

Abstract
Mitochondria are dynamic organelles, which couple the various cellular processes that regulate metabolism, cell proliferation and survival. Environmental stress can cause mitochondrial dysfunction and dynamic changes including reduced mitochondrial biogenesis, oxidative phosphorylation and ATP production, as well as mitophagy impairment, which leads to increased ROS, inflammatory responses and cellular senescence. Oxidative stress, inflammation and cellular senescence all have important roles in the pathogenesis of chronic lung diseases, such as chronic obstructive pulmonary disease, pulmonary fibrosis and bronchopulmonary dysplasia. In this review, we discuss the current state on how mitochondrial dysfunction affects inflammatory responses and cellular senescence, the mechanisms of mitochondrial dysfunction underlying the pathogenesis of chronic lung diseases and the potential of mitochondrial transfer and replacement as treatments for these diseases.
Abbreviations
- AMPK
AMP‐activated protein kinase
- α‐SMA
α‐smooth muscle actin
- BPD
bronchopulmonary dysplasia
- COPD
chronic obstructive pulmonary disease
- DRP1, DNM1L
dynamin 1‐like
- Δψm
mitochondrial membrane potential
- ETC
electron transport chain
- FIS1
fission 1
- MDVs
mitochondria‐derived vesicles
- Mfn
mitofusin
- MiDAS
mitochondrial dysfunction‐associated senescence
- MPTP
mitochondrial permeability transition pore
- mtDNA
mitochondrial DNA
- mtROS
mitochondrial ROS
- O2.−
superoxide anion
- OPA1
optic atrophy 1
- PINK1
PTEN‐induced putative kinase 1
- SASP
senescence‐associated secretory phenotype
- UCP
mitochondrial uncoupling protein
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes d |
| TNF | AMPK |
| GPCRs b | PINK1 |
| CXCR2 | SIRT1 |
| P2Y receptors | Transporters e |
| Catalytic receptors c | UCP |
| PRRs | |
| TNFRs |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,eAlexander et al., 2015a, 2015b, 2015c, 2015d, 2015e).
Introduction
Mitochondria are highly dynamic organelles which contain outer and inner membranes that separate the inter‐membrane space and matrix. The inner membrane is folded many times and it is these folds that are known as the cristae, which harbors the electron transport chain (ETC) that contains five multiprotein complexes named complex I–V. Mitochondria act as a cytosolic calcium reservoir, and mitochondrial calcium regulates ATP synthesis (Tarasov et al., 2012). However, calcium overload in the mitochondrial matrix enhances the generation of mitochondrial ROS (mtROS) and the release of cytochrome c, resulting in apoptosis and cell death (Brookes et al., 2004; Finkel et al., 2015). Sustained levels of ROS cause the oxidation of DNA, lipid and protein, which play important roles in the pathogenesis of chronic lung diseases. However, moderate levels of mtROS act as a physiological signal to protect against the adverse effects of various cellular stresses and infection (West et al., 2011; Al‐Mehdi et al., 2012; Katz et al., 2014). For instance, suppression of perinuclear mitochondrial clustering reduces mtROS‐mediated vascular endothelial growth factor gene transcription in response to hypoxia (Al‐Mehdi et al., 2012). Therefore, mtROS is a double‐edged sword with regard to the regulation of cell function (Sena and Chandel, 2012; Kalogeris et al., 2014).
Mitochondria are considered to be the powerhouse of the cell as they have the ability to generate ATP through oxidative phosphorylation (Figure 1); this provides the energy for cell survival and functions. Also, there is accumulating evidence that the mitochondria has a non‐energetic role in regulating metabolism, apoptosis, innate immunity, inflammatory responses and aging (Nunnari and Suomalainen, 2012; Friedman and Nunnari, 2014). All of these processes are involved in the pathogenesis of chronic lung diseases, such as chronic obstructive pulmonary disease (COPD), pulmonary fibrosis and bronchopulmonary dysplasia (BPD). In this review, we focus on how the dysfunction of mitochondria can affect inflammatory responses and cellular senescence, as well as their involvement in the development of chronic lung diseases and their co‐morbidities.
Figure 1.
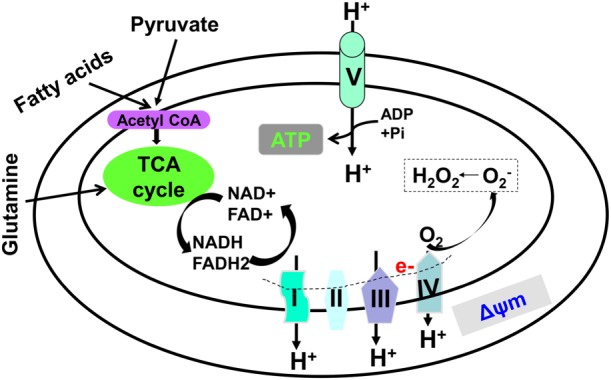
Tricarboxylic acid (TCA) cycle and oxidative phosphorylation. ATP generation starts in the TCA cycle and ends with oxidative phosphorylation. During the TCA cycle, NADH and FADH2 are generated, which are oxidized to provide electrons through the ETC. Mitochondrial O2 .− is produced when oxygen molecules gain electrons leaked from the ETC. Through oxidative phosphorylation, free energy is used to pump protons out of the matrix, forming Δψm, which ultimately provides the energy needed to generate ATP.
Mitochondrial dynamics and quality control
Upon stress, mitochondria develop a protective mechanism to control and maintain their quality (Figure 2). This starts as mitochondrial biogenesis, which is the growth and division of pre‐existing mitochondria rather than de novo synthesis. Once stressed, mitochondria fuse together so as to separate the healthy and damaged/stress mitochondria through a fission mechanism. At the molecular level, both dynamin 1‐like (DNM1L or DRP1) and fission 1 (FIS1) regulate mitochondrial fission, while optic atrophy 1 (OPA1) and mitofusin 1 and 2 (Mfn1 and Mfn2) modulate the fusion process. Healthy mitochondria enter the mitochondrial life cycle to produce energy and take part in cell signalling, whereas damaged mitochondria are mainly degraded via the mitochondrial autophagy (mitophagy) pathway (Youle and Narendra, 2011; Ashrafi and Schwarz, 2013). The timely elimination of damaged, dysfunctional and aged mitochondria plays a crucial a role in preventing the release of pro‐apoptotic proteins, mtROS and mitochondrial DNA (mtDNA), which cause apoptosis and inflammasome activation (Kepp et al., 2011; Nakahira et al., 2011; Zhou et al., 2011; Shimada et al., 2012; Alfonso‐Loeches et al., 2014).
Figure 2.
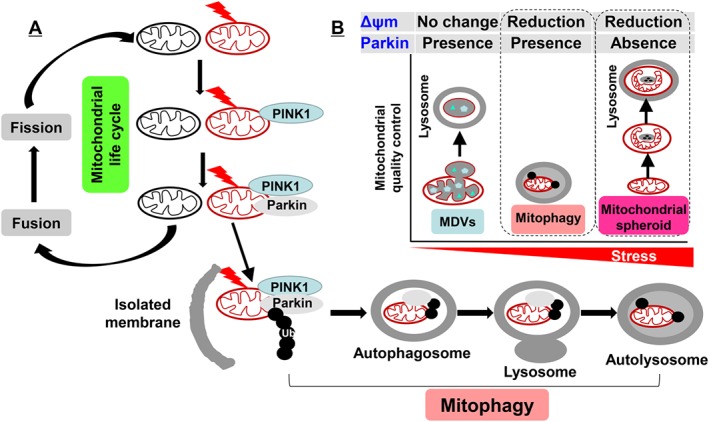
Mitochondrial quality control. (A) Once mitochondria encounter stress and the Δψm is reduced, damaged mitochondria are separated, and Pink1 is recruited into the mitochondrial out membrane. This leads to the translocation of Parkin to mitochondria, which triggers the ubiquitination of mitochondrial proteins including Mfns. Finally, autophagosomes enclose damaged mitochondria and fuse with lysosome for degradation. The intact mitochondria enter the mitochondrial life cycle through fusion and fission for biogenesis. (B) There are several approaches for mitochondrial quality control, including MDVs, mitophagy and mitochondrial spheroid formation. Based on the requirement of Δψm reduction and Parkin recruitment, we surmise that MDVs, mitophagy and mitochondrial spheroid formation are employed in different degrees of mitochondrial stress.
Mitophagy is initiated when the mitochondrial membrane potential (Δψm) is reduced by mild stress. This causes the recruitment of a PTEN‐induced putative kinase 1 (PINK1) on the mitochondrial out membrane, which further recruits and phosphorylates an E3 ubiquitin ligase, Parkin, from the cytosol (Springer and Kahle, 2011; Vincow et al., 2013). The phosphorylated Parkin (Ser65) triggers the ubiquitination and degradation of mitochondrial proteins including Mfn1 and Mfn2, hence preventing mitochondrial fusion. This triggers the translocation of damaged mitochondria on the isolated membranes containing microtubule‐associated protein light chain 3, leading to the formation of autophagosomes, which finally fuse with lysosomes leading to the clearance of damaged mitochondria from the cells (Youle and Narendra, 2011; Hattori et al., 2014). However, recent studies have reported Smurf1‐ and Mul1‐dependent mitophagy, which is independent of Parkin (Orvedahl et al., 2011; Lokireddy et al., 2012; Fu et al., 2013; Chen et al., 2014). Therefore, further investigations on Parkin‐dependent and ‐independent mitophagy would enhance the understanding of mitochondrial biology and pathophysiology of mitochondrial dysfunction‐associated diseases. However, the approaches used to detect mitophagy in vivo are limited. Traditional methods such as electronic microscopy are able to capture and determine the mitochondrial components or remnants within the autophagosome (Zhu et al., 2011). However, these methods are not quantitative as they only measure a small fraction of cell and tissue. Keima is a coral‐derived protein that has pH‐dependent fluorescent properties, which allows the determination of its localization in mitochondria (pH ~8.0) or lysosome (pH ~4.5). A newly developed transgenic mouse model, in which a mitochondrial‐targeted form of the fluorescent reporter Keima is overexpressed, provides a real‐time monitor of mitophagy in vivo (Sun et al., 2015).
Recent studies have shown that mitochondria‐derived vesicles (MDVs) can be formed as an alternative pathway for mitochondrial quality control. MDVs carry the oxidized mitochondrial proteins and lipids into peroxisomes or fuse with endosomes, which are finally degraded by lysosome, and this process is independent of a reduction in the Δψm or mitophagy (Soubannier et al., 2012a; Du Toit, 2014; Sugiura et al., 2014). In other words, MDVs can occur in cells with an intact mitochondrial network with no reduction in the Δψm. It is interesting to note that MDVs do not contain the mitochondrial ETC or nucleoids (Soubannier et al., 2012b), which suggests the constituents of MDVs are specific. Both PINK1 and Parkin have been shown to involved in the biogenesis of MDVs (McLelland et al., 2014; Sugiura et al., 2014). However, how PINK1 and Parkin have differential effects on mitophagy and MDV formation remains to be determined.
Mitochondrial spheroid formation is a reversible process, which has also been shown to control mitochondrial quality and can be induced in vivo in the liver by acetaminophen‐mediated severe oxidative mitochondrial damage in the absence of Parkin (Ding et al., 2012; Ni et al., 2013; Cook et al., 2014; Ding and Eskelinen, 2014). In contrast to mitophagy or MDVs, Parkin suppresses mitochondrial spheroid formation by ubiquitin and degradation of Mfn, whereas Mfn promotes the formation of mitochondrial spheroids (Ding et al., 2012). Overall, we surmise that mitochondria will employ either MDVs, mitophagy or spheroid formation according to the extent of their damage (from mild to severe) to maintain their homoeostasis (Figure 2). Further studies are required to determine the regulation, interaction and crosstalk among mitophagy, MDVs and mitochondrial spheroids needed to maintain mitochondrial quality control under stress conditions.
Mitochondrial dysfunction in inflammation
Mitochondrial dysfunction is linked to inflammatory responses (Lopez‐Armada et al., 2013). Accumulating evidence shows that increased mtROS generation, extracellular ATP and mtDNA release cause inflammatory responses, which, in turn, aggravate mitochondrial dysfunction (Figure 3).
Figure 3.
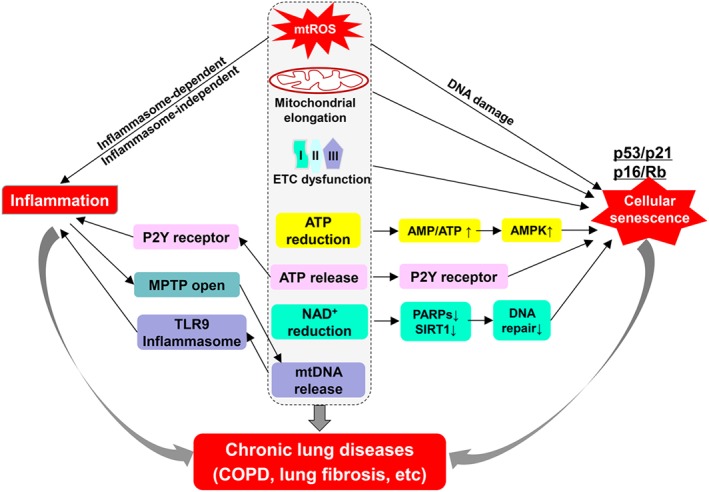
Mitochondrial dysfunction in inflammatory responses and cellular senescence. Abnormal changes in mitochondrial morphology and function play pivotal roles in stress‐induced inflammatory responses and cellular senescence. Increased mtROS, mitochondrial elongation and impaired oxidative phosphorylation cause cell cycle arrest and cellular senescence. ATP and NAD+ reduction due to dysfunctional oxidative respiration also result in cellular senescence by regulating AMPK, sirtuin1 and PARPs. Meanwhile, mtROS, extracellular ATP and mtDNA can cause inflammatory responses in inflammasome‐dependent or independent manners. Inflammation is able to induce the opening of MPTP, leading to mtDNA release from mitochondria. Both cellular senescence and inflammatory responses participate in the pathogenesis of chronic lung diseases.
mtROS are generated during the transportation of electrons provided by the oxidation of NADH and FADH2 produced during the tricarboxylic acid cycle. The main site of electron leakage is the complex I (NADH dehydrogenase), which oxidizes oxygen to generate superoxide anion (O2 .−). There is approximately 0.2%–2% oxygen consumed during oxidative phosphorylation generating O2 .−, which is further dismutated into hydrogen peroxide (H2O2). Due to the difference in their electrophilic and electrophobic properties as well as their half‐life, the concentrations of H2O2 in mitochondria are 100 times greater than that of O2.− (Cadenas and Davies, 2000; Li et al., 2013). This makes mitochondrial H2O2 an ideal signalling molecule in cells (Al‐Mehdi et al., 2012), whereas O2.− causes oxidative stress leading to DNA damage and an impairment of mitochondrial integrity through the generation of peroxynitrite within mitochondria. Under severe mitochondrial damage, high amounts of mtROS are generated which causes deleterious effects including abnormal inflammatory responses (Bulua et al., 2011; Naik and Dixit, 2011). There are several mechanisms for mtROS‐mediated inflammatory responses, including RIG‐I‐like receptors, MAPK, TNFRs and NLRP3 inflammasome signals (Emre et al., 2007; Tal et al., 2009; Bulua et al., 2011; Zhou et al., 2011). This is reflected by the findings that inhibition of mtROS production reduces the release of pro‐inflammatory mediators including IL‐6 and TNF in cells from patients with TNFR1‐associated periodic syndrome (Bulua et al., 2011). A separate study demonstrated that NLRP3 deletion abolishes mtROS elevation‐induced IL‐1β expression (Zhou et al., 2011). Mitochondrial uncoupling is a physiological process that dissipates the proton gradient, which allows protons to bypass ATP synthase. Mitochondrial uncoupling is modulated by uncoupling proteins (UCPs) located in the mitochondrial inner membrane. Mild mitochondrial uncoupling significantly reduces mtROS generation, thereby decreasing inflammatory responses (Lee et al., 2005; Toime and Brand, 2010; Romaschenko et al., 2015).
There are three mechanisms for ATP release: (i) vesicular (exocytotic) release; (ii) diffusion through membrane pores or damaged membranes; and (iii) active transport, which occurs in a variety of cells including neurons, epithelial cells, fibroblasts, macrophages and neutrophils (Fitz, 2007). Released ATP regulates cell functions by activating purine receptors (P2X and P2Y) in the plasma membrane in an autocrine and paracrine manner. A persistent increase in extracellular ATP serves as a danger signal is associated with the development of inflammation and cell death (Riteau et al., 2011, 2010). It has been shown that activation of purine receptors results in the generation of ROS, chemokines and pro‐inflammatory mediators (Bours et al., 2011; Cauwels et al., 2014). Additionally, inflammasome is involved in extracellular ATP‐mediated aseptic inflammation (Iyer et al., 2009; Weber et al., 2013, 2010). Therefore, the blockade of purine receptors may represent a potential therapeutic for abnormal inflammatory responses to extracellular ATP.
The mitochondrion has its own circular DNA, like bacterial DNA, which encodes 13 polypeptide subunits essential for the oxidative phosphorylation, 22 tRNAs and 2 rRNAs. Recent studies have shown that mtDNA can be released into cytosol or the circulation. It then acts as mitochondrial damage‐associated molecular patterns (DAMPs) in inducing activation of inflammasome (Kepp et al., 2011; Nakahira et al., 2011; Oka et al., 2012; Shimada et al., 2012) or engages the DNA sensor cGMP–AMP synthase promoting interferon responses and viral resistance (West et al., 2015). Further studies have revealed that oxidized mtDNA is the actual element that binds and activates the NLRP3 inflammasome (Shimada et al., 2012). mtDNA digestion by DNase II protects against tissue inflammation, while IL‐1β production is significantly reduced in macrophages lacking mtDNA (Shimada et al., 2013, 2012). The mechanisms underlying mtDNA‐mediated inflammasome activation are associated with the cell‐autonomous activation of specific pattern recognition receptors such as TLR9 (Oka et al., 2012). One of the mechanisms for mtDNA release is the formation of exosomes and the opening of the mitochondrial permeability transition pore (MPTP) (Patrushev et al., 2010, 2004). It should be noted that inflammation itself induces the opening of MPTP. This is demonstrated by the findings that the pro‐inflammatory cytokines, including TNF‐α, IFN‐γ and IL‐17, cause the opening of MPTP in human conjunctival epithelial cells and platelets (Gao et al., 2013; Yuan et al., 2015). Cyclosporine A, an MPTP inhibitor, prevents mtDNA release (Gao et al., 2013). Hence, MPTP serves as a checkpoint to determine the fate of cells. These findings suggest a robust feedback exists between mtDNA and inflammatory responses, and inhibition of exosome formation and MPTP would protect against mtDNA release‐induced inflammatory responses.
Mitochondrial dysfunction in cellular senescence
Cellular senescence is a status of irreversible growth arrest, which is a biological term for aging. There are two p53/p21cip1 and p16INK4/Rb pathways that mediate cellular senescence. According to the mitochondrial free radical theory of aging, mitochondrial stress is considered as an inducer of cellular senescence whereas mtROS is the factor most studied factor through persistent DNA damage (Figure 3). Scavenging mtROS with the mitochondria‐targeted antioxidant MitoQ or acetyl‐l‐carnitine delays replicative senescence and/or stress‐induced premature senescence (Saretzki et al., 2003; Wu et al., 2014). Furthermore, replicative senescence of human fibroblasts is delayed by mild mitochondrial uncoupling, which is associated with reduced production of mtROS (Passos et al., 2007). However, overproduction of UCP2 causes senescent‐like morphology in a kidney fibroblast cell line (Nishio and Ma, 2016), suggesting the existance of mtROS‐independent cellular senescence. Controversial studies have suggested that mtROS increase is just a consequence of cellular senescence. High levels of oxidative damage to mtDNA are observed in the longest‐living rodent, the naked mole rat (Andziak et al., 2006). Additionally, overexpression of mitochondrial SOD2 and catalase are not sufficient to block stress‐induced cellular senescence (Klimova et al., 2009; Lawless et al., 2012). Nevertheless, whether mtROS triggers cellular senescence independently of its role in inducing oxidative damage is still not known.
Abnormal alterations in mitochondrial dynamics affects mitochondria, leading to cellular senescence (Figure 3). Genetic disruption of membrane‐associated ring finger C3HC4 5 (MARCH5), a mitochondrial E3 ubiquitin ligase, induces cellular senescence by blocking DRP1 activity and causing mitochondrial elongation (Park et al., 2010). This is in agreement with our findings that mitochondria are elongated during smoke stress‐induced senescence in lung epithelial cells and fibroblasts (Ahmad et al., 2015). In contrast, depletion of both hFIS1 and OPA1 leads to extensive mitochondrial fragmentation and significantly protects against senescence‐associated phenotypic changes (Lee et al., 2007). These findings indicate that mitochondrial elongation promotes cell cycle arrest and subsequent senescence, which is associated with reduced Δψm, increased mtROS and DNA damage. We and others have shown that mitophagy impairment triggers stress‐induced cellular senescence (Ahmad et al., 2015; Ito et al., 2015). This may be due to the reduction in PINK1 and Parkin levels, as knockdown of PINK1 or Parkin enhances smoke stress‐induced premature senescence. Further investigations are required to determine how impairment of PINK1/Parkin‐dependent mitophagy up‐regulates p53‐ and p16‐dependent cellular senescence.
Mitochondrial dysfunction decreases ATP synthesis, which induces cellular senescence (Figure 3). The mechanism underlying this involves AMP‐activated protein kinase (AMPK) activation via increased AMP (or ADP) to ATP ratio. Indeed, AMPK directly up‐regulates p53 and p16 at the transcription and post‐translational levels leading to cellular senescence (Wang et al., 2013, 2003). AMPK has been shown to up‐regulate sirtuin1, an anti‐aging/senescence molecule, by augmenting mitochondrial metabolite NAD+ levels (Canto et al., 2009). However, under conditions of mitochondrial stress, complex I is not capable of oxidizing the NADH into NAD+, which may provoke AMPK to induce cellular senescence.
Link between inflammation and senescence in chronic lung diseases
During proliferation arrest, senescence cells are metabolically active and are more likely to secrete inflammatory mediators such as IL‐1, IL‐6, IL‐8 and MMPs. This phenomenon is termed senescence‐associated secretory phenotype (SASP). The mechanisms underlying SASP are associated with activation of p38 MAPK and NF‐κB (Chien et al., 2011; Freund et al., 2011). In turn, these SASP mediators can reinforce senescence in neighbouring cells through CXCR2 receptors (Acosta et al., 2008). This is evident from findings showing that NF‐κB inhibition delays cellular senescence (Tilstra et al., 2012). Interestingly, mitochondrial defects also trigger proliferation of neighbouring cells via a SASP in Drosophila (Nakamura et al., 2014). Hence, the SASP mediators from senescent cells may have different components, which pose differential influences on neighbouring cells, such as senescence, proliferation or differentiation. Indeed, a recent study has shown that mitochondrial dysfunction‐associated senescence (MiDAS) exhibits a modified SASP that lacks the IL‐1‐dependent inflammatory arm but shows high levels of the anti‐inflammatory cytokine IL‐10 (Wiley et al., 2016). Further studies on SASP components from senescent epithelial cells, fibroblasts and inflammatory cells along with mitochondrial function may reveal the role of mitochondria in diverse paracrine effects of SASP.
Both senescence and inflammatory responses play important roles in the pathogenesis of chronic lung diseases including COPD and lung fibrosis, which we and others have extensively discussed (Aoshiba and Nagai, 2009; MacNee and Tuder, 2009; Faner et al., 2012; Yao and Rahman, 2012a; Yao and Rahman, 2012b; Kumar et al., 2014). In the present review, we are not deliberating this topic. However, senescence in lung epithelial cells and fibroblasts is observed in both COPD and lung fibrosis, which raises the question of how senescence in both cell types leads to different pulmonary phenotypes? One of the mechanisms may be associated with the different senescence phenotypes (DNA damage‐initiated senescence vs. MiDAS), leading to differential paracrine effects of SASP on neighbouring cells.
Mitochondrial dysfunction in chronic lung diseases
There is accumulating evidence showing that mitochondrial function is abnormal during the development of chronic lung diseases including COPD, pulmonary fibrosis and BPD (Mizumura et al., 2014; Schumacker et al., 2014; Ahmad et al., 2015; Ito et al., 2015) (Table 1). Although these chronic lung diseases share a lot of similar mitochondrial abnormalities, the mechanisms by which similar mitochondrial modifications result in different pulmonary phenotypes are unknown. This may be due to cell‐specific damage of mitochondrial function [e.g. epithelial cells (type I and II), (myo)fibroblasts, endothelial cells and muscle cells] in response to different stimuli. Various approaches including in vitro, in vivo and ex vivo experiments are employed to determine the casual relationship between mitochondrial dysfunction and chronic lung diseases.
Table 1.
Changes in mitochondrial morphology and function in chronic lung diseases
| Mitochondrial dysfunction | COPD | Pulmonary fibrosis | BPD* |
|---|---|---|---|
| Morphology | Fragmentation and elongation | Fragmented, enlarged and swollen mitochondria | Abnormal |
| Biogenesis | Reduction of PGC1α‐dependent biogenesis | Reduced mtDNA copy number | Impaired biogenesis |
| mtROS | Increased | Increased | Increased |
| Oxidative phosphorylation | Damaged | Impaired complex I and IV activity | Decreased complex I and II function |
| Mitophagy | Impaired mitophagy | Impairment of PINK1‐mediated mitophagy | — |
| mtDAMP | Increased release of mtDNA and ATP | Increased release of mtDNA | — |
All findings were obtained from a rodent BPD model of hyperoxia.
Mitochondrial dysfunction in COPD
COPD is characterized by a persistent airflow limitation and lung function decline, where cigarette smoke is the main etiological factor. The pathogenesis of COPD involves a variety of cellular processes, including oxidative stress, inflammatory responses and cellular senescence.
It has been shown that primary bronchial epithelial cells from COPD patients have swollen mitochondrial elongation and fragmentation, swelling and cristae depletion (Hoffmann et al., 2013). Furthermore, chronic cigarette smoke exposure causes mitochondrial dysfunction in lung epithelial cells (Hoffmann et al., 2013). This is in agreement with the findings that cigarette smoke hampers exercise‐induced an increase in mitochondria density through a biogenesis response in the brain (Speck et al., 2011). This may be due to the reduction in PGC1α, a master regulator of mitochondrial biogenesis in the cells of COPD patients compared with cells from non‐smokers and smokers (Hoffmann et al., 2013). Interestingly, acute cigarette smoke up‐regulates the expression of genes involved in energy metabolism, ETC, and oxidative phosphorylation in mouse lungs (Agarwal et al., 2012). Similarly, cytochrome c oxidase activity and mtDNA‐related gene (12S rRNA) are increased in skeletal muscle in COPD patients, which are inversely associated with the degree of arterial hypoxaemia (Sauleda et al., 1998). This may be the compensatory mechanisms and adaptive responses needed to maintain mitochondrial homeostasis. Mice deficient in the synthesis of cytochrome c oxidase are protected from cigarette smoke‐induced pulmonary inflammation (Cloonan et al., 2016). Mitochondrial genome sequencing will further reveal the changes in mitochondria‐related genes and their roles in the development of COPD. In fact, whole genome analysis in rat COPD is ongoing (Jiang et al., 2014).
Mitochondrial dysfunction in muscle cells may contribute to the loss of muscle strength, leading to a decline in physical function, which is a systemic manifestation in COPD patients. Indeed, reduced mitochondrial biogenesis, impaired oxidative phosphorylation and increased mtROS generation, along with a reduction in UCP3, are also observed in skeletal muscles and airway smooth muscle cells from COPD patients (Gosker et al., 2003; Russell et al., 2004; Gosker et al., 2006; Meyer et al., 2013; Puente‐Maestu et al., 2013; Wiegman et al., 2015; Wiley et al., 2016). However, mitochondrial ETC function (i.e. oxidative capacity) in the inspiratory muscles is enhanced in patients with COPD (Ribera et al., 2003). A further causal relationship between mitochondrial function and muscle atrophy as well as the development of COPD should be established to reveal the role of mitochondria in this disease.
Mitophagy, one of the methods by which the quality of mitochondria is controlled, has been shown to be involved in the pathogenesis of COPD. We and others have shown that cigarette smoke impairs mitophagy, leading to incomplete degradation of damaged mitochondria (Ahmad et al., 2015; Ito et al., 2015). These damaged mitochondria may accumulate in the perinuclear region where generating excessive mtROS leads to nuclear DNA damage and subsequent cellular senescence. This is corroborated by the finding that restoration of mitophagy by the overexpression of Parkin reduces cigarette smoke‐induced DNA damage and cellular senescence when used in combination with a mtROS scavenger (Ahmad et al., 2015). Indeed, the levels of Parkin are reduced in lung tissues of COPD patients compared with those of non‐smokers and smokers (Ahmad et al., 2015; Ito et al., 2015), which further confirms the impairment of mitophagy in COPD. It is interesting to note that severe stress derived from high concentrations of cigarette smoke induces necroptosis (i.e. necrosis and apoptosis), which is mediated by PINK1‐induced mitophagy (Mizumura et al., 2014). During the mitophagy, lipofuscin accumulates within the lysosomes, which reduces the efficiency of the endosomal/lysosomal pathway, thereby rendering mitophagy incomplete. Therefore, it is possible that during stress induced by high concentrations of cigarette smoke, mitophagy is incomplete due to lipofuscin accumulation, which causes the release of undegraded pro‐apoptotic proteins such as cytochrome c from mitochondria, resulting in necroptosis.
Morphologically, mitochondrial fragmentation is observed in cigarette smoke‐exposed lung epithelial cells and airway smooth muscle cells, as well as in lungs from COPD patients (Hara et al., 2013; Hoffmann et al., 2014, 2013). This is due to the increased expression of Drp1 and Mfn2. Interestingly, we have observed the mixed morphological changes of mitochondrial fragmentation and elongation in lung fibroblasts (Ahmad et al., 2015). These discrepancies may be due to cell‐specific responses to different concentrations and durations of cigarette smoke exposure.
mtDNA is considered to be a mitochondrial DAMP once it is released from mitochondria, thereby leading to inflammatory responses. As compared with that in non‐smokers, mtDNA copy number in the circulation is increased in smokers and COPD patients (Pouwels et al., 2014). Similarly, the mtDNA of cytochrome c oxidase‐I and cytochrome c oxidase‐II is increased in exfoliated cells in saliva from smokers as compared with non‐smokers, and this increase positively correlates with number of years spent smoking and number of cigarettes smoked (Masayesva et al., 2006). This is in agreement with the findings that cigarette smoke exposure causes mtDNA release from neutrophils, which may amplify airway inflammation (Heijink et al., 2015). An increase in mtDNA release may be associated with cigarette smoking‐induced impairment of mitophagy (Oka et al., 2015, 2012) and the opening of a MPTP (Naserzadeh et al., 2015); this needs to be investigated further. In addition to mtDNA, the opening of a MPTP causes cytochrome C release and ATP depletion, leading to apoptosis and inflammation.
Extracellular ATP acts as a DAMP and activates P2 nucleotide receptors, which are increased in bronchoalveolar lavage fluids of smokers and COPD patients as well as in mouse lungs with emphysema (Mortaz et al., 2009; Lommatzsch et al., 2010). Moreover, ATP neutralization or non‐specific P2 receptor blockade significantly attenuates cigarette smoke‐induced lung inflammation and emphysema (Cicko et al., 2010). A recent study revealed the important role of P2Y14 receptors in stress‐induced stem cell senescence (Cho et al., 2014), which raises the question of whether P2Y receptors modulate cigarette smoke‐induced lung cellular senescence in COPD. These findings suggest potential therapeutic avenues using adenosine P2 receptor antagonists or inhibitors of ATP or the inflammasome for COPD.
In addition to tobacco smoking, other factors, such as hypoxia, hypercapnia, corticosteroid therapy, infection and a sedentary lifestyle, are all also able to cause mitochondrial dysfunction (Gayan‐Ramirez and Decramer, 2013; Meyer et al., 2013), despite their mechanisms being not fully understood. This may contribute to the pathogenesis and exacerbation of COPD as well as its co‐morbidities such as skeletal muscle atrophy (Gifford et al., 2015; Liu et al., 2016). Overall, the targeting damaged mitochondria would be a promising strategy to attenuate the progression and exacerbation of COPD.
Mitochondrial dysfunction in pulmonary fibrosis
The main feature of pulmonary fibrosis is the excessive accumulation and deposit of extracellular matrix, thereby leading to the scar and loss of elasticity in lungs. Both fibroblasts and myofibroblasts are the principal effector cells of the lung for the generation of extracellular matrix (Yao and Li, 2015).
Recent studies have highlighted the importance of mitochondrial function and mitophagy in the pathogenesis of lung fibrosis (Bueno et al., 2015; Patel et al., 2015). It has been shown that damaged or dysfunctional mitochondria accumulate in alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. mtDNA copy numbers are significantly reduced in mouse lungs with bleomycin‐induced fibrosis (Gazdhar et al., 2014). The mtDNA encoded respiratory chain, such as cytochrome c oxidase subunit I, is reduced in mouse lungs with fibrosis and in lung epithelial cells treated with TGF‐β (Sohn et al., 2012; Gazdhar et al., 2014). These findings suggest that both mitochondrial biogenesis and oxidative phosphorylation are reduced during lung fibrosis.
Further investigations have revealed that the accumulation of these dysfunctional mitochondria is due to the impairment of PINK1‐mediated mitophagy, but not mitochondrial biogenesis (Bueno et al., 2015). This is in contrast to the findings that vascular mitochondrial biogenesis is activated in response to asbestos and bleomycin inhalation (Carraway et al., 2008). Furthermore, PINK1 deletion enhances bleomycin‐induced pulmonary fibrosis in mice (Patel et al., 2015). Hence, PINK1‐mediated mitophagy is important for the maintainance of mitochondrial homeostasis (i.e. Ppargc1a, Tfam and cytochrome c) and dynamics in epithelial cells, thereby protecting against the development of lung fibrosis. Fibroblasts and myofibroblasts are the effector cells for the development of lung fibrosis. However, it is not clear whether mitochondrial dysfunction and mitophagy impairment occur in (myo)fibroblasts during pulmonary fibrosis. Also further studies are required to determine how mitochondrial dysfunction in lung epithelial cells interacts with (myo)fibroblasts, leading to fibrogenesis.
It has been shown that once released mtDNA recruits peripheral blood mononuclear cells and stimulates epithelial cells to generate TGF‐β1 (Li et al., 2015). Targeting mtDNA by DNase I protects against paraquat‐induced pulmonary fibrosis (Li et al., 2015). Interestingly, TGF‐β1 increases the number of mitochondria, mitochondria‐specific proteins, voltage‐dependent anion channels, adenine nucleotide transporter and mtDNA content, whereas mitochondrial oxidative phosphorylation and mitophagy are impaired during fibroblast differentiation (Negmadjanov et al., 2015; Sosulski et al., 2015). We propose that mitochondrial biogenesis is needed for fibroblast differentiation that this effect is further promoted by mitophagy impairment and damaged mitochondria during fibrogenesis. However, it is not clear how TGF‐β1 alters mitochondrial homeostasis, inducing the accumulation of damaged mitochondria and subsequent fibrogenesis.
Aging has been shown to participate in the pathogenesis of lung fibrosis as both mitochondria respiration and mitophagy are compromised in aging, whereas the levels of the mitochondrial biogenesis marker PGC1α, mitochondrial transcription factor A Tfam and mitochondrial gene cytochrome c are comparable between young and old mouse lungs (Bueno et al., 2015; Sosulski et al., 2015). Interestingly, there is no significant difference in the severity of fibrosis, in response to bleomycin, between young and aged mice, whereas fibrosis resolution is impaired in aged mice (Hecker et al., 2014). This is ascribed to the accumulation of senescent myofibroblasts that are resistant to apoptosis in aged mice. Recent studies have shown that fibroblasts that resist cigarette smoke‐induced cellular senescence acquire a pro‐fibrotic phenotype (Kanaji et al., 2014; Ahmad et al., 2015), which may explain the fibrotic lesions occurring in COPD. Interestingly, primary lung fibroblasts derived from patients with idiopathic pulmonary fibrosis exhibited accelerated cellular senescence, where senescent fibroblasts express increased levels of the myofibroblast marker α‐smooth muscle actin (α‐SMA) (Yanai et al., 2015). The mechanisms underlying these discrepancies in the causal relationships between fibroblast senescence and differentiation into myofibroblasts are not clear. Removing senescent fibroblasts would reveal whether senescence promotes their differentiation into myofibroblasts. In addition to (myo)fibroblasts, lung epithelial cells undergo senescence during the development of pulmonary fibrosis (Minagawa et al., 2011; Shivshankar et al., 2012). Further studies are required to determine their contributions to the pathogenesis of lung fibrosis and how these senescent cells interact with each other (autocrine and paracrine) to induce fibrogenesis.
Mitochondrial dysfunction in BPD
BPD is a chronic lung disorder of infants with low birth weight and in those who receive prolonged mechanical ventilation with oxygen to treat respiratory distress syndrome (McEvoy et al., 2014). Antenatal and/or post‐natal exposure to stress including mechanical ventilation, oxygen toxicity and pulmonary and systemic infection disrupts pulmonary development, thereby leading to inflammation and damage to the highly vulnerable premature lung in BPD. New BPD develops at the canalicular and early saccular phases, whereas old BPD starts at the saccular and alveolar phases of lung development. New BPD is pathologically characterized by the simplified alveolarization and vascularization, which is in contrast to severe lung injury with fibrosis in old BPD (Kramer, 2008).
Accumulating evidence shows that mitochondria are dysfunctional and respiration rates, ATP‐production rate and complex I activity reduced in hyperoxia‐exposed lungs in a mouse model of BPD (Ratner et al., 2009). This is consistent with the reduced mitochondrial aconitase activity observed in lungs of baboons exposed to hyperoxia (Morton et al., 1998). In vitro, hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in mouse lung epithelial cells and inhibits complex I and II function, but not complex IV activity in mouse isolated lung mitochondria (Das, 2013). Furthermore, a complex I inhibitor delays alveolarization in neonate mice exposed to hyperoxia (Das, 2013). Impaired mitochondrial biogenesis may be responsible for hyperoxia‐induced delayed alveolarization, which is the most common feature of BPD.
Hyperoxia‐induced impairment of branching morphogenesis is associated with increased mtROS generation accompanied by increased mtDNA damage and mutation (Gebb et al., 2013). Compared with nuclear DNA, mtDNA is prone to stress‐induced damage and mutation due to lack of compact chromatin structure or DNA repair mechanisms. Indeed, the transmission of mtDNA mutations occurs in one in every 200 newborns, which potentially may cause disease or increase disease susceptibility (Elliott et al., 2008). Treatment with a mitochondria‐specific antioxidant, mitoTEMPO, during early post‐natal hyperoxia protected against compromised alveolarization (Datta et al., 2015), which suggests that targeted antioxidant therapy could be used to prevent or treat BPD. Hyperoxic exposure induces UCP2 expression in pulmonary macrophages and enhanced UCP3 levels in skeletal muscles in mice (Flandin et al., 2005; Steer et al., 2013), which may be compensatory responses to increased oxidative stress and mtROS. Further studies are required to determine the role of UCP in the development of BPD using genetic manipulation approaches.
Recently, it has been shown that the NLRP3–PINK1 axis plays a pivotal role in hyperoxia‐induced cell and tissue death in adult mice (Zhang et al., 2014). Indeed, the NLRP3 inflammasome is critically involved in the development of BPD, as demonstrated using in vivo mouse and baboon models as well as ex vivo tracheal aspirates from BPD patients (Liao et al., 2015). PINK1 deletion exaggerates hyperoxia‐induced mtROS and apoptosis as well as lung injury (Zhang et al., 2014). It is still not known whether PINK1‐mediated mitophagy is impaired and how it is regulated during NLRP3‐mediated inflammasome formation and subsequent BPD development. Additionally, there are no studies showing the changes in mitochondrial morphology and function in BPD patients; this needs to be investigated further.
A recent study has shown that 23 genes are differentially methylated with reciprocal changes in expression in patients with BPD compared with preterm or term lungs (Cuna et al., 2015). Mitoepigenetics such as mitochondrial methylation occur in a variety of cultured mammalian cells, and this process is modulated by DNA methyltransferase 1 in mitochondria (Shock et al., 2013, 2014, 2011). Therefore, it is possible that exposure to environmental factors including smoke, particulate metals and diet in utero leads to mtDNA methylation (Byun et al., 2013), which renders newborns more susceptible to the development of BPD.
Conclusion and future directions
Mitochondria can function as a signal in addition to generating the energy needed for the survival of cells and their functions. Mitochondrial dysfunction plays an important role in the stress‐induced inflammation and cellular senescence that occur in chronic lung diseases, such as COPD, pulmonary fibrosis and BPD. This is reflected by the findings that mitochondrial biogenesis, oxidative phosphorylation and mitophagy are reduced in chronic lung diseases.
A study using a rodent model has demonstrated that disruption of normal lung morphogenesis by lung‐specific epithelial deletion of the CCAAT/enhancer‐binding protein α in foetal life subsequently drives the onset of spontaneous pulmonary emphysema in adult mice (Didon et al., 2010). This has leads to the hypothesis that the origins of COPD occur in early life. It is well known that mtDNA is inherited from the maternal side. Therefore, it would be interesting to study whether environmental stress causes dysfunctional mtDNA (e.g. mutation or epigenetic modifications) that are passed onto offspring, leading to the development of BPD and subsequently COPD. Employment of a mitochondrial–nuclear exchange technique will help to reveal the role of mtDNA in the pathogenesis of chronic lung diseases (Fetterman et al., 2013; Betancourt et al., 2014).
Mitochondrial transfer and replacement have recently been approved in the UK for use in the prevention and treatment of mitochondrial diseases (Mitalipov and Wolf, 2014; Dunham‐Snary and Ballinger, 2015). It has been shown that mitochondrial transfer of induced pluripotent stem cell‐derived mesenchymal stem cells to airway epithelial cells ameliorates cigarette smoke‐induced airspace enlargement in mice (Li et al., 2014). Similarly, mitochondrial transfer from stem cells protects against injurious and inflammatory responses in the lung evoked by LPS and allergens (Islam et al., 2014, 2012). Tunneling nanotubes are considered as a duct to transfer healthy mitochondria to damaged cells, which are controlled by a mitochondrial Rho‐GTPase (Miro1 encoding by Rhot 1 gene) (Ahmad et al., 2014; Wang and Gerdes, 2015). However, it remains unclear how tunneling nanotubes are regulated particularly in the condition of lung injury. Further studies on mitochondrial dysfunction and transfer as well as their regulation would enhance the understanding of the molecular pathogenesis of chronic lung diseases and provide potential novel ways for treating them.
Author contributions
L.Y. and H.Y. drafted and edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Yue, L. , and Yao, H. (2016) Mitochondrial dysfunction in inflammatory responses and cellular senescence: pathogenesis and pharmacological targets for chronic lung diseases. British Journal of Pharmacology, 173: 2305–2318. doi: 10.1111/bph.13518.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. [DOI] [PubMed] [Google Scholar]
- Agarwal AR, Zhao L, Sancheti H, Sundar IK, Rahman I, Cadenas E (2012). Short‐term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am J Physiol Lung Cell Mol Physiol 303: L889–L898. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 33: 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H et al. (2015). Impaired mitophagy leads to cigarette smoke stress‐induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J 29: 2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC et al. (2012). Perinuclear mitochondrial clustering creates an oxidant‐rich nuclear domain required for hypoxia‐induced transcription. Sci Signal 5: ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso‐Loeches S, Urena‐Peralta JR, Morillo‐Bargues MJ, Oliver‐De La Cruz J, Guerri C (2014). Role of mitochondria ROS generation in ethanol‐induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci 8: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andziak B, O'Connor TP, Qi W, DeWaal EM, Pierce A, Chaudhuri AR et al. (2006). High oxidative damage levels in the longest‐living rodent, the naked mole‐rat. Aging Cell 5: 463–471. [DOI] [PubMed] [Google Scholar]
- Aoshiba K, Nagai A (2009). Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc 6: 596–601. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R et al. (2014). Cigarette smoke‐induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306: L840–L854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi D, D'Aquila P, Scafone T, Giordano M, Riso V, Riccio A et al. (2013). The control region of mitochondrial DNA shows an unusual CpG and non‐CpG methylation pattern. DNA Res 20: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AM, King AL, Fetterman JL, Millender‐Swain T, Finley RD, Oliva CR et al. (2014). Mitochondrial‐nuclear genome interactions in non‐alcoholic fatty liver disease in mice. Biochem J 461: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours MJ, Dagnelie PC, Giuliani AL, Wesselius A, Di Virgilio F (2011). P2 receptors and extracellular ATP: a novel homeostatic pathway in inflammation. Front Biosci (Schol Ed) 3: 1443–1456. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS (2004). Calcium, ATP, and ROS: a mitochondrial love–hate triangle. Am J Physiol Cell Physiol 287: C817–C833. [DOI] [PubMed] [Google Scholar]
- Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C et al. (2015). PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 125: 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY et al. (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 208: 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P et al. (2013). Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol 10: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29: 222–230. [DOI] [PubMed] [Google Scholar]
- Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway MS, Suliman HB, Kliment C, Welty‐Wolf KE, Oury TD, Piantadosi CA (2008). Mitochondrial biogenesis in the pulmonary vasculature during inhalational lung injury and fibrosis. Antioxid Redox Signal 10: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P (2014). Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis 5: e1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BB, Coon TA, Glasser JR, Zou C, Ellis B, Das T et al. (2014). E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep 7: 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K et al. (2013). ATP‐P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol 45: 932–943. [DOI] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE et al. (2011). Control of the senescence‐associated secretory phenotype by NF‐kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Yusuf R, Kook S, Attar E, Lee D, Park B et al. (2014). Purinergic P2Y(1)(4) receptor modulates stress‐induced hematopoietic stem/progenitor cell senescence. J Clin Invest 124: 3159–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicko S, Lucattelli M, Muller T, Lommatzsch M, De Cunto G, Cardini S et al. (2010). Purinergic receptor inhibition prevents the development of smoke‐induced lung injury and emphysema. J Immunol 185: 688–697. [DOI] [PubMed] [Google Scholar]
- Cloonan SM, Glass K, Laucho‐Contreras ME, Bhashyam AR, Cervo M, Pabon MA et al. (2016). Mitochondrial iron chelation ameliorates cigarette smoke‐induced bronchitis and emphysema in mice. Nat Med 22: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KL, Soto‐Pantoja DR, Jin L, Abu‐Asab M, Clarke R (2014). When is a vesicle not just a vesicle: mitochondrial spheroids and mitochondrial autophagosomes. Cell Biosci 4: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuna A, Halloran B, Faye‐Petersen O, Kelly D, Crossman DK, Cui X et al. (2015). Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am J Respir Cell Mol Biol 53: 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KC (2013). Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE‐12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One 8: e73358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Kim GA, Taylor JM, Gugino SF, Farrow KN, Schumacker PT et al. (2015). Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS dependent. Am J Physiol Lung Cell Mol Physiol 309: L369–L377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didon L, Roos AB, Elmberger GP, Gonzalez FJ, Nord M (2010). Lung‐specific inactivation of CCAAT/enhancer binding protein alpha causes a pathological pattern characteristic of COPD. Eur Respir J 35: 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Eskelinen EL (2014). Do mitochondria donate membrane to form autophagosomes or undergo remodeling to form mitochondrial spheroids? Cell Biosci 4: 65–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Guo F, Ni HM, Bockus A, Manley S, Stolz DB et al. (2012). Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem 287: 42379–42388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL (2013). Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep 3: 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Toit A (2014). Protein degradation: an alternative route for mitochondrial quality control. Nat Rev Mol Cell Biol 15: 150–151. [DOI] [PubMed] [Google Scholar]
- Dunham‐Snary KJ, Ballinger SW (2015). GENETICS. Mitochondrial–nuclear DNA mismatch matters. Science 349: 1449–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF (2008). Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83: 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre Y, Hurtaud C, Nubel T, Criscuolo F, Ricquier D, Cassard‐Doulcier AM (2007). Mitochondria contribute to LPS‐induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J 402: 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faner R, Rojas M, Macnee W, Agusti A (2012). Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186: 306–313. [DOI] [PubMed] [Google Scholar]
- Fetterman JL, Zelickson BR, Johnson LW, Moellering DR, Westbrook DG, Pompilius M et al. (2013). Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. Biochem J 455: 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J et al. (2015). The ins and outs of mitochondrial calcium. Circ Res 116: 1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz JG (2007). Regulation of cellular ATP release. Trans Am Clin Climatol Assoc 118: 199–208. [PMC free article] [PubMed] [Google Scholar]
- Flandin P, Donati Y, Barazzone‐Argiroffo C, Muzzin P (2005). Hyperoxia‐mediated oxidative stress increases expression of UCP3 mRNA and protein in skeletal muscle. FEBS Lett 579: 3411–3415. [DOI] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J (2011). p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J 30: 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J (2014). Mitochondrial form and function. Nature 505: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, St‐Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR (2013). Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24: 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Sana R, Calder V, Calonge M, Lee W, Wheeler LA et al. (2013). Mitochondrial permeability transition pore in inflammatory apoptosis of human conjunctival epithelial cells and T cells: effect of cyclosporin A. Invest Ophthalmol Vis Sci 54: 4717–4733. [DOI] [PubMed] [Google Scholar]
- Gayan‐Ramirez G, Decramer M (2013). Mechanisms of striated muscle dysfunction during acute exacerbations of COPD. J Appl Physiol (1985)114: 1291–1299. [DOI] [PubMed] [Google Scholar]
- Gazdhar A, Lebrecht D, Roth M, Tamm M, Venhoff N, Foocharoen C et al. (2014). Time‐dependent and somatically acquired mitochondrial DNA mutagenesis and respiratory chain dysfunction in a scleroderma model of lung fibrosis. Sci Rep 4: 5336–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebb SA, Decoux A, Waggoner A, Wilson GL, Gillespie MN (2013). Mitochondrial DNA damage mediates hyperoxic dysmorphogenesis in rat fetal lung explants. Neonatology 103: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Sengupta S, Scaria V (2014). Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 18: 58–62. [DOI] [PubMed] [Google Scholar]
- Gifford JR, Trinity JD, Layec G, Garten RS, Park SY, Rossman MJ et al. (2015). Quadriceps exercise intolerance in patients with chronic obstructive pulmonary disease: the potential role of altered skeletal muscle mitochondrial respiration. J Appl Physiol (1985)119: 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosker HR, Schrauwen P, Broekhuizen R, Hesselink MK, Moonen‐Kornips E, Ward KA et al. (2006). Exercise training restores uncoupling protein‐3 content in limb muscles of patients with chronic obstructive pulmonary disease. Am J Physiol Endocrinol Metab 290: E976–E981. [DOI] [PubMed] [Google Scholar]
- Gosker HR, Schrauwen P, Hesselink MK, Schaart G, van der Vusse GJ, Wouters EF et al. (2003). Uncoupling protein‐3 content is decreased in peripheral skeletal muscle of patients with COPD. Eur Respir J 22: 88–93. [DOI] [PubMed] [Google Scholar]
- Guescini M, Guidolin D, Vallorani L, Casadei L, Gioacchini AM, Tibollo P et al. (2010). C2C12 myoblasts release micro‐vesicles containing mtDNA and proteins involved in signal transduction. Exp Cell Res 316: 1977–1984. [DOI] [PubMed] [Google Scholar]
- Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y et al. (2013). Mitochondrial fragmentation in cigarette smoke‐induced bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol 305: L737–L746. [DOI] [PubMed] [Google Scholar]
- Hattori N, Saiki S, Imai Y (2014). Regulation by mitophagy. Int J Biochem Cell Biol 53: 147–150. [DOI] [PubMed] [Google Scholar]
- Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T et al. (2014). Reversal of persistent fibrosis in aging by targeting Nox4–Nrf2 redox imbalance. Sci Transl Med 6: 231ra247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink IH, Pouwels SD, Leijendekker C, de Bruin HG, Zijlstra GJ, van der Vaart H et al. (2015). Cigarette smoke‐induced damage‐associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 52: 554–562. [DOI] [PubMed] [Google Scholar]
- Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S et al. (2013). Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res 14: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K et al. (2012). Mitochondrial transfer from bone‐marrow‐derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18: 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M et al. (2015). PARK2‐mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11: 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK et al. (2009). Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106: 20388–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X (2013). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZM, Zhao QS, Xu YQ, Li T, Jie J, Zhang ZH (2014). Whole mitochondrial genome sequencing and analysis for chronic obstructive pulmonary disease rat strain. Mitochondrial DNA : 1–2. [DOI] [PubMed] [Google Scholar]
- Kalogeris T, Bao Y, Korthuis RJ (2014). Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2: 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaji N, Basma H, Nelson A, Farid M, Sato T, Nakanishi M et al. (2014). Fibroblasts that resist cigarette smoke‐induced senescence acquire profibrotic phenotypes. Am J Physiol Lung Cell Mol Physiol 307: L364–L373. [DOI] [PubMed] [Google Scholar]
- Katz A, Hernandez A, Caballero DM, Briceno JF, Amezquita LV, Kosterina N et al. (2014). Effects of N‐acetylcysteine on isolated mouse skeletal muscle: contractile properties, temperature dependence, and metabolism. Pflugers Arch 466: 577–585. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Kroemer G (2011). Mitochondrial control of the NLRP3 inflammasome. Nat Immunol 12: 199–200. [DOI] [PubMed] [Google Scholar]
- Klimova TA, Bell EL, Shroff EH, Weinberg FD, Snyder CM, Dimri GP et al. (2009). Hyperoxia‐induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB J 23: 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H (2011). The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL‐33 release and innate Th2‐type responses. J Immunol 186: 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer BW (2008). Antenatal inflammation and lung injury: prenatal origin of neonatal disease. J Perinatol 28 (Suppl 1): S21–S27. [DOI] [PubMed] [Google Scholar]
- Kumar M, Seeger W, Voswinckel R (2014). Senescence‐associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 51: 323–333. [DOI] [PubMed] [Google Scholar]
- Lawless C, Jurk D, Gillespie CS, Shanley D, Saretzki G, von Zglinicki T et al. (2012). A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS One 7: e32117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS et al. (2005). Effects of recombinant adenovirus‐mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res 96: 1200–1207. [DOI] [PubMed] [Google Scholar]
- Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X et al. (2007). Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem 282: 22977–22983. [DOI] [PubMed] [Google Scholar]
- Li G, Yuzhen L, Yi C, Xiaoxiang C, Wei Z, Changqing Z et al. (2015). DNaseI protects against Paraquat‐induced acute lung injury and pulmonary fibrosis mediated by mitochondrial DNA. Biomed Res Int 2015: 386952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF (2013). Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y et al. (2014). Mitochondrial transfer of induced pluripotent stem cell‐derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke‐induced damage. Am J Respir Cell Mol Biol 51: 455–465. [DOI] [PubMed] [Google Scholar]
- Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D et al. (2015). The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun 6: 8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi L et al. (2016). Mitochondrial dysfunction launches dexamethasone‐induced skeletal muscle atrophy via AMPK/FOXO3 signaling. Mol Pharm 13: 73–84. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C et al. (2012). The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle‐wasting stimuli. Cell Metab 16: 613–624. [DOI] [PubMed] [Google Scholar]
- Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, Stoll P et al. (2010). Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 181: 928–934. [DOI] [PubMed] [Google Scholar]
- Lopez‐Armada MJ, Riveiro‐Naveira RR, Vaamonde‐Garcia C, Valcarcel‐Ares MN (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13: 106–118. [DOI] [PubMed] [Google Scholar]
- MacNee W, Tuder RM (2009). New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc 6: 527–531. [DOI] [PubMed] [Google Scholar]
- Masayesva BG, Mambo E, Taylor RJ, Goloubeva OG, Zhou S, Cohen Y et al. (2006). Mitochondrial DNA content increase in response to cigarette smoking. Cancer Epidemiol Biomarkers Prev 15: 19–24. [DOI] [PubMed] [Google Scholar]
- McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL (2014). Bronchopulmonary dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann Am Thorac Soc 11 (Suppl 3): S146–S153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Zoll J, Charles AL, Charloux A, de Blay F, Diemunsch P et al. (2013). Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: central actor and therapeutic target. Exp Physiol 98: 1063–1078. [DOI] [PubMed] [Google Scholar]
- Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y et al. (2011). Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF‐beta‐induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 300: L391–L401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitalipov S, Wolf DP (2014). Clinical and ethical implications of mitochondrial gene transfer. Trends Endocrinol Metab 25: 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T et al. (2014). Mitophagy‐dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortaz E, Braber S, Nazary M, Givi ME, Nijkamp FP, Folkerts G (2009). ATP in the pathogenesis of lung emphysema. Eur J Pharmacol 619: 92–96. [DOI] [PubMed] [Google Scholar]
- Morton RL, Ikle D, White CW (1998). Loss of lung mitochondrial aconitase activity due to hyperoxia in bronchopulmonary dysplasia in primates. Am J Physiol 274: L127–L133. [DOI] [PubMed] [Google Scholar]
- Naik E, Dixit VM (2011). Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 208: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Ohsawa S, Igaki T (2014). Mitochondrial defects trigger proliferation of neighbouring cells via a senescence‐associated secretory phenotype in Drosophila. Nat Commun 5: 5264. [DOI] [PubMed] [Google Scholar]
- Naserzadeh P, Hosseini MJ, Arbabi S, Pourahmad J (2015). A comparison of toxicity mechanisms of cigarette smoke on isolated mitochondria obtained from rat liver and skin. Iran J Pharm Res 14: 271–277. [PMC free article] [PubMed] [Google Scholar]
- Negmadjanov U, Godic Z, Rizvi F, Emelyanova L, Ross G, Richards J et al. (2015). TGF‐beta1‐mediated differentiation of fibroblasts is associated with increased mitochondrial content and cellular respiration. PLoS One 10: e0123046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Jaeschke H, Ding WX (2013). Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen‐induced necrosis in the liver. Redox Biol 1: 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio K, Ma Q (2016). Effect of overproduction of mitochondrial uncoupling protein 2 in Cos7 cells: induction of senescent‐like morphology and oncotic cell death. Curr Aging Sci. DOI: 10.2174/1874609809666160211125332. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012). Mitochondria: in sickness and in health. Cell 148: 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T et al. (2012). Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y et al. (2011). Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 480: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H (2010). Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin‐related protein 1 and mitofusin 1. J Cell Sci 123: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H et al. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere‐dependent senescence. PLoS Biol 5: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y et al. (2015). Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor‐beta1 in pulmonary fibrosis. PLoS One 10: e0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A (2004). Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61: 3100–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC et al. (2014). DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol 7: 215–226. [DOI] [PubMed] [Google Scholar]
- Puente‐Maestu L, Lazaro A, Humanes B (2013). Metabolic derangements in COPD muscle dysfunction. J Appl Physiol (1985)114: 1282–1290. [DOI] [PubMed] [Google Scholar]
- Ratner V, Starkov A, Matsiukevich D, Polin RA, Ten VS (2009). Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia‐exposed mice. Am J Respir Cell Mol Biol 40: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribera F, N'Guessan B, Zoll J, Fortin D, Serrurier B, Mettauer B et al. (2003). Mitochondrial electron transport chain function is enhanced in inspiratory muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 167: 873–879. [DOI] [PubMed] [Google Scholar]
- Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L et al. (2010). Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 182: 774–783. [DOI] [PubMed] [Google Scholar]
- Romaschenko VP, Zinovkin RA, Galkin II, Zakharova VV, Panteleeva AA, Tokarchuk AV et al. (2015). Low concentrations of uncouplers of oxidative phosphorylation prevent inflammatory activation of endothelial cells by tumor necrosis factor. Biochemistry (Mosc) 80: 610–619. [DOI] [PubMed] [Google Scholar]
- Russell AP, Somm E, Debigare R, Hartley O, Richard D, Gastaldi G et al. (2004). COPD results in a reduction in UCP3 long mRNA and UCP3 protein content in types I and IIa skeletal muscle fibers. J Cardiopulm Rehabil 24: 332–339. [DOI] [PubMed] [Google Scholar]
- Saretzki G, Murphy MP, von Zglinicki T (2003). MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2: 141–143. [DOI] [PubMed] [Google Scholar]
- Sauleda J, Garcia‐Palmer F, Wiesner RJ, Tarraga S, Harting I, Tomas P et al. (1998). Cytochrome oxidase activity and mitochondrial gene expression in skeletal muscle of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 157: 1413–1417. [DOI] [PubMed] [Google Scholar]
- Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA et al. (2014). Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306: L962–L974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS (2012). Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Le Saux CJ (2012). Caveolin‐1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol 47: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A 108: 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn EJ, Kim J, Hwang Y, Im S, Moon Y, Kang DM (2012). TGF‐beta suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cell Mol Biol (Noisy‐le‐grand) 58 (Suppl): OL1763–OL1767. [PubMed] [Google Scholar]
- Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG (2015). Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1. Aging Cell 14: 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA et al. (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22: 135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7: e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck AE, Fraga D, Soares P, Scheffer DL, Silva LA, Aguiar AS Jr et al. (2011). Cigarette smoke inhibits brain mitochondrial adaptations of exercised mice. Neurochem Res 36: 1056–1061. [DOI] [PubMed] [Google Scholar]
- Springer W, Kahle PJ (2011). Regulation of PINK1–Parkin‐mediated mitophagy. Autophagy 7: 266–278. [DOI] [PubMed] [Google Scholar]
- Steer JH, Mann TS, Lo SZ, Inglis JJ, Yap HS, Henry PJ et al. (2013). Early induction of uncoupling protein‐2 in pulmonary macrophages in hyperoxia‐associated lung injury. Inhal Toxicol 25: 544–552. [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM (2014). A new pathway for mitochondrial quality control: mitochondrial‐derived vesicles. EMBO J 33: 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II et al. (2015). Measuring in vivo mitophagy. Mol Cell 60: 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A (2009). Absence of autophagy results in reactive oxygen species‐dependent amplification of RLR signaling. Proc Natl Acad Sci U S A 106: 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI, Griffiths EJ, Rutter GA (2012). Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 52: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP et al. (2012). NF‐kappaB inhibition delays DNA damage‐induced senescence and aging in mice. J Clin Invest 122: 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toime LJ, Brand MD (2010). Uncoupling protein‐3 lowers reactive oxygen species production in isolated mitochondria. Free Radic Biol Med 49: 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ et al. (2013). The PINK1–Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110: 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang X, Lopez de Silanes I, Carling D, Gorospe M (2003). Increased AMP:ATP ratio and AMP‐activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem 278: 27016–27023. [DOI] [PubMed] [Google Scholar]
- Wang X, Gerdes HH (2015). Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22: 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber FC, Esser PR, Muller T, Ganesan J, Pellegatti P, Simon MM et al. (2010). Lack of the purinergic receptor P2X(7) results in resistance to contact hypersensitivity. J Exp Med 207: 2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P et al. (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE et al. (2015). Oxidative stress‐induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 136: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Niu J, Li X, Wang X, Guo Z, Zhang F (2014). TGF‐beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev Biol 14: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Shteinberg A, Porat Z, Budovsky A, Braiman A, Zeische R et al. (2015). Cellular senescence‐like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 7: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Rahman I (2012a). Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem Pharmacol 84: 1332–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Rahman I (2012b). Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol 303: L557–L566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HW, Li J (2015). Epigenetic modifications in fibrotic diseases: implications for pathogenesis and pharmacological targets. J Pharmacol Exp Ther 352: 2–13. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP (2011). Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ding PW, Yu M, Zhang SS, Long Q, Cheng X et al. (2015). IL‐17 Induces MPTP opening through ERK2 and P53 signaling pathway in human platelets. J Huazhong Univ Sci Technolog Med Sci 35: 679–683. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sauler M, Shinn AS, Gong H, Haslip M, Shan P et al. (2014). Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J Immunol 192: 5296–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225. [DOI] [PubMed] [Google Scholar]
- Zhu J, Dagda RK, Chu CT (2011). Monitoring mitophagy in neuronal cell cultures. Methods Mol Biol 793: 325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]