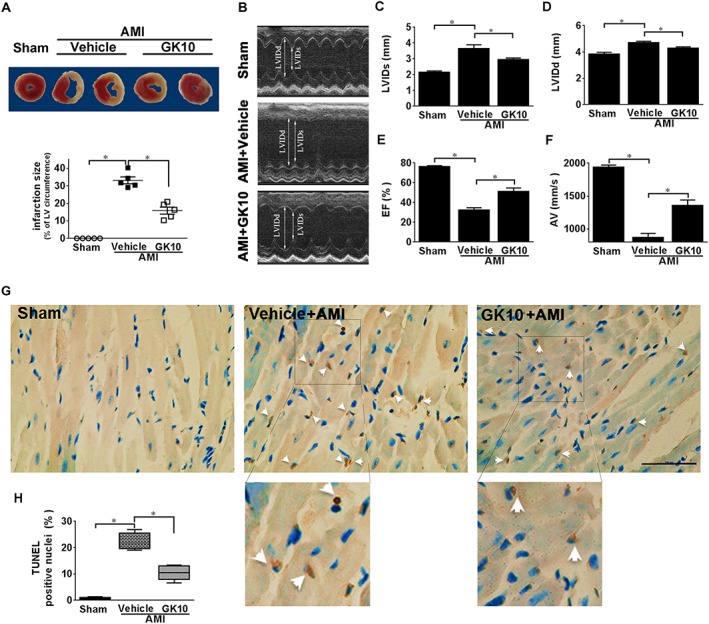
GK exerted cardioprotection on mice subjected to AMI. Mice were subjected to AMI by permanent LCA occlusion. (A) After 24 h of coronary ligation, myocardial infarct size was determined by TTC staining. The quantitative analysis confirmed a diminished infarction size in GK treatment group. (B–F) Echocardiographic assessment of LVIDd and LVIDs and left ventricular (LV) EF (%) were detected by M‐mode images, AV was detected by pulse wave velocity and GK treatment group exhibited a significantly decreased LVIDd and LVIDs, as well as increased EF and AV, compared with AMI model group. (G, H) TUNEL staining was performed on sections from the border zone of heart infarction (scale bar, 100 μm). Lower images are 1.9× magnifications of the boxed areas in the upper images. Arrows indicate TUNEL positive nuclei. Quantitative analysis indicated decreased apoptosis by GK treatment. Data are presented as means ± SEM (n = 5); *P < 0.05.
GK activated IRE1α in ER‐stressed myocardium of AMI mice
The in vitro results described above led us to examine the effects of GK on ER stress in the AMI mice. Expansion of the ER was investigated using TEM of samples of cardiac tissue. This analysis demonstrated normal ER and mitochondria in sham hearts, while more dilated ER was observed in hearts from AMI + Vehicle mice (Figure 8A). In contrast, the ER in myocardium from mice treated with 10 mg·kg−1 GK appeared fairly normal with no significant expansion (Figure 8A). Western blot analysis demonstrated a significantly raised level of the ER stress marker proteins GRP78 and CHOP, total and phosphorylation of IRE1α, XBP1s and autophagy marker proteins LC3‐II and p62 in AMI mice (Figure 8B–I). As compared with the AMI vehicle group, 10 mg·kg−1 GK treatment considerably decreased CHOP and p62 but augmented phosphorylation of IRE1α, XBP1s and LC3‐II. These results demonstrate the beneficial effects of GK on relieving ER stress through the IRE1α branch in an animal model of AMI.
Figure 8.
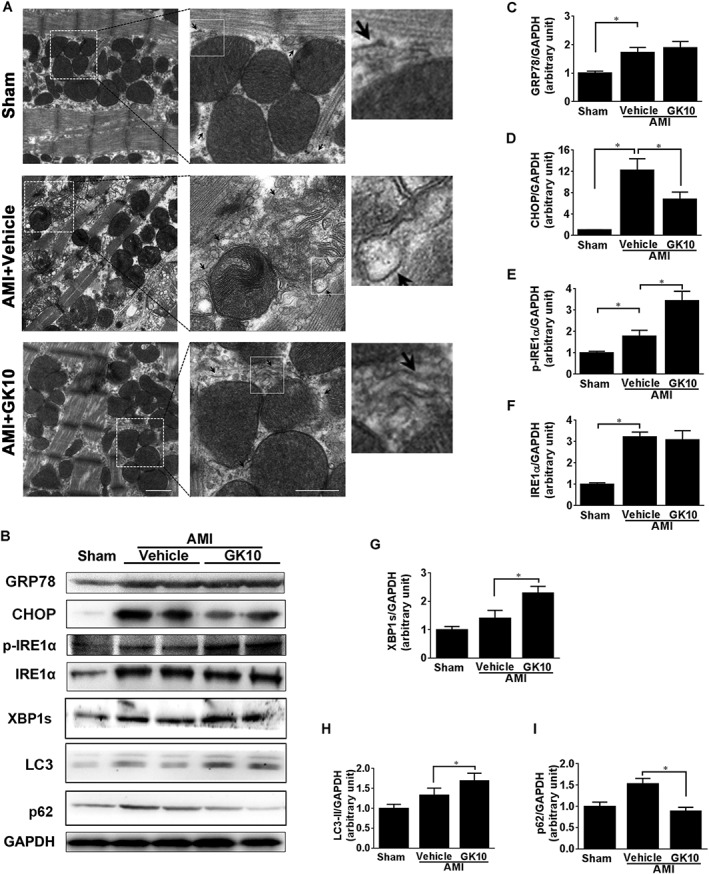
GK attenuated ER stress and ER expansion in myocardium of mice subjected to AMI. (A) Ultrastructure of ER and mitochondria of left ventricular papillary muscle was examined by TEM. Middle images (scare bar, 500 nm) are magnifications of boxed areas in the left images (scale bar, 1 μm). Right images are 3× magnifications of boxed areas in the middle images. Representative images showed dilated ER morphology in myocardium from vehicle mice. The ER structure in myocardium from mice treated with 10 mg·kg−1 GK appeared fairly normal with no significant expansion. The arrow heads show the ER morphology. (B) Levels of ER stress maker proteins GRP78, CHOP, total and phosphorylated IRE1α, XBP1s and autophagy marker proteins LC3 and p62, were assessed using western blot. (C–I) Quantitative analysis showed that 10 mg·kg−1 GK treatment considerably decreased CHOP and p62, whereas phosphorylation of IRE1α, XBP1s and LC3‐II was augmented. Data are presented as means ± SEM (n = 5); *P < 0.05.
Discussion and conclusion
Extracts of G. biloba, especially those containing the ginkgolides including ginkgolides A, B, C and J, exhibit potent cardioprotective properties, by inhibiting PAF or scavenging free radicals, and thus protecting isolated heart preparations against ischaemia (Koltai et al., 1989; Tosaki et al., 1996; Pietri et al., 1997; Varga et al., 1999). In the present study, we demonstrated that GK, a newly isolated constituent of the ginkgolide family, protected NRCMs in vitro and mouse myocardium in vivo against ER stress‐induced apoptosis. The selective activation of the IRE1α/XBP1 pathway in the UPR played a pivotal role in the beneficial effects of GK on ER stress. These results suggest that GK provided cytoprotection in response to ER stress.
As the most ancient of three ER transmembrane sensors, the IRE1α protein plays a central role in the UPR. IRE1α senses unfolded proteins either directly or indirectly through its ER luminal domain that becomes oligomerized during stress (Han et al., 2009; Chen and Brandizzi, 2013). The oligomerization allows trans‐autophosphorylation, and the resultant conformational change leads to IRE1α activation. In our experiments, GK markedly activated the IRE1α arm of the UPR in both in vivo and in vitro models, as shown by higher level of phosphorylated IRE1α and XBP1 mRNA splicing, thus increasing the protein expression of XBP1s, which is a transcriptional factor with target genes, including GRP78 (Yoshida et al., 2001; Lee et al., 2003; Hosoi et al., 2012). In this study, we observed an increase in GRP78 level that may be due to de novo synthesis as a result of activating the IRE1α/XBP1s pathway.
In yeast, IRE1 is activated through a two‐step mechanism, namely, dissociation of GRP78 and direct interaction with unfolded proteins at its luminal domain (Bravo et al., 2013). In the past, the dissociation of GRP78 had been thought to equally regulate the activation of these three sensors. However, differences in the responses of these sensors to the GRP78 dissociation have been recognized, and distinct activation kinetics for each sensor were also reported (DuRose et al., 2006). It remains unclear whether a similar mechanism is applicable to IRE1α in mammalian cells. Some studies suggested that mammalian IRE1α activation strongly depends on the dissociation of GRP78 rather than a direct interaction with unfolded proteins (Oikawa et al., 2009; Oikawa et al., 2012). Preferential activation of UPR signalling branches by alternative types of ER stress have also been studied (DuRose et al., 2006). The structural differences between IRE1α and PERK play a role in their differential activation kinetics (DuRose et al., 2006). In contrast to IRE1α and PERK, less structure–function information is available for the lumenal portion of the type II transmembrane protein ATF6. Apart from the dissociation of GRP78, ATF6 activation is negatively regulated by intramolecular and intermolecular disulphide bridge formation (Nadanaka et al., 2007; Tsukumo et al., 2007). Therefore, individual branches may be specialized to respond to particular stressors.
In contrast to its pro‐survival role through splicing XBP1, activated IRE1α can interact with TRAF2, which leads to activation of ASK1 and downstream JNK to induce pro‐death signalling. The IRE1α–JNK connection provides mechanistic insight that the UPR is able to regulate anti‐apoptotic and pro‐apoptotic molecules to determine divergent fates of stressed cells. c‐Jun N‐terminal inhibitory kinase and Jun activation domain‐binding protein 1 have been identified as IRE1α‐interacting molecules, and they might influence the shift of the UPR to either pro‐survival or pro‐apoptosis signalling, by association with or dissociation from IRE1α (Oono et al., 2004). Moreover, low MW compounds might interrupt the IRE1α/TRAF2 interaction to selectively utilize the beneficial effects following IRE1α activation. These low MW compounds might not interfere with XBP1 mRNA splicing but could prevent the activation of the pro‐apoptotic processes (Szegezdi et al., 2006). The present study shows that GK treatment did not affect the phosphorylation of ASK1 but it did significantly decrease the level of phospho‐JNK. The mechanism underlying reduced phospho‐JNK by GK treatment requires further investigation. A reasonable hypothesis would be that GK might have effects on other pathways, such as JNK phosphatases, which can negatively regulate JNK phosphorylation, thus blunting phospho‐JNK level whereas phospho‐ASK level remaining unchanged.
Furthermore, IRE1 is able to exert protective or pro‐apoptotic effects via splicing of XBP1 mRNA and the induction of RIDD respectively (Chen and Brandizzi, 2013). It is believed that modulating different sites of IRE1 can differentially regulate XBP1 splicing and RIDD activity. IRE1 activation requires its dimerization, oligomerization and subsequent trans‐autophosphorylation, which lead to a conformational change on its RNase domain and an increased RNase activity. Our study demonstrated that GK augmented IRE1α autophosphorylation in response to ER stress stimuli and this increased autophosphorylation correlated with enhanced XBP1 mRNA splicing. Conversely, RIDD activation and JNK phosphorylation were decreased by GK treatment. Akin to the beneficial effects of GK, recent studies also reported that IRE1 modulators, such as quercetin (Wiseman et al., 2010), 1NM‐PP1 (Han et al., 2009) and F6 peptide (Bouchecareilh et al., 2011), were able to enhance XBP1 mRNA splicing but attenuate ER stress‐mediated RIDD and JNK activation. The study by Wiseman et al., (2010) showed that binding of quercetin at the dimer interface of IRE1's kinase extension nuclease domain enhances IRE1 dimerization, which activates its RNase activity, leading to increasing XBP1 mRNA splicing. Moreover, Han et al., (2009) pointed out that an ATP‐competitive inhibitor, 1NM‐PP1, was able to facilitate dimerization/oligomerization of a mutant form of IRE1 with an altered kinase domain, thus increasing IRE1 RNase activity and XBP1 mRNA splicing, but not affecting the degradation of RIDD substrates. In addition, it has been suggested that IRE1 dimerization/oligomerization could influence its RNA substrate specificity (Tam et al., 2014). However, the extent to which oligomerization dictates the substrate specificity remains unknown. Overall, we hypothesize that GK is likely to interact with IRE1α, which induces IRE1α phosphorylation and subsequent conformational change of its RNase domain. This allosteric change could enhance RNase activity of IRE1α for XBP1 mRNA splicing. Nevertheless, the interaction between GK and IRE1α remains to be investigated.
In summary, this study presents evidence that GK selectively activated the IRE1α/XBP1 pathway, in vitro and in vivo, leading to amelioration of ER stress. These actions of GK demonstrate its potential for treating cardiovascular diseases (Figure 9).
Figure 9.
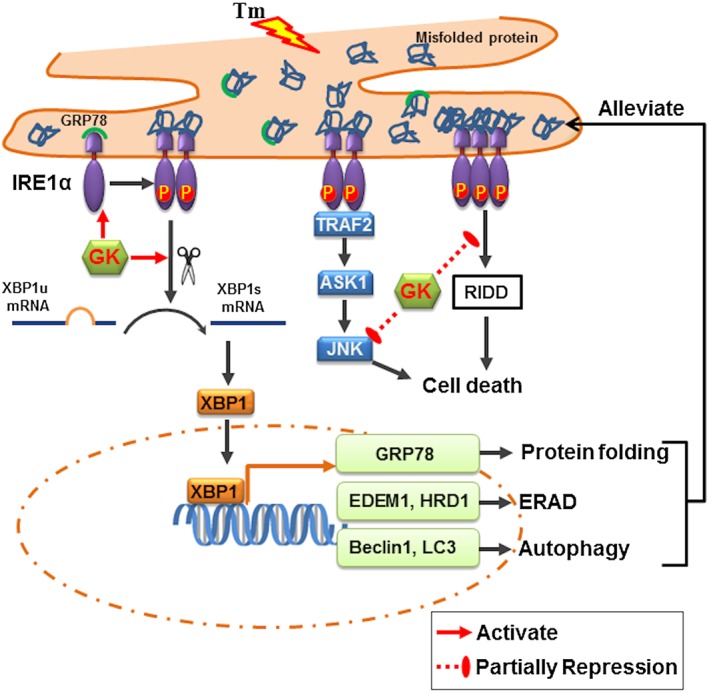
Proposed scheme of the effects of GK in our model. GK selectively activates the IRE1α/XBP1 pathway, which in turn leads to up‐regulation of GRP78, ERAD and autophagy influx. In addition, GK is able to partly repress the pro‐apoptotic action of RIDD and the JNK. Thus, GK can ameliorate ER stress and protect cardiomyocytes. Tm, tunicamycin.
Author contributions
S.W., Z.W. and X.W. designed the research study. X.W., G.D. and W.X. supervised the project. S.W., Z.W. and J.G. performed the experiments. S.W., J.G. and Q.F. analysed the data. X.W., G.D., W.X. and Q.F. contributed essential techniques, reagents or tools. X.W., S.W., J.G. and G.G. drafted the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. The effects of GK alone on cell death and apoptosis signalling pathways in NRCMs. NRCMs were treated with GK at the indicated concentrations for up to 36 h; then cell death was detected by LDH assays, and the apoptosis marker proteins were examined by Western blot. (A) GK alone did not affect the viability of NRCMs. (B) GK alone had no effect on levels of apoptosis‐relevant proteins Bax, Bcl‐2, cytochrome c and cleaved caspase 3 in NRCMs. Data are presented as means ± SEM (n = 5). Cyt c, cytochrome c; cyto, cytosolic; mito, mitochondrial.
Figure S2. The effects of GK alone on ER stress, autophagy and ERAD signalling pathways. (A–C) NRCMs were pretreated with or without 10 mΜ GK for 36 h. Western blot showed that GK alone had no effects on ER stress (A), ERAD (B) and autophagy (C) signalling. (D) NRCMs were pretreated with E64d and pepstatin A for 2 h to inhibit lysosomal proteases followed by incubation with or without 10 μM GK for 12 h and then with or without tunicamycin for additional 24 h incubation. At the end point, LC3‐II and p62 levels were analysed by Western blot. (D–F) Quantitative analysis of LC3 (D) and p62 (F). Data are presented as means ± SEM (n = 5); *P < 0.05.
Figure S3. The effects of siIRE1 and siXBP1 on apoptosis in NRCMs. NRCMs were transfected with negative control siRNA (scramble), specific siRNA of IRE1α (siIRE1) and XBP1 (siXBP1), respectively, for 72 h, and then stimulated with or without tunicamycin for 24 h. Apoptosis was detected using the TUNEL assay. Quantitative analysis showed that siIRE1 or siXBP1 increased TUNEL positive cells in the presence or absence of tunicamycin. Data are presented as means ± SEM (n = 5); *P < 0.05.
Figure S4. The effect of GK alone or with thapsigargin on RIDD in NRCMs. NRCMs were pretreated with or without 10 mΜ GK for 36 h. (A) qPCR results showed that GK alone had no effect on the transcription of Blos1, Scara3, Hgnat, Pdgfrb, Col6 and Pmp22. (B) Blos1, Hgnat, Pdgfrb and Col6 were reduced in thapsigargin‐treated NRCMs.
Data S1 Supporting info item
Acknowledgements
This study was supported by the grants from National Major Scientific and Technological Special Project for ‘Significant New Drugs Development’ during the Twelfth Five‐year Plan Period (2013ZX09402203) and the National Natural Science Foundation of China (81202538).
Wang, S. , Wang, Z. , Fan, Q. , Guo, J. , Galli, G. , Du, G. , Wang, X. , and Xiao, W. (2016) Ginkgolide K protects the heart against endoplasmic reticulum stress injury by activating the inositol‐requiring enzyme 1α/X box‐binding protein‐1 pathway. British Journal of Pharmacology, 173: 2402–2418. doi: 10.1111/bph.13516.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amm I, Sommer T, Wolf DH (2014). Protein quality control and elimination of protein waste: the role of the ubiquitin‐proteasome system. Biochim Biophys Acta 1843: 182–196. [DOI] [PubMed] [Google Scholar]
- Bouchecareilh M, Higa A, Fribourg S, Moenner M, Chevet E (2011). Peptides derived from the bifunctional kinase/RNase enzyme IRE1α modulate IRE1α activity and protect cells from endoplasmic reticulum stress. FASEB J 25: 3115–3129. [DOI] [PubMed] [Google Scholar]
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F et al. (2013). Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol 301: 215–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AE, Brewer JW (2013). Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life 65: 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YN, Brandizzi F (2013). IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV (2014). Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. Biomed Res Int 2014: 610140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y (2014). Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21: 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickhout JG, Carlisle RE, Austin RC (2011). Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res 108: 629–642. [DOI] [PubMed] [Google Scholar]
- DuRose JB, Tam AB, Niwa M (2006). Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell 17: 3095–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan ZY, Liu XG, Guo RZ, Dong X, Gao W, Li P et al. (2015). Pharmacokinetic studies of ginkgolide K in rat plasma and tissues after intravenous administration using ultra‐high performance liquid chromatography–tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 988: 1–7. [DOI] [PubMed] [Google Scholar]
- Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M et al. (2010). A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S et al. (2014). Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158: 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendyk J, Agellon LB, Michalak M (2013). Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol 75: 49–67. [DOI] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A et al. (2009). IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138: 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009). Regulated Ire1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Korematsu K, Horie N, Suezawa T, Okuma Y, Nomura Y et al. (2012). Inhibition of casein kinase 2 modulates XBP1–GRP78 arm of unfolded protein responses in cultured glial cells. PLoS One 7: e40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D et al. (2010). Targeted deletion of the extracellular signal‐regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload‐induced apoptosis in the heart. Circ Res 106: 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai M, Tosaki A, Hosford D, Braquet P (1989). Ginkgolide B protects isolated hearts against arrhythmias induced by ischemia but not reperfusion. Eur J Pharmacol 164: 293–302. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH (2003). XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23: 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yan Y, Bao L, Chen B, Zhao Y, Qi R (2014). Ginkgolide B inhibits platelet release by blocking Syk and p38 MAPK phosphorylation in thrombin‐stimulated platelets. Thromb Res 134: 1066–1073. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhao G, Yan Y, Bao L, Chen B, Qi R (2012). Ginkgolide B reduces atherogenesis and vascular inflammation in ApoE(−/−) mice. PLoS One 7: e36237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Kim JK, Chen B, Abdel‐Latif A, Kitakaze M, Yan L (2015). Attenuation of ER stress prevents post‐infarction‐induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem Biol Interact 225: 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Liu H, Jiao H, Wang L, Chen L, Liang J et al. (2012a). Neuroprotective effect of ginkgolide K on glutamate‐induced cytotoxicity in PC 12 cells via inhibition of ROS generation and Ca(2+) influx. Neurotoxicology 33: 59–69. [DOI] [PubMed] [Google Scholar]
- Ma S, Liu X, Xun Q, Zhang X (2014). Neuroprotective effect of ginkgolide K against H2O2‐induced PC12 cell cytotoxicity by ameliorating mitochondrial dysfunction and oxidative stress. Biol Pharm Bull 37: 217–225. [DOI] [PubMed] [Google Scholar]
- Ma S, Yin H, Chen L, Liu H, Zhao M, Zhang X (2012b). Neuroprotective effect of ginkgolide K against acute ischemic stroke on middle cerebral ischemia occlusion in rats. J Nat Med 66: 25–31. [DOI] [PubMed] [Google Scholar]
- Maclennan KM, Darlington CL, Smith PF (2002). The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog Neurobiol 67: 235–257. [DOI] [PubMed] [Google Scholar]
- Margariti A, Li H, Chen T, Martin D, Vizcay‐Barrena G, Alam S et al. (2013). XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN‐1 transcriptional activation. J Biol Chem 288: 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel M, Dejeans N, Taouji S, Chevet E, Grosset CF (2013). MicroRNA‐1291‐mediated silencing of IRE1α enhances glypican‐3 expression. RNA 19: 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine CS, Werstuck GH (2013). The development and progression of atherosclerosis: evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc Hematol Disord Drug Targets 13: 158–164. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Zheng Z, Fan Z, Rajagopalan S, Sun Q, Zhang K (2013). Exposure to fine airborne particulate matter induces macrophage infiltration, unfolded protein response, and lipid deposition in white adipose tissue. Am J Transl Res 5: 224–234. [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S (2013). Role of alpha‐crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis 4: e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T (2007). How to interpret LC3 immunoblotting. Autophagy 3: 542–545. [DOI] [PubMed] [Google Scholar]
- Montes P, Ruiz‐Sanchez E, Rojas C, Rojas P (2015). Ginkgo biloba Extract 761: a review of basic studies and potential clinical use in psychiatric disorders. CNS Neurol Disord Drug Targets 14: 132–149. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Okada T, Yoshida H, Mori K (2007). Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol 27: 1027–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K, Iwawaki T (2009). Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res 315: 2496–2504. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kitamura A, Kinjo M, Iwawaki T (2012). Direct association of unfolded proteins with mammalian ER stress sensor, IRE1beta. PLoS One 7: e51290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oono K, Yoneda T, Manabe T, Yamagishi S, Matsuda S, Hitomi J et al. (2004). JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem Int 45: 765–772. [DOI] [PubMed] [Google Scholar]
- Pietri S, Maurelli E, Drieu K, Culcasi M (1997). Cardioprotective and anti‐oxidant effects of the terpenoid constituents of Ginkgo biloba extract (EGb 761). J Mol Cell Cardiol 29: 733–742. [DOI] [PubMed] [Google Scholar]
- Reinstein E, Frentz S, Morgan T, Garcia‐Minaur S, Leventer RJ, McGillivray G et al. (2013). Vascular and connective tissue anomalies associated with X‐linked periventricular heterotopia due to mutations in filamin A. Eur J Hum Genet: EJHG 21: 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozen E, Karademir B, Ozer NK (2015). Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic Biol Med 78: 30–41. [DOI] [PubMed] [Google Scholar]
- Su H, Wang X (2010). The ubiquitin‐proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res 85: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A (2006). Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep 7: 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam AB, Koong AC, Niwa M (2014). Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep 9: 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosaki A, Pali T, Droy‐Lefaix MT (1996). Effects of Ginkgo biloba extract and preconditioning on the diabetic rat myocardium. Diabetologia 39: 1255–1262. [DOI] [PubMed] [Google Scholar]
- Tsukumo Y, Tomida A, Kitahara O, Nakamura Y, Asada S, Mori K et al. (2007). Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem 282: 29264–29272. [DOI] [PubMed] [Google Scholar]
- Varga E, Bodi A, Ferdinandy P, Droy‐Lefaix MT, Blasig IE, Tosaki A (1999). The protective effect of EGb 761 in isolated ischemic/reperfused rat hearts: a link between cardiac function and nitric oxide production. J Cardiovasc Pharmacol 34: 711–717. [DOI] [PubMed] [Google Scholar]
- Vekich JA, Belmont PJ, Thuerauf DJ, Glembotski CC (2012). Protein disulfide isomerase‐associated 6 is an ATF6‐inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion‐mediated cell death. J Mol Cell Cardiol 53: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Liu JL, Wu L, Yu Z, Yang HT (2014). Concentration‐dependent wrestling between detrimental and protective effects of H2O2 during myocardial ischemia/reperfusion. Cell Death Dis 5: e1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Hendershot LM (1996). Protein folding and assembly in the endoplasmic reticulum. EXS 77: 41–55. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J et al. (2010). Flavonol activation defines an unanticipated ligand‐binding site in the kinase‐RNase domain of IRE1. Mol Cell 38: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen X, Sharma P, Moon M, Sheftel AD, Dawood F et al. (2014). HACE1‐dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun 5: 3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen B, Wu W, Bao L, Qi R (2011). Ginkgolide B reduces inflammatory protein expression in oxidized low‐density lipoprotein‐stimulated human vascular endothelial cells. J Cardiovasc Pharmacol 57: 721–727. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yao H, Yin HL, Zhu QL, Sun JL, Ma W et al. (2013). Ginkgo biloba extract and ginkgolide antiarrhythmic potential by targeting hERG and ICa‐L channel. J Pharmacol Sci 123: 318–327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effects of GK alone on cell death and apoptosis signalling pathways in NRCMs. NRCMs were treated with GK at the indicated concentrations for up to 36 h; then cell death was detected by LDH assays, and the apoptosis marker proteins were examined by Western blot. (A) GK alone did not affect the viability of NRCMs. (B) GK alone had no effect on levels of apoptosis‐relevant proteins Bax, Bcl‐2, cytochrome c and cleaved caspase 3 in NRCMs. Data are presented as means ± SEM (n = 5). Cyt c, cytochrome c; cyto, cytosolic; mito, mitochondrial.
Figure S2. The effects of GK alone on ER stress, autophagy and ERAD signalling pathways. (A–C) NRCMs were pretreated with or without 10 mΜ GK for 36 h. Western blot showed that GK alone had no effects on ER stress (A), ERAD (B) and autophagy (C) signalling. (D) NRCMs were pretreated with E64d and pepstatin A for 2 h to inhibit lysosomal proteases followed by incubation with or without 10 μM GK for 12 h and then with or without tunicamycin for additional 24 h incubation. At the end point, LC3‐II and p62 levels were analysed by Western blot. (D–F) Quantitative analysis of LC3 (D) and p62 (F). Data are presented as means ± SEM (n = 5); *P < 0.05.
Figure S3. The effects of siIRE1 and siXBP1 on apoptosis in NRCMs. NRCMs were transfected with negative control siRNA (scramble), specific siRNA of IRE1α (siIRE1) and XBP1 (siXBP1), respectively, for 72 h, and then stimulated with or without tunicamycin for 24 h. Apoptosis was detected using the TUNEL assay. Quantitative analysis showed that siIRE1 or siXBP1 increased TUNEL positive cells in the presence or absence of tunicamycin. Data are presented as means ± SEM (n = 5); *P < 0.05.
Figure S4. The effect of GK alone or with thapsigargin on RIDD in NRCMs. NRCMs were pretreated with or without 10 mΜ GK for 36 h. (A) qPCR results showed that GK alone had no effect on the transcription of Blos1, Scara3, Hgnat, Pdgfrb, Col6 and Pmp22. (B) Blos1, Hgnat, Pdgfrb and Col6 were reduced in thapsigargin‐treated NRCMs.
Data S1 Supporting info item