

Abstract
Background and Purpose
Leptin, an important regulator of the energy balance, acts on the brain to inhibit feeding. However, the mechanisms involved in leptin signalling have not yet been fully elucidated. Heat shock protein 90 (HSP90) is a molecular chaperone that is involved in regulating cellular homeostasis. In the present study, we investigated the possible involvement of HSP90 in leptin signal transduction.
Experimental Approach
HEK293 and SH‐SY5Y cell lines stably transfected with the Ob‐Rb leptin receptor (HEK293 Ob‐Rb, SH‐SY5Y Ob‐Rb) were used in the present study. Phosphorylation of JAK2 and STAT3 was analysed by western blotting. An HSP90 inhibitor was administered i.c.v. into rats and their food intake was analysed.
Key Results
The knock‐down of HSP90 in the HEK293 Ob‐Rb cell line attenuated leptin‐induced JAK2 and STAT3 signalling. Moreover, leptin‐induced JAK2/STAT3 phosphorylation was markedly attenuated by the HSP90 inhibitors geldanamycin, radicicol and novobiocin. However, these effects were not mediated through previously known factors, which are known to be involved in the development of leptin resistance, such as suppressor of cytokine signalling 3 or endoplasmic reticulum stress. The infusion of an HSP90 inhibitor into the CNS blunted the anorexigenic actions of leptin in rats (male Wister rat).
Conclusions and Implications
HSP90 may be a novel factor involved in leptin‐mediated signalling that is linked to anorexia.
Abbreviations
- ER stress
endoplasmic reticulum stress
- HSP90
heat shock protein 90
- POMC
proopiomelanocortin
- PTP1B
protein tyrosine phosphatase‐1B
- SOCS3
suppressor of cytokine signalling 3
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| GPR78 | JAK2 |
| Catalytic receptors b | |
| Leptin receptor |
| LIGANDS | |
|---|---|
| HSP90α1 | Leptin |
| HSP90β | POMC (ACTH) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Obesity is a complex condition associated with an increased risk of diseases, such as diabetes, cardiovascular disease and hypertension. Therefore, the underlying mechanisms involved in the development of obesity need to be elucidated in more detail. Leptin is an anti‐obesity hormone that was identified by Friedman's group in 1994 (Zhang et al., 1994). It is mostly secreted from adipose tissue, circulates in the blood, and acts on the brain. Leptin activates the Ob‐Rb leptin receptor, which is expressed on neurons within the hypothalamus, and induces the JAK2/STAT3 signalling pathway (Vaisse et al., 1996; Hosoi et al., 2002). By activating the JAK2/STAT3 pathway, leptin inhibits food intake and body weight gain (Campfield et al., 1995). The activation of STAT3 in proopiomelanocortin (POMC) neurons has been shown to increase the production of α‐melanocyte‐stimulating hormone (αMSH), which, in turn, activates melanocortin receptors. The activation of melanocortin receptors activates catabolic pathways by inhibiting food intake. Furthermore, leptin has been shown to inhibit anabolic pathways by inhibiting neuropeptide Y/agouti‐related neuropeptide (NPY/AgRP) neurons (Schwartz et al., 2000). Although leptin has anti‐obesity effects and was initially expected to be useful as an anti‐obesity drug, most obese patients were found to be unresponsive to leptin, which indicated that leptin resistance was involved in the development of obesity (Münzberg and Myers, 2005). Therefore, elucidation of the underlying mechanisms involved in the development of leptin resistance is of importance (Friedman, 2003). Until now, suppressors of cytokine signalling 3 (SOCS3) (Endo et al., 1997; Naka et al., 1997; Starr et al., 1997; Bjørbaek et al., 1998), protein tyrosine phosphatase‐1B (PTP1B) (Cheng et al., 2002; Zabolotny et al., 2002) and endoplasmic reticulum (ER) stress (Hosoi et al., 2008; Zhang et al., 2008; Ozcan et al., 2009; Hosoi et al., 2014) were the only pathways thought to be involved in the development of leptin resistance (Hosoi and Ozawa, 2010). SOCS3 is up‐regulated in the arcuate nucleus of the hypothalamus from diet‐induced obese mice (Münzberg et al., 2004) and inhibits leptin signalling by binding to Ob‐Rb leptin receptors (Bjørbaek et al., 1998, Bjorbak et al., 2000). PTP1B‐deficient mice are resistant to high‐fat diet‐induced obesity (Elchebly et al., 1999; Klaman et al., 2000), and the activation of PTP1B inhibits leptin signalling by dephosphorylating the leptin receptor‐associated kinase, JAK2 in the hypothalamus (Zabolotny et al., 2002, Cheng et al., 2002). Endoplasmic reticulum stress was found to be activated in the hypothalamus of obese mice (Zhang et al., 2008; Ozcan et al., 2009), and leptin resistance developed under ER‐stressed conditions (Hosoi et al., 2008; Ozcan et al., 2009). Taken together, these findings indicate that several mechanisms are involved in the development of leptin resistance. However, the mechanisms underlying leptin resistance are still unclear and need to be examined in more detail.
HSP90 is a molecular chaperone that is involved in regulating cellular homeostasis and stress responses. Several studies have demonstrated its important role in regulating CNS function. HSP90 was previously shown to promote tau solubility and binding to microtubules in the CNS, thereby preventing tau aggregation in Alzheimer's disease (AD) (Dou et al., 2003). HSP90 was also shown to inhibit amyloid β‐(1–42) aggregation, which has been implicated in the progression of AD (Evans et al., 2006). These findings suggested the critical involvement of HSP90 in neuronal function. In addition to its function as a molecular chaperone, HSP90 has been reported to regulate signal transduction. Previous studies suggested that HSP90 interacts with STAT3 and regulates its activation in IL‐6 stimulated cells (Shah et al., 2002; Sato et al., 2003). However, the detailed relationships between the two molecular functions of HSP90: as a chaperone and as a mediator of STAT3 signalling had not yet been elucidated. HSP90 is expressed in the hypothalamus (Olazábal et al., 1992), and immunohistochemical analyses revealed that its expression is primarily neuronal (Itoh et al., 1993; Gass et al., 1994). The actions of leptin on the regulation of feeding behavior are known to be mediated through neuronal cells; therefore, we hypothesized that HSP90 may regulate leptin‐induced signal transduction, activating leptin‐induced STAT3, which has been linked to anorexia. In the present study, we demonstrated that HSP90 plays a key role in regulating the leptin‐induced activation of the JAK2/STAT3 signalling. Furthermore, we showed that HSP90 inhibitors attenuate leptin‐induced anorexia, suggesting the important role of HSP90 in regulating the actions of leptin.
Methods
Generation of Ob‐Rb leptin receptor‐transfected cells
The Ob‐Rb leptin receptor is a long isoform of the leptin receptor, which plays an important role in activating leptin‐induced JAK2‐STAT3 signalling (Ghilardi et al., 1996). The human Ob‐Rb leptin receptor construct, a gift from Genetech Inc., was transfected into SH‐SY5Y and HEK293 cell lines using LipofectAMINE PLUS Reagent (Life Technologies Inc.) according to the manufacturer's instructions. Stable transfectants (SH‐SY5Y‐Ob‐Rb and HEK293‐Ob‐Rb cells) were obtained by selection with the antibiotic G418 (Hosoi et al., 2006).
Transient transfection of Ob‐Rb plasmid into GT1–7 cell line
The human Ob‐Rb leptin receptor construct was transfected into GT1–7 cells using Lipofectamine 2000 Reagent (Life Technologies Inc.) according to the manufacturer's instructions. Forty‐eight hours after the transfection, the cells were stimulated with leptin.
Experimental design
The analyses were not performed blind. However, we made an effort to be close to the conditions of blinded assays. All the samples were obtained using the same procedures, and the samples were treated in the same way. Each of the samples was numbered, and the identity of each sample was not obvious during the experiment. The analysis was not performed with randomization. However, the experiments were carried out using the same conditions (the room temperature was same for all of the experiments), and we made an effort to keep the conditions close to those of a randomization assay.
RNAi experiment
Lipofectamine RNAiMAX (Life Technologies) was used to transfect siRNA at a final concentration of 25 nM (at Santa Cruz siRNAs) or 10 nM (at Life Technologies siRNAs) according to the manufacturer's directions. An Opti‐MEM1 medium was used for the transfection. HSP90 α/β siRNA (h) (SantaCruz, sc‐35608) and control siRNA‐A (Santa Cruz, sc‐37007) were used for the siRNA transfection. HSP 90α/β siRNA (h) (sc‐35608) is a pool of four different siRNA duplexes. Sequence of these siRNAs are as follows: sc‐35608A: sense; CGUGAGAUGUUGCAACAAAtt, antisense; UUUGUUGCAACAUCUCACGtt, sc‐35608B: sense; CCUGUUCAGUACUCUACAAtt, antisense; UUGUAGAGUACUGAACAGGtt, sc‐35608C: sense; GAAGACAAGGAGAAUUACAtt, antisense; UGUAAUUCUCCUUGUCUUCtt, sc‐35608D: sense; GCAAGGCAAAGUUUGAGAAtt, antisense; UUCUCAAACUUUGCCUUGCtt. Strands A and B are specific to HSP 90α. Strands C and D are specific to HSP 90β. We also used HSP90 siRNA and Silencer® select negative control siRNA #1 from Life technologies (Supporting Information Fig. S1). The sequence of the siRNA is as follows: sense: CUAUGGGUCGUGGAACAAAtt, antisense: UUUGUUCCACGACCCAUAGgt. Cells were harvested 72 h after the transfection.
Western blotting
Western blotting was performed as described previously (Hosoi et al., 2012). Cells were washed with ice‐cold PBS and lysed in a buffer containing 10 mM HEPES‐NaOH (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 10 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin, 1 mM PMSF and 1% NP‐40 for 20 min. The lysates were centrifuged at 20630 × g for 20 min at 4°C, and the supernatants were collected. The samples were boiled with Laemmli buffer for 3 min, fractionated by SDS‐PAGE and transferred at 4°C to nitrocellulose membranes. The membranes were incubated with anti‐KDEL (StressGen; diluted to 1:1000), anti‐CHOP (Santa Cruz; diluted to 1:500), anti‐HSP90 (Sigma or Santa Cruz; diluted to 1:1000), anti‐JAK2 (Santa Cruz; diluted to 1:500), anti‐Phospho (Tyr1007/1008)‐JAK2 (Cell Signaling or upstate; diluted to 1:1000), anti‐Phospho (Tyr705)‐STAT3 (Cell Signaling; diluted to 1:1000), anti‐STAT3 (Cell Signaling; diluted to 1:1000), anti‐Phospho‐Tyr (upstate; diluted to 1:2000), and anti‐GAPDH (Chemicon; diluted to 1:2000) antibodies followed by anti‐horseradish peroxidase‐linked antibody. Peroxidase was detected by chemiluminescence using an enhanced chemiluminescence system.
Immunoprecipitation
Cells were lysed in lysis buffer (10 mM HEPES‐NaOH (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 10 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin, 1 mM PMSF and 0.1% NP‐40), and samples were homogenized using a 21G needle. The lysates were centrifuged at 20630 × g for 20 min at 4°C, and the supernatants were collected. An antibody was added to the lysate and rotated at 4°C. Dynabeads Protein G (Invitrogen) was then added and rotated at 4°C for 20 min. Immunoprecipitates were washed three times with lysis buffer. The immunoprecipitates from cell lysates were resolved on SDS‐PAGE and transferred to a nitrocellulose transfer membrane. The filters were then immunoblotted with each antibody. Immunoreactive proteins were visualized using an enhanced chemiluminescence detection system. Immunohistochemistry
Cells were fixed with methanol for 10 min at −20°C. After being washed with PBS, the cells were incubated with 5% normal bovine serum at 37°C for 1 h and allowed to react with anti‐HSP90 (Santa Cruz; diluted to 1:200) and anti‐phospho‐STAT3 (Cell Signaling; diluted to 1:50) antibodies at 4°C overnight. The cells were then incubated with anti‐mouse Alexa 488 (1:2000) and anti‐rabbit Alexa 488 (1:2000) at 37°C for 1 h. The cells were visualized using confocal laser scanning microscopy. The confocal laser scanning microscopy was carried out at the Analysis Center of Life Science, Natural Science Center for Basic Research and Development, Hiroshima University.
I.c.v. injections and measurement of food intake in rats
To install a stainless‐steel guide cannula for the i.c.v. injection of leptin and geldanamycin, the male Wister rat was anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.) and placed in a stereotaxic apparatus. A 24G stainless‐steel guide cannula was inserted into the brain (1.0 mm posterior to the bregma, 1.5 mm to the right lateral side and 3.7 mm below the surface). The guide cannula was secured with dental cement anchored by two stainless steel screws fixed on the dorsal surface of the skull. After surgery, a dummy cannula (30G) was inserted into the guide cannula. Animals were allowed to recover for at least 10 days after this operation. Four days before the leptin and geldanamycin injection, the dummy cannula was replaced by a microinjection cannula, all rats were injected with saline and their food intake was measured. Food was removed at 17:30, and saline was injected at 18:00. Food was placed back in the cage and at 19:30 and their food intake was measured after 4, 14 and 24 h. On the day of the experiment, the dummy cannula was replaced by a microinjection cannula. The food was removed at 17:30. Geldanamycin (100 nmol·3 μL−1) and leptin 5 μg·3 μL−1 was injected at 18:00. As the geldanamycin was dissolved in DMSO, we injected DMSO (3 μL) for the control experiments. Food was placed back in the cage at 19:30, and their food intake was measured after 4, 14 and 24 h. The placement of the cannula was verified at the end of the experiment by injecting 10 μL dye (5 μg·mL−1 Evans Blue). All animal experiments were carried out in accordance with the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals. Considering welfare and ethics, the present experiments were approved by the animal care and use committee at Hiroshima University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilly, 2015). The sex and ages of the mice and rats were the same and the room temperature was the same for all of the experiments.
I.c.v. injections and measurement of STAT3 phosphorylation in mice
Food‐deprived (16 h) male C57BL/6 mice were anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.). Novobiocin (0.5 μmol, 1 μL) or saline was injected i.c.v into the skull 0.3 mm caudal from the bregma, 0.9 mm right lateral side from the midline and 2.0 mm below the dura matter. Five minutes after the injection, leptin (2.25 μg, 2 μL) or saline was injected i.c.v. Thirty minutes after this injection, the animal was killed by decapitation and the hypothalamus was snap‐frozen in liquid nitrogen and stored in −80°C until use. Samples were homogenized using Precellys 24 (Bertin Technologies, Orleans, France) in a buffer containing 10 mM HEPES‐NaOH (pH 7.5), 150 mM NaCl, 1 mM EGTA, 21 mM Na3VO4, 10 mM NaF, 10 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin, 1 mM PMSF and 1% Nonidet P‐40. Samples were then centrifuged at 20630 × g for 45 min at 4°C, and the supernatants were collected for western blot analysis.
Data and statistical analysis
The present studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are expressed as the means ± SEM. Statistical analysis was performed using Student's t‐test, paired t‐test or Dunnett's test. ANOVA analysis and post hoc multiple comparison tests (Holm's test) were applied to compare groups. Before applying ANOVA, we checked homoscedasticity by Bartlett test. Statistical significance was assumed when the P value was <0.05. We measured the density of each of the bands using image j software (Wayne Rasband, NIH). Each set of data is expressed as fold increase over control cells. Then, we performed statistical analysis using each of the data expressed as fold intensity.
Materials
Tunicamycin (Tm) and novobiocin were obtained from Wako Pure Chemical Ltd. (Japan). Geldanamycin and radicicol were obtained from Sigma (St. Louis, MO, USA). Leptin was obtained from Sigma. GT1–7 cell line was a kind gift from Mellon PL (University of California, San Diego, CA, USA) (Wetsel et al., 1991).
Results
Knocking down HSP90 blunted leptin signalling
To determine whether HSP90 had any effect on the leptin‐induced activation of JAK2‐STAT3, we knocked down HSP90 with siRNA and analysed leptin‐induced signal transduction. Because HEK293 cells do not express a sufficient number of leptin receptors, we used an HEK293 cell line stably transfected with the Ob‐Rb leptin receptor (HEK293‐Ob‐Rb cells). The phosphorylation status of JAK2 was analysed using a phospho‐specific antibody of JAK2 following the imunoprecipitation of JAK2 from the HEK293‐Ob‐Rb cells. As shown in Figure 1A, leptin increased the activation of JAK2 and STAT3 in HEK293‐Ob‐Rb cells, which stably express the Ob‐Rb leptin receptor. This leptin‐induced JAK2/STAT3 activation was markedly suppressed by knocking down HSP90 (Figure 1). Similar results were obtained using other sequences of siRNA (Supporting Information Fig. S1). Knock‐down efficiency was confirmed by western blotting using an HSP90‐specific antibody (Figure 1C and Supporting Information Fig. S1A). These results suggest that HSP90 inhibits leptin‐induced JAK2/STAT3 signal transduction. Because HSP90 is involved in regulating leptin‐induced signals, we next examined whether leptin itself can affect the expression levels of HSP90. HEK293‐Ob‐Rb cells were treated with leptin, and the expression levels of HSP90 were analysed. Although leptin markedly increased the phosphorylation of STAT3, HSP90 did not appear to increase the expression of leptin in our investigation (Figure 2). Therefore, leptin itself may not have induced the expression of HSP90.
Figure 1.
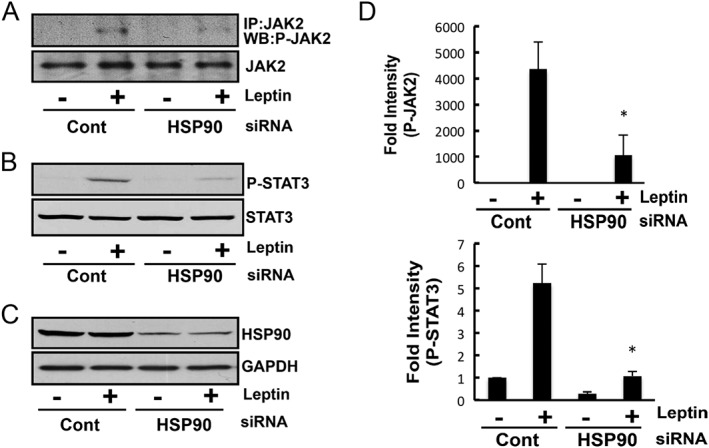
HSP90 knock‐down attenuated leptin‐induced signal transduction. Western blotting analysis of leptin‐induced JAK2/STAT3 activation in the HSP90‐knocked down HEK293‐Ob‐Rb cell line. Cells were stimulated with leptin (0.5 μg·mL−1) for 15 min, and Western blotting was then performed. The leptin‐induced phosphorylation of (A) JAK2 and (B) STAT3 was significantly inhibited in HSP90‐knocked down cells. (C) Expression level of HSP90 was analysed in HSP90‐knocked down cells. (D) A densitometoric analysis of phospho‐JAK2 and phospho‐STAT3 was conducted using image j analysis software. Each set of data was expressed as fold increase over control cells. *P < 0.05 versus control cells treated with leptin. Student's t‐test. n = 5 for phospho‐JAK2 and phospho‐STAT3 experiments.
Figure 2.
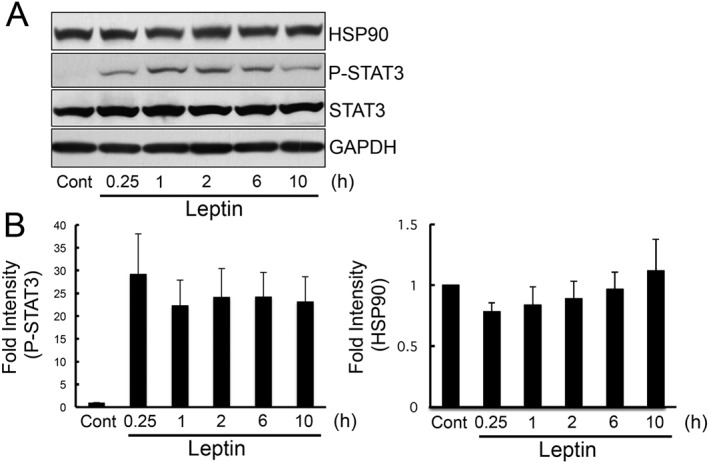
Leptin did not induce HSP90 expression. HEK 293‐Ob‐Rb cells were stimulated with leptin (0.5 μg·mL−1) for 0.25–10 h. (A) Western blotting was performed using specific antibodies for HSP90, GAPDH, phospho‐STAT3 (Tyr705) and STAT3. (B) A densitometoric analysis of phospho‐STAT3 and HSP90 was conducted using image j analysis software. Each set of data was expressed as fold increase over control cells. n = 5 for phospho‐STAT3 and HSP90 experiments.
HSP90 inhibitors blunted leptin signalling
To further elucidate whether HSP90 had any effects on the leptin‐induced activation of JAK2/STAT3, we examined the effect several different types of HSP90‐specific inhibitors, including geldanamycin, radicicol and novobiocin on the leptin‐induced phosphorylation of JAK2/STAT3. Geldanamycin and radicicol had previously been shown to inhibit HSP90 activity by binding to the N‐terminal ATP/ADP‐binding domain of HSP90 (Roe et al., 1999). Novobiocin inhibits its function by binding to the C‐terminal dimerization domain of HSP90 (Marcu et al., 2000). Leptin induced the phosphorylation of JAK2, and this effect was significantly inhibited by treatment of the HEK293‐Ob‐Rb cell line with the HSP90 inhibitors geldanamycin, radicicol and novobiocin (Figure 3A). The inhibitory effects of these drugs were dose‐dependent (Figure 3A). Similar results were obtained for the leptin‐induced activation of STAT3 (Figure 3B). Therefore, all types of HSP90 inhibitors, which have different pharmacological properties, can inhibit leptin‐mediated signal transduction. Because leptin exerts its effects through neurons, we next used a neuronal cell line to check the effect of HSP90 on leptin's action. In the present study, the SH‐SY5Y human neuroblastoma cell line stably transfected with the Ob‐Rb leptin receptor (SH‐SY5Y‐Ob‐Rb) was used as neuronal model. The HSP90 inhibitors, geldanamycin, radicicol and novobiocin significantly inhibited the leptin‐induced phosphorylation of STAT3 in SH‐SY5Y‐Ob‐Rb cells (Figure 4A and Supporting Information Fig. S2). Similar results were also observed using the Ob‐Rb receptor transfected GT1–7 hypothalamic neuronal cell line (Figure 4C). Furthermore, STAT3 phosphorylation induced by a lower dose of leptin (0.03 μg·mL−1) was also inhibited by the HSP90 inhibitors in SH‐SY5Y‐Ob‐Rb cells (Figure 4B). Thus, HSP90 may be involved in leptin‐mediated signal transduction in neurons. We also performed immunohistochemistry to elucidate the effects of HSP90 on leptin‐mediated signals in SH‐SY5Y‐Ob‐Rb cells. The treatment with leptin increased nuclear phopho‐STAT3 staining, as indicated by a merged photo of phopho‐STAT3 staining and nuclear PI staining (Figure 5). This effect was markedly attenuated by the HSP90 inhibitor geldanamycin (Figure 5). Thus, these results suggest that geldanamycin inhibits the leptin‐induced nuclear localization of phospho‐STAT3. To elucidate the functional significance of HSP90 on leptin‐induced signalling, we analysed the leptin‐induced expression of SOCS3 in HSP90 inhibitor‐treated cells. SOCS3 is a feedback regulator of JAK–STAT pathways, which were previously shown to be induced by leptin (Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). As shown in Figure 6, leptin increased SOCS3 levels in SH‐SY5Y‐Ob‐Rb cells. This leptin‐induced increase in SOCS3 was suppressed to undetectable levels by the addition of HSP90 inhibitors (Figure 6). These results demonstrate that HSP90 inhibitors suppress the leptin‐induced STAT3‐mediated induction of SOCS3, indicating that HSP90 may functionally regulate STAT3 signalling.
Figure 3.
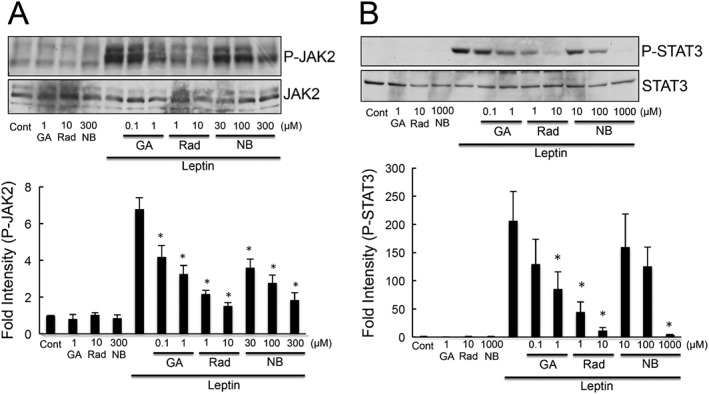
HSP90 inhibitors inhibited leptin‐induced activation of JAK2/STAT3. HEK293‐Ob‐Rb cells were pretreated with geldanamycin (GA: 0.1–1 μM), radicicol (Rad: 1–10 μM) and novobiocin (NB: 30–300 μM) for 1 h, and then stimulated with leptin (0.5 μg·mL−1) for 15 min. Western blotting was performed using specific antibodies for phospho‐JAK2 and phospho‐STAT3. Phospho‐JAK2 is shown in panel (A), and phospho‐STAT3 in panel (B). Each set of data was expressed as fold increase over control cells. Dunnett's test was performed to compare control cells treated with leptin (defined as control group of Dunnett's test) and other groups. n = 5 for phospho‐JAK2 and phospho‐STAT3 experiments.
Figure 4.

An HSP90 inhibitor inhibited leptin‐induced activation of STAT3 in neuronal cells. (A) SH‐SY5Y‐Ob‐Rb cells were treated with the HSP90 inhibitor, geldanamycin (GA: 0.1–10 μM), and leptin (0.5 μg·mL−1)‐induced STAT3 phosphorylation was analysed. The HSP90 inhibitor inhibited leptin‐induced STAT3 phosphorylation. Dunnett's test was performed to compare control cells treated with leptin (defined as control group of Dunnett's test) and other groups. *P < 0.05 versus control cells treated with leptin. n = 5. (B) STAT3 phosphorylation induced by a lower dosage of leptin (0.03 μg·mL−1) was attenuated by the HSP90 inhibitor, geldanamycin (GA: 10 μM), in SH‐SY5Y‐Ob‐Rb cells. *P < 0.05 versus control cells treated with leptin. Student's t‐test. n = 5. (C) Ob‐Rb transfected GT1–7 cells were treated with the HSP90 inhibitor, geldanamycin (GA: 10 μM), and leptin (0.1 μg·mL−1, 15 min)‐induced STAT3 phosphorylation was analysed. The HSP90 inhibitor inhibited leptin‐induced STAT3 phosphorylation. Each set of data was expressed as fold increase over control cells. *P < 0.05 versus control cells treated with leptin. Student's t‐test. n = 5.
Figure 5.
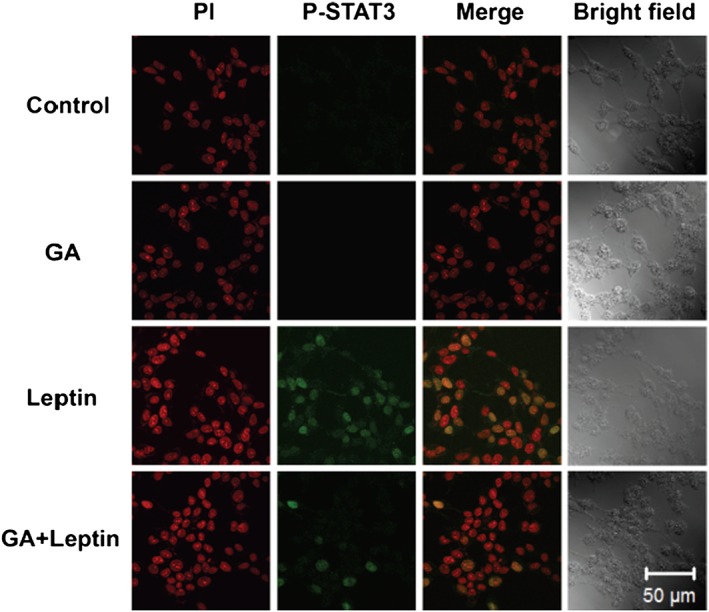
An HSP90 inhibitor inhibited leptin‐induced increases in nuclear phospho‐STAT3 staining. Fluorescence images of phospho‐STAT3 staining in SH‐SY5Y‐Ob‐Rb cells. Cells were treated with geldanamycin (GA: 10 μM) for 1 h and then stimulated with leptin (0.5 μg·mL−1) for 15 min. Left panels show PI nucleus staining (red). Second panels show phospho‐STAT3 staining (green). Third panels show merged photo of PI and phospho‐STAT3 staining. Geldanamycin inhibited leptin‐induced increases in nuclear phospho‐STAT3 staining. PI, propidium iodide. Original magnification: ×63.
Figure 6.
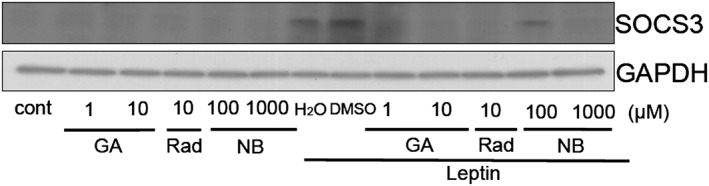
HSP90 inhibitors inhibited the leptin‐induced induction of SOCS3. SH‐SY5Y‐Ob‐Rb cells were pretreated with geldanamycin (GA: 1–10 μM), radicicol (Rad: 10 μM) or novobiocin (NB: 100–1000 μM) for 1 h and were then stimulated with leptin (0.5 μg·mL−1) for 6 h. Western blotting was performed using specific antibodies for SOCS3.
SOCS3 and ER stress were not involved in HSP90 inhibitor‐induced leptin resistance
We treated cells with the HSP90 inhibitors geldanamycin, radicicol and novobiocin, and analysed the expression levels of SOCS3 in SH‐SY5Y‐Ob‐Rb cells. However, we could not observe a detectable level of SOCS3 (Figure 6). Therefore, the HSP90 inhibitors themselves are unlikely to induce the expression of SOCS3.
We and other groups recently reported that ER stress is involved in the development of leptin resistance, an insensitivity to leptin‐induced signalling, in obesity (Hosoi et al., 2008; Zhang et al., 2008; Ozcan et al., 2009; Hosoi et al., 2014). Because HSP90 is a chaperone, its knock‐down may induce ER stress, which can indirectly inhibit the actions of leptin. However, this was not the case because the ER stress markers, GRP78 and CHOP, were not up‐regulated in HSP90‐knocked down cells (Figure 7).
Figure 7.
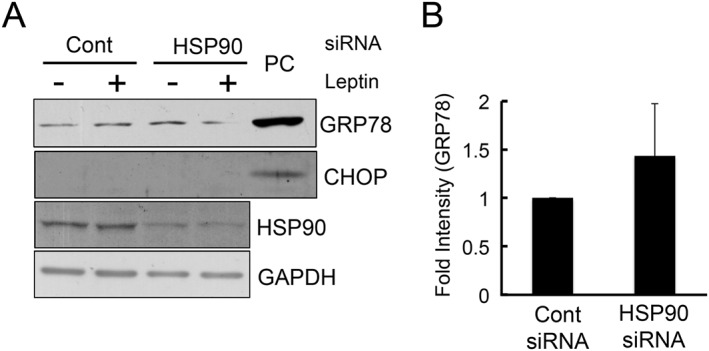
Knocking down HSP90 did not affect ER stress. HEK 293‐Ob‐Rb cells were transfected with HSP90 siRNA (25 nM) for 72 h, and Western blotting was performed using specific antibodies for HSP90, GAPDH, GRP78 and CHOP. PC, positive control: HEK293 Ob‐Rb cells treated with Tm (1 μg·mL−1) for 24 h. Each set of data was expressed as fold increase over control cells.
Therefore, these results suggest that SOCS3 and ER stress were not involved in HSP90 inhibitor‐induced leptin resistance.
Infusion of an HSP90 inhibitor blunted the anorexigenic actions of leptin
To examine the functional significance of the HSP90 pathway in leptin signalling in vivo, we determined whether inhibiting the HSP90 pathway impaired the ability of leptin to inhibit food intake. We injected the selective HSP90 inhibitor, geldanamycin, i.c.v. into rats and analysed the inhibitory effects of leptin on food intake. As expected, the i.c.v. injection of leptin markedly reduced food intake at 4, 14, and 24 h. In contrast, leptin did not reduce food intake in rats pretreated with an HSP90 inhibitor (Figure 8A). However, geldanamycin was unable to completely attenuate leptin's action. It is possible that geldanamycin only partially inhibited the effect of HSP90. Alternatively, it is also possible that a pathway other than HSP90 is involved in the regulation of food intake mediated by leptin. Overall, these results suggest that HSP90 was involved in regulating leptin‐induced anorexia. To further evaluate the role of HSP90 in leptin's action in vivo, we injected an HSP90 inhibitor i.c.v. and analysed leptin‐induced STAT3 phosphorylation in the mouse hypothalamus. As shown in Figure 8B, the HSP90 inhibitor novobiocin significantly inhibited the leptin‐induced phosphorylation of STAT3. Therefore, these results suggest that HSP90 is involved in leptin's action in the mouse hypothalamus.
Figure 8.

An HSP90 inhibitor ameliorated the anorexic effect of leptin. (A) Rats were treated with geldanamycin (GA: 100 nmol·3 μL−1) for 1 min and then with leptin (5 μg·3 μL−1) i.c.v. at the onset of the dark cycle. Food intake was subsequently measured. Changes in food intake were analysed in each rat. Homoscedasticity of groups was confirmed by Bartlett test (P > 0.05). From the result of ANOVA and Holm's test, there are significant difference among groups (ANOVA: P < 0.05) and significant difference between pairs of 0–14 h and 0–24 h were confirmed by Holm's test (all P < 0.05). *P < 0.05 n = 10–13 per group of five independent experiments. In each experiment, we used the following rat numbers: control group: 1–4 rats, leptin treatment group: 1–5 rats and GA + leptin treatment group: 1–5 rats. In total, control group 10 rats, leptin treatment group 12 rats and GA + leptin treatment group 13 rats were used for the overall experiments. (B) HSP90 inhibitor (novobiocin, NB: 0.5 μmol) and leptin (2.25 μg) were administered through an intracerebroventricular route to male mice. Animals were pretreated with novobiocin (5 min) prior to leptin injection. Thirty minutes after the treatment, the hypothalamus was removed, and the phosphorylation level of STAT3 was analysed by western blotting. Student's t‐test, *P < 0.05 versus control mice treated with leptin. n = 8 per group of three independent days of experiments. In each experiment, we used the following mouse numbers: control group two mice, leptin treatment group four mice, NB treatment group two mice and NB + leptin treatment group four mice for two independent experiments. Control group of four mice and NB treatment group of four mice were used for another experimental group. In total, control group eight mice, leptin treatment group eight mice, NB treatment group eight mice and NB + leptin treatment group eight mice were used for the overall experiments. Each set of data was expressed as fold increase over control rodents.
Discussion
Obesity has been associated with an increased risk of diseases, including diabetes, cardiovascular disease and hypertension. Therefore, the underlying mechanisms need to be elucidated in more detail. In the present study, we demonstrated that HSP90 plays a key role in leptin‐induced anorexia. This mechanism was mediated by an effect on the leptin‐induced signal transduction via the JAK2/STAT3 pathway. The nuclear import of phosphorylated STAT3 is an important process for the leptin‐induced inhibition of food intake. Following this nuclear import, leptin stimulates the POMC catabolic pathway, resulting in anorexia (Schwartz et al., 2000; Bates et al., 2003). We observed that leptin‐induced STAT3 phosphorylation and nuclear phospho‐STAT3 staining were inhibited by an HSP90 inhibitor. To the best of our knowledge, this is the first time that HSP90 has been demonstrated to be involved in the physiological actions of leptin.
Geldanamycin and radicicol inhibit HSP90 activity by binding to the N‐terminal ATP/ADP‐binding domain of HSP90 (Roe et al., 1999), whereas novobiocin inhibits its function by binding to its C‐terminal dimerization domain (Marcu et al., 2000). In the present study, we found that all of these compounds inhibited leptin‐induced STAT3 phosphorylation. It is interesting that all of these different compounds inhibited leptin‐induced STAT3 phosphorylation. It is possible that these HSP90 inhibitors destabilize the HSP90‐interacting proteins, thereby reducing STAT3 phosphorylation. Further analysis is required to elucidate the mechanism of these inhibitors.
There is increasing evidence that leptin resistance, an insensitivity to the actions of leptin, plays a major role in the development of obesity (Friedman, 2003). SOCS3 is a feedback regulator of JAK–STAT pathways involved in inhibiting leptin‐induced signal transduction. It has also been reported to be involved in the development of leptin resistance (Bjørbaek et al., 1998). Therefore, HSP90 inhibitors may induce SOCS3 levels, thereby inhibiting JAK2/STAT3 signals. In the present study, the HSP90 inhibitors did not induce the expression of detectable levels of SOCS3. Therefore, the inhibitory effects of HSP90 on the actions of leptin may not have been mediated through the induction of SOCS3. In contrast, the HSP90 inhibitors inhibited leptin‐induced SOCS3 induction. As the SOCS3 induction is mediated through STAT3, the HSP90 inhibitors may attenuate a STAT3‐regulated gene. Meanwhile, inhibiting the leptin‐mediated induction of SOCS3 should potentiate leptin signalling because SOSC3 is a negative regulator of STAT3 signalling. The leptin‐induced increase in SOCS3 at the protein level takes about 6 h. In the present study, we did not investigate the effect of an HSP90 inhibitor on leptin‐induced phosphorylation of STAT3 for longer times, when the SOCS3 protein is likely to be increased. Future studies are required to evaluate these possibilities by analysing the effects of the inhibitors at later time points. In addition to SOCS3, recent studies have suggested that ER stress plays a key role in the development of leptin resistance (Hosoi et al., 2008; Zhang et al., 2008; Ozcan et al., 2009; Hosoi et al., 2014). Therefore, the inhibition of HSP90 may induce ER stress, thereby attenuating leptin‐induced signalling. However, this is unlikely to be the case in the present study, because we did not observe an increase in ER stress markers in the HSP90 knocked‐down cells. Therefore, the inhibition of HSP90 is unlikely to induce the expression of a previously identified factor, which is then involved in the development of leptin resistance. Thus, HSP90 may be critically involved in the leptin‐induced activation of JAK2/STAT3 signalling, and inhibiting the action of HSP90 may result in an impairment of leptin‐induced signals. Future experiments are warranted to investigate HSP90 levels and/or activation in the hypothalamus in in vivo obesity models. Recent evidence has suggested that HSP90 plays a key role in regulating transcription and signal transduction (Taipale et al., 2010). HSP90 can bind to the various client proteins (Taipale et al., 2010). In the present study, we found that HSP90 is involved in leptin‐induced STAT3 phosphorylation. Therefore, it may be possible that one of these client proteins functions to regulate STAT3 activation. Future studies are required to test these possibilities.
Obesity has been suggested as a risk factor for several brain disorders, including Parkinson's disease (PD) and AD (Spielman et al., 2014). A previous study reported that the weights of ob/ob mouse brains were less than those of wild‐type mice (van der Kroon and Speijers, 1979). However, ob/ob mice treated with leptin had higher brain weights (Ahima et al., 1999). These findings indicate that leptin is involved in neuronal and glial maturation. In addition, ther is increasing evidence that leptin has neuroprotective properties. Leptin can protect against neuronal cell death through JAK2/STAT3 signalling (Russo et al., 2004; Guo et al., 2008) as well as in several types of neurodegenerative diseases such as ischaemic injury (Zhang et al., 2008), PD (Weng et al., 2007), and AD (Fewlass et al., 2004; Pérez‐González et al., 2011). Furthermore, HSP90 has also been shown to protect against ischaemia, PD and AD (Mailhos et al., 1994; Kakimura et al., 2002; Evans et al., 2006; Daturpalli et al., 2013). In the present study, we identified a link between leptin and HSP90, which suggests that HSP90 plays a role in leptin‐induced signal transduction. Therefore, the modulation of HSP90 by leptin may be one of the mechanisms involved in neuroprotection, and, as such, HSP90 may be a potential therapeutic target for leptin‐related neurodegenerative diseases.
Leptin receptors were previously reported to be overexpressed in several types of cancer cells (Attoub et al., 2000; Ishikawa et al., 2004) and enhance cancer cell growth (Dieudonne et al., 2002; Yin et al., 2004; Mauro et al., 2007; Birmingham et al., 2009). Leptin is known to be involved in cancer progression in several types of cells. Leptin was shown to induce the expression of HSP90 through a STAT3‐mediated pathway in a breast cancer cell line (Giordano et al., 2013). HSP90 was also found to be involved in tumour cell progression (Maloney and Workman, 2002). 17‐Allylaminogeldanamycin, a derivative of geldanamycin, exerts its antitumour activity by binding to HSP90 in tumour cells (Solit et al., 2002; Kamal et al., 2003). In the present study, we showed that HSP90 is involved in the leptin‐induced activation of JAK2/STAT3. Therefore, HSP90 inhibitors, by inhibiting the actions of leptin, could be beneficial for the treatment of several types of cancer.
In conclusion, we identified a novel mechanism for the actions of leptin; the involvement of HSP90. Our results suggest that HSP90 is involved in the leptin‐mediated signalling linked to anorexia. HSP90 has multiple functions related to the actions of leptin. Advancing our understanding of the link between HSP90 and leptin is important as it may provide a novel pharmacological strategy for treating diseases.
Author contributions
T.H. conceived the hypothesis, designed and performed experiments, analysed the data and wrote the manuscript. K.O. conceived the hypothesis and analysed the data. T.K., S.M., M.I. and A.K. designed and performed experiments and analysed the data. T.A. and J.T. analysed the data.
Conflict of interest
The authors have no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 HSP90 knockdown attenuated leptin‐induced STAT3 phosphorylation. Western blotting analysis of leptin‐induced STAT3 activation in the HSP90‐knocked down HEK293‐Ob‐Rb cell line. Cells were stimulated with leptin (0.5 μg/mL) for 15 min and Western blotting was then performed. (A) Expression level of HSP90 was attenuated by HSP90 siRNA. (B) The leptin‐induced phosphorylation of STAT3 was inhibited in HSP90‐knocked down cells. Typical blot among three independent experiments was shown.
Figure S2 A HSP90 inhibitors inhibited leptin‐induced activation of STAT3 in neuronal cells. SH‐SY5Y‐Ob‐Rb cells were treated with the HSP90 inhibitor, radicicol (Rad: 10 μM) and novobiocin (NB: 1000 μM), and leptininduced STAT3 phosphorylation was analyzed. *P < 0.05 v.s. control cells treated with leptin. Dunnett's test. n = 4. © 2016 The British Pharmacological Society.
Figure 1 Supporting info item
Acknowledgements
Part of this study was carried out at the Analysis Center of Life Science, Natural Science Center for Basic Research and Development, Hiroshima University. Animal experiments were supported by the Institute of Laboratory Animal Science (Hiroshima University). This research was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and Takeda Science Foundation.
Hosoi, T. , Kohda, T. , Matsuzaki, S. , Ishiguchi, M. , Kuwamura, A. , Akita, T. , Tanaka, J. , and Ozawa, K. (2016) Key role of heat shock protein 90 in leptin‐induced STAT3 activation and feeding regulation. British Journal of Pharmacology, 173: 2434–2445. doi: 10.1111/bph.13520.
References
- Alexander SPH , Davenport AP , Kelly E , Marrion N , Peters JA , Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744‐5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH,Fabbro D,Kelly E,Marrion N,Peters JA,Benson HEet al.(2015b).The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors .Br J Pharmacol 172:5979.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH , Fabbro D , Kelly E , Marrion N , Peters J A , Benson H E et al. ( 2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes . Br J Pharmacol 172 : 6024 – 6109 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Bjorbaek C, Osei S, Flier JS. (1999). Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology 140: 2755‐2762. [DOI] [PubMed] [Google Scholar]
- Attoub S, Noe V, Pirola L, Bruyneel E, Chastre E, Mareel M et al. (2000). Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3‐kinase‐, rho‐, and rac‐dependent signaling pathways. FASEB J 14: 2329‐2338. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y et al. (2003). STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421: 856‐859. [DOI] [PubMed] [Google Scholar]
- Birmingham JM, Busik JV, Hansen‐Smith FM, Fenton JI. (2009). Novel mechanism for obesity‐induced colon cancer progression. Carcinogenesis 30: 690‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. (1998). Identification of SOCS‐3 as a potential mediator of central leptin resistance. Mol Cell 1: 619‐625. [DOI] [PubMed] [Google Scholar]
- Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS et al (2000). SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem 275: 40649‐40657. [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269: 546‐549. [DOI] [PubMed] [Google Scholar]
- Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee‐Loy A, McGlade CJ et al (2002). Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2: 497‐503. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al (2015) Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461‐3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daturpalli S, Waudby CA, Meehan S, Jackson SE. (2013). Hsp90 inhibits α‐synuclein aggregation by interacting with soluble oligomers. J Mol Biol 425: 4614‐4628. [DOI] [PubMed] [Google Scholar]
- Dieudonne MN, Machinal‐Quelin F, Serazin‐Leroy V, Leneveu MC, Pecquery R, Giudicelli Y. (2002). Leptin mediates a proliferative response in human MCF7 breast cancer cells. Biochem Biophys Res Commun 293: 622‐628. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A et al (2003). Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A 100: 721‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL et al. (1999). Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase‐1B gene. Science 283: 1544‐1548. [DOI] [PubMed] [Google Scholar]
- Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K et al. (1997). A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387: 921‐924. [DOI] [PubMed] [Google Scholar]
- Evans CG, Wisén S, Gestwicki JE. (2006). Heat shock proteins 70 and 90 inhibit early stages of amyloid β‐(1‐42) aggregation in vitro. J Biol Chem 281: 33182‐33191. [DOI] [PubMed] [Google Scholar]
- Fewlass DC, Noboa K, Pi‐Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. (2004). Obesity‐related leptin regulates Alzheimer's Aβ. FASEB J 18: 1870‐1878. [DOI] [PubMed] [Google Scholar]
- Friedman JM. (2003). A war on obesity, not the obese. Science 299: 856‐858. [DOI] [PubMed] [Google Scholar]
- Gass P, Schröder H, Prior P, Kiessling M. (1994). Constitutive expression of heat shock protein 90 (HSP90) in neurons of the rat brain. Neurosci Lett 182: 188‐192. [DOI] [PubMed] [Google Scholar]
- Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. (1996). Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A 93: 6231‐6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C, Vizza D, Panza S, Barone I, Bonofiglio D, Lanzino M et al. (2013). Leptin increases HER2 protein levels through a STAT3‐mediated up‐regulation of Hsp90 in breast cancer cells. Mol Oncol 7: 379‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Jiang H, Xu X, Duan W, Mattson MP. (2008). Leptin‐mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem 283: 1754‐1763. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Kawagishi T, Okuma Y, Tanaka J, Nomura Y. (2002). Brain stem is a direct target for leptin's action in the central nervous system. Endocrinology 143: 3498‐3504. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Matsunami N, Nagahama T, Okuma Y, Ozawa K, Takizawa T et al (2006). 2‐Aminopurine inhibits leptin receptor signal transduction. Eur J Pharmacol 553: 61‐66. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Ozawa K. (2010). Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 118: 19‐29. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Korematsu K, Horie N, Suezawa T, Okuma Y, Nomura Y et al (2012). Inhibition of casein kinase 2 modulates XBP1‐GRP78 arm of unfolded protein responses in cultured glial cells. PLoS One 7: e40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M et al. (2008). Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol 74: 1610‐1619. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Yamaguchi R, Noji K, Matsuo S, Baba S, Toyoda K et al. (2014). Flurbiprofen ameliorated obesity by attenuating leptin resistance induced by endoplasmic reticulum stress. EMBO Mol Med 6: 335‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Kitayama J, Nagawa H. (2004). Enhanced expression of leptin and leptin receptor (OB‐R) in human breast cancer. Clin Cancer Res 10: 4325‐4331. [DOI] [PubMed] [Google Scholar]
- Itoh H, Tashima Y, Eishi Y, Okeda R. (1993). Localization of HSP90 in rat brain. Int J Biochem 25: 93‐99. [DOI] [PubMed] [Google Scholar]
- Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K et al (2002). Microglial activation and amyloid‐beta clearance induced by exogenous heat‐shock proteins. FASEB J 16: 601‐603. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC et al. (2003). A high‐affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425: 407‐410. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C , Browne, W , Cuthill, IC , Emerson, M , Altman, DG ; NC3Rs Reporting Guidelines Working Group . (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM et al (2000). Increased energy expenditure, decreased adiposity, and tissue‐specific insulin sensitivity in protein‐tyrosine phosphatase 1B‐deficient mice. Mol Cell Biol 20: 5479‐5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailhos C, Howard MK, Latchman DS. (1994). Heat shock proteins hsp90 and hsp70 protect neuronal cells from thermal stress but not from programmed cell death. J Neurochem 63: 1787‐1795. [DOI] [PubMed] [Google Scholar]
- Maloney A, Workman P. (2002). HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2: 3‐24. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. (2000). The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP‐binding domain in the carboxyl terminus of the chaperone. J Biol Chem 275: 37181‐37186. [DOI] [PubMed] [Google Scholar]
- Mauro L, Catalano S, Bossi G, Pellegrino M, Barone I, Morales S et al. (2007). Evidences that leptin up‐regulates E‐cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res 67: 3412‐3421. [DOI] [PubMed] [Google Scholar]
- McGrath J C, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzberg H, Myers MG Jr. (2005). Molecular and anatomical determinants of central leptin resistance. Nat Neurosci 8: 566‐570. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. (2004). Region‐specific leptin resistance within the hypothalamus of diet‐induced obese mice. Endocrinology 145: 4880‐4889. [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A et al (1997). Structure and function of a new STAT‐induced STAT inhibitor. Nature 387: 924‐929. [DOI] [PubMed] [Google Scholar]
- Olazábal UE, Pfaff DW, Mobbs CV. (1992). Estrogenic regulation of heat shock protein 90 kDa in the rat ventromedial hypothalamus and uterus. Mol Cell Endocrinol 84: 175‐183. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D et al. (2009). Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9: 35‐51. [DOI] [PubMed] [Google Scholar]
- Pérez‐González R, Antequera D, Vargas T, Spuch C, Bolós M, Carro E. (2011). Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer's disease. J Alzheimers Dis 2: 17‐25. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. (1999). Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42: 260‐266. [DOI] [PubMed] [Google Scholar]
- Russo VC, Metaxas S, Kobayashi K, Harris M, Werther GA. (2004). Antiapoptotic effects of leptin in human neuroblastoma cells. Endocrinology 145: 4103‐4112. [DOI] [PubMed] [Google Scholar]
- Sato N, Yamamoto T, Sekine Y, Yumioka T, Junicho A, Fuse H et al (2003). Involvement of heat‐shock protein 90 in the interleukin‐6‐mediated signaling pathway through STAT3. Biochem Biophys Res Commun 300: 847‐852. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. (2000). Central nervous system control of food intake. Nature 404: 661‐671. [DOI] [PubMed] [Google Scholar]
- Shah M, Patel K, Fried VA, Sehgal PB. (2002). Interactions of STAT3 with caveolin‐1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J Biol Chem 277: 45662‐45669. [DOI] [PubMed] [Google Scholar]
- Solit DB, Zheng FF, Drobnjak M, Münster PN, Higgins B, Verbel D et al (2002). 17‐Allylamino‐17‐demethoxygeldanamycin induces the degradation of androgen receptor and HER‐2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8: 986‐993. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al; NC‐IUPHAR (2016) The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44:D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman LJ, Little JP, Klegeris A. (2014). Inflammation and insulin/IGF‐1 resistance as the possible link between obesity and neurodegeneration. J Neuroimmunol 273: 8‐21. [DOI] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ et al. (1997). A family of cytokine‐inducible inhibitors of signalling. Nature 387: 917‐921. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11: 515‐528. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr., Stoffel M, Friedman JM. (1996). Leptin activation of Stat3 in the hypothalamus of wild‐type and ob/ob mice but not db/db mice. Nat Genet 14: 95‐97. [DOI] [PubMed] [Google Scholar]
- van der Kroon PH, Speijers GJ. (1979). Brain deviations in adult obese‐hyperglycemic mice (ob/ob). Metabolism 28: 1‐3. [DOI] [PubMed] [Google Scholar]
- Weng Z, Signore AP, Gao Y, Wang S, Zhang F, Hastings T et al. (2007). Leptin protects against 6‐hydroxydopamine‐induced dopaminergic cell death via mitogen‐activated protein kinase signaling. J Biol Chem 282: 34479‐34491. [DOI] [PubMed] [Google Scholar]
- Wetsel WC, Mellon PL, Weiner RI, Negro‐Vilar A (1991). Metabolism of pro‐luteinizing hormone‐releasing hormone in immortalized hypothalamic neurons. Endocrinology 129: 1584‐1595. [DOI] [PubMed] [Google Scholar]
- Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B et al. (2004). Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res 64: 5870‐5875. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Bence‐Hanulec KK, Stricker‐Krongrad A, Haj F, Wang Y, Minokoshi Y et al (2002). PTP1B regulates leptin signal transduction in vivo. Dev Cell 2: 489‐495. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. (2008). Hypothalamic IKKβ/NF‐κB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372: 425‐432. [DOI] [PubMed] [Google Scholar]
- Zhang F, Chen J. (2008). Leptin protects hippocampal CA1 neurons against ischemic injury. J Neurochem 107: 578‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 HSP90 knockdown attenuated leptin‐induced STAT3 phosphorylation. Western blotting analysis of leptin‐induced STAT3 activation in the HSP90‐knocked down HEK293‐Ob‐Rb cell line. Cells were stimulated with leptin (0.5 μg/mL) for 15 min and Western blotting was then performed. (A) Expression level of HSP90 was attenuated by HSP90 siRNA. (B) The leptin‐induced phosphorylation of STAT3 was inhibited in HSP90‐knocked down cells. Typical blot among three independent experiments was shown.
Figure S2 A HSP90 inhibitors inhibited leptin‐induced activation of STAT3 in neuronal cells. SH‐SY5Y‐Ob‐Rb cells were treated with the HSP90 inhibitor, radicicol (Rad: 10 μM) and novobiocin (NB: 1000 μM), and leptininduced STAT3 phosphorylation was analyzed. *P < 0.05 v.s. control cells treated with leptin. Dunnett's test. n = 4. © 2016 The British Pharmacological Society.
Figure 1 Supporting info item