

Pharmacokinetic/pharmacodynamic studies of artemether-lumefantrine and 3 antiretroviral regimens were conducted in malaria-infected Ugandan children. Efavirenz-based treatment was associated with significant reductions in antimalarial exposure and higher risks of recurrent malaria. Caution in their concurrent use is warranted.
Keywords: malaria, HIV, antimalarial, antiretroviral, pharmacokinetics
Abstract
Background. The optimal treatment of malaria in human immunodeficiency virus (HIV)–infected children requires consideration of critical drug–drug interactions in coinfected children, as these may significantly impact drug exposure and clinical outcomes.
Methods. We conducted an intensive and sparse pharmacokinetic/pharmacodynamic study in Uganda of the most widely adopted artemisinin-based combination therapy, artemether-lumefantrine. HIV-infected children on 3 different first-line antiretroviral therapy (ART) regimens were compared to HIV-uninfected children not on ART, all of whom required treatment for Plasmodium falciparum malaria. Pharmacokinetic sampling for artemether, dihydroartemisinin, and lumefantrine exposure was conducted through day 21, and associations between drug exposure and outcomes through day 42 were investigated.
Results. One hundred forty-five and 225 children were included in the intensive and sparse pharmacokinetic analyses, respectively. Compared with no ART, efavirenz (EFV) reduced exposure to all antimalarial components by 2.1- to 3.4-fold; lopinavir/ritonavir (LPV/r) increased lumefantrine exposure by 2.1-fold; and nevirapine reduced artemether exposure only. Day 7 concentrations of lumefantrine were 10-fold lower in children on EFV vs LPV/r-based ART, changes that were associated with an approximate 4-fold higher odds of recurrent malaria by day 28 in those on EFV vs LPV/r-based ART.
Conclusions. The choice of ART in children living in a malaria-endemic region has highly significant impacts on the pharmacokinetics and pharmacodynamics of artemether-lumefantrine treatment. EFV-based ART reduces all antimalarial components and is associated with the highest risk of recurrent malaria following treatment. For those on EFV, close clinical follow-up for recurrent malaria following artemether-lumefantrine treatment, along with the study of modified dosing regimens that provide higher exposure, is warranted.
(See the Editorial Commentary by Fehintola, Adedeji, and Morse on pages 423–4.)
Malaria and human immunodeficiency virus infection (HIV) impose extensive and overlapping burdens in sub-Saharan Africa. The World Health Organization (WHO) estimated 198 million malaria cases and 584 000 malaria-related deaths in 2013, with 90% occurring in sub-Saharan Africa [1]. Although malaria deaths have declined, a substantial burden remains [1], reflected in malaria infection rates of up to 6 episodes per person year in HIV-uninfected children in Eastern Uganda [2–4].
Sub-Saharan Africa is also home to 25 million people with HIV [5, 6]; 2.9 million are children aged <15 years [6]. Access to antiretroviral therapy (ART) has expanded, and the WHO recommends that all children receive ART [7, 8]. Currently, a lopinavir/ritonavir (LPV/r)–based regimen is recommended as first-line in children <3 years, or, if this is not feasible, a nevirapine (NVP)–based regimen [9]. For children aged ≥3 years, including adolescents, efavirenz (EFV) is preferred, and NVP is the alternative [9]. Despite reduced mother-to-child transmission, an estimated 2 million HIV-infected children will reside in sub-Saharan Africa in 2020 [8].
Optimally treating malaria and HIV in children requires consideration of complex biological and pharmacological factors that impact artemisinin-based combination therapy (ACT). Developmental changes in pharmacokinetics in children are often ignored, and concomitant ACT and ART results in drug–drug interactions that may have significant treatment effects. HIV infection impairs antimalarial immunity, and if untreated, increases malaria morbidity and mortality [10]. In addition, use of trimethoprim-sulfamethoxazole (TMP-SMX) prophylaxis further impacts one's risk of malaria [11]. Overlooking these factors places children at greater risk of poor treatment outcomes, including increasing rates of reinfection in highly endemic regions, and of greater concern, the potential for treatment failure and ACT resistance [1, 4, 12, 13].
ACTs are adopted in 79 of 88 Plasmodium falciparum–endemic countries [1]. ACTs combine a short-acting artemisinin that rapidly reduces parasite load, with a long-acting partner drug that eradicates parasites and helps prevent new infections, minimizing the emergence and spread of resistance. Artemether-lumefantrine is the most widely used ACT, and is metabolized by cytochrome P450 (CYP), including CYP3A4/2B6 for artemether (converted to the active metabolite dihydroartemisinin [DHA]) [14] and CYP3A4 for lumefantrine [15]; thus, both are susceptible to CYP inhibition/induction with ART.
Our group previously reported that the incidence of recurrent malaria following artemether-lumefantrine in HIV-infected children was lower in children on LPV/r- vs nonnucleoside reverse transcriptase inhibitor (NNRTI)–based ART [4]. We now report results from a large intensive pharmacokinetic/pharmacodynamic (PK/PD) study, aimed at fully delineating the pharmacological basis of these differences. The PK/PD of artemether-lumefantrine for treatment of malaria in HIV-infected children stabilized on 3 first-line ART regimens were compared both between ART regimens and to concurrently enrolled HIV-uninfected children, with the goal of informing artemether-lumefantrine treatment guidelines.
METHODS
Study Area and Patients
This prospective PK/PD study of artemether-lumefantrine for the treatment of uncomplicated malaria in HIV-infected children and HIV-uninfected children was conducted from 5 August 2011 to 5 November 2014 in the high-transmission-intensity district of Tororo, Uganda. Children aged 0.5–8 years were enrolled if eligibility criteria were met (Supplementary Data). HIV-infected children were typically on standard weight-based-dosed lamivudine and zidovudine with either LPV/r, or the NNRTIs NVP or EFV, and 96% were on daily TMP-SMX prophylaxis [9].
Clinical Management and Pharmacokinetic Methods
Children enrolled had uncomplicated P. falciparum malaria confirmed by thick blood smear (regardless of parasite density), and documented or 24-hour fever history (≥38.0°C). Active follow-up occurred on days 0 (diagnosis) through 28, with passive follow-up to day 42. Recurrent malaria episodes were genotyped to distinguish recrudescent from new infections [16].
A standard 6-dose treatment of weight-based artemether-lumefantrine (Coartem Dispersible 20 mg/120 mg, Novartis Pharma AG, Basel, Switzerland) was administered with milk or breastfeeding in the clinic or at home, to enhance and control for lumefantrine absorption [17]. For the intensive PK cohort, sampling was pre–first dose (day 0) and pre/post–sixth dose (7 venous samples on day 3 at 0, 0.5, 1, 2, 3, 4, and 8 hours post–last dose), and days 4, 7, 14, and 21 (capillary) (Supplementary Data). For the sparse PK cohort, lumefantrine capillary sampling occurred on days 7, 14, and 21. Concentrations of artemether, DHA, and lumefantrine were determined using liquid chromatography–tandem mass spectrometry, as previously described [18, 19]. For artemether and DHA, the calibration range was 0.5–200 ng/mL, the lower limit of quantification (LLOQ) was 0.5 ng/mL [18], and the coefficient of variation (CV%) was <10% for each analyte. For lumefantrine, calibration range was 50–20 000 ng/mL, LLOQ was 50 ng/mL, and CV% was <5% [19]. Primary outcome was plasma PK parameters for artemether, DHA, and lumefantrine. For intensive studies, this included the area under the concentration-time curve (AUC0–8 hours for artemether and DHA; AUC0-∞ for lumefantrine), maximal concentration (Cmax), time to Cmax (Tmax), elimination half-life (T1/2), DHA/artemether AUC ratio, C24h (artemether), and C8h (DHA). Noncompartmental analysis was utilized (Supplementary Data).
Treatment Outcomes
Secondary safety and tolerability including adverse events were assessed using National Institute of Allergy and Infectious Diseases Division of AIDS criteria [20]. Secondary treatment outcomes included polymerase chain reaction–unadjusted/adjusted recurrent malaria at 28 and 42 days using standard WHO criteria (Table 4; Supplementary Data) [21]. Gametocytemia was assessed on day 0 and each day of follow-up.
Table 4.
World Health Organization 28-Day Treatment Outcomes Stratified by Antiretroviral Therapy Group
| Outcome | Day 28 Outcomesa |
|||
|---|---|---|---|---|
| HIV-Uninfected Children (n = 181)b | HIV-Infected Children |
|||
| EFV-Based ART (n = 50) | LPV/r-Based ART (n = 70) | NVP-Based ART (n = 61) | ||
| Adequate clinical and parasitological response | 51.4 (93) | 62.0 (31) | 85.7 (60) | 68.9 (42) |
| Late parasitological failure | 39.2 (71) | 30.0 (15) | 14.3 (10) | 19.7 (12) |
| Late clinical failure | 9.4 (17) | 8.0 (4) | 0 (0) | 11.5 (7) |
Data are presented as percentage (No.) with polymerase chain reaction–unadjusted treatment outcome.
Abbreviations: ART, antiretroviral therapy; EFV, efavirenz; HIV, human immunodeficiency virus; LPV/r, lopinavir/ritonavir; NVP, nevirapine.
a Eight children not included in World Health Organization day 28 outcomes analysis due to taking other antimalarials over the course of follow-up and lost to follow-up. One child died due to pneumonia on day 23.
b Significant differences (P < .05) in any treatment failure (late parasitological failure + late clinical failure) outcome between LPV/r-treated children and both the HIV-uninfected and EFV-treated children.
Statistical Analysis
For intensive PK, it was necessary to use 30 subjects for each ART regimen to detect a 35% AUC difference of all analytes (80% power; α = .05). Data were presented as geometric mean (GM) or median (range) as appropriate. A mixed-models repeated-measures analysis was performed for group comparisons. Odds of recurrent malaria were assessed using generalized estimating equations (GEEs) with covariate adjustment for age, parasite density, hemoglobin, sex, and lumefantrine exposure. Mediation analysis was performed to investigate whether associations between treatment regimens and outcomes were transmitted by lumefantrine exposure, using Sobel test on the product of coefficients for the mediation effect [22]. Mediation was presented as the proportion of the effect of the regimen (exposure) on the outcome that acts through the path of the drug concentration (mediator). Risk of recurrent malaria was analyzed using proportional Cox regression with robust sandwich estimator that accounts for repeated episodes adjusted for above covariates. Statistical significance was a 2-sided P value < .05, except for PK parameter pairwise group comparisons where Bonferroni correction was used. Stata version SE12.1 (StataCorp, College Station, Texas) and SAS version 9.4 (SAS Institute, Cary, North Carolina) software programs were used for analyses.
RESULTS
Study Profile
Participants were enrolled into intensive or sparse PK cohorts (Table 1; Supplementary Figure 1). Cohorts were combined for analyses except for intensive PK parameters, as specified. For the intensive cohort, children were screened for enrollment over the course of 219 episodes of malaria; 166 met enrollment criteria, and 145 were included in the final PK/PD analysis (54 HIV-uninfected/91 HIV-infected children). For the sparse PK cohort, children were screened over the course of 448 episodes of malaria; 232 were eligible, with 225 included in the final analysis (134 HIV-uninfected/91 HIV-uninfected children). Children were eligible for enrollment for a single episode of intensive PK sampling, and multiple episodes of sparse PK sampling. Parasite densities were comparable between ART groups, but lower in LPV/r-treated vs HIV-uninfected children (GM, 6917 vs 16 189 parasites/µL; P = .002). Baseline gametocytemia was higher in children on EFV (22.0%) than in those on LPV/r (7.1%) (P = .03).
Table 1.
Demographics of Study Participants
| Variable | HIV-Uninfected | HIV-Infected |
||
|---|---|---|---|---|
| EFV-Based ART | LPV/r-Based ART | NVP-Based ART | ||
| Total episodes, No. | 188 | 182 | ||
| Intensive PK sampling episodes (n = 145) | 54 | 31 | 30 | 30 |
| Sparse PK sampling episodes (n = 225) | 134 | 19 | 40 | 32 |
| Age, y, median (range) | 3.5 (1.1–7.9) | 5.6 (3.1–8.6) | 4.6 (1.4–8.0) | 4.6 (1.4–8.0) |
| Weight, kg, median (range) | 13.8 (9.8–27) | 17.6 (11.4–25.1) | 15.1 (7.7–23.7) | 16.3 (8.5–30.0) |
| Parasite density at diagnosis, geometric mean µL−1 (95% CI)a | 16 189 (12 042–21 763) | 11 671 (6389–21 321) | 6917 (3839–12 463) | 10 568 (5746–19 435) |
| Gametocytes present at diagnosisb | 11.2% | 22.0% | 7.1% | 14.8% |
| Gametocytes present at any point from day 1 to 28c | 17.0% | 36.0% | 20.0% | 29.6% |
| Hemoglobin at diagnosis, g/dL, median (IQR) | 10.8 (9.7–11.4) | 10.5 (9.7–11.3) | ||
| Total lumefantrine dose, mg/kg, median (range) | 65.4 (48.3–96.0) | 74.0 (48.3–96.4) | ||
| Total artemether dose, mg/kg, median (range) | 10.9 (8.1–16.0) | 12.3 (8.1–16.1) | ||
There were no differences in demographic parameters between the intensive and sparse PK sampling cohorts.
Abbreviations: ART, antiretroviral therapy; CI, confidence interval; EFV, efavirenz; HIV, human immunodeficiency virus; IQR, interquartile range; LPV/r, lopinavir/ritonavir; NVP, nevirapine; PK, pharmacokinetic.
a Geometric mean parasite density on the day of diagnosis was significantly different in those on LPV/r vs HIV-uninfected children (P = .002; adjusted for repeated measures); differences not significant for other group comparisons.
b P = .032 for gametocytemia on the day of diagnosis in those on EFV- vs LPV/r-based ART; adjusted for repeated measures.
c Gametocytemia newly developing on days 1–28 was significantly more common in both children on EFV- or NVP-based ART compared with HIV-uninfected children (P = .008; adjusted for repeated measures).
Pharmacokinetics of Artemether and DHA
Pharmacokinetic parameters for the intensive cohort are summarized in Table 2 and Figure 1. Compared to results for HIV-uninfected children, both NNRTIs significantly reduced artemether Cmax and AUC0–8h (2.3- and 2.5-fold, respectively, for EFV; 3.5- and 3.3-fold, respectively, for NVP). DHA AUC0–8h was reduced by 3.4-fold with EFV and approached significance with NVP. Cmax was only reduced (2.8-fold) in the context of EFV. In contrast, LPV/r did not impact artemisinin parameters.
Table 2.
Artemisinin Pharmacokinetics Following a 6-Dose Regimen of Artemether-Lumefantrine in Human Immunodeficiency Virus (HIV)-Uninfected and HIV-Infected Children
| Pharmacokinetic Parameter | HIV-Uninfected |
HIV-Infected |
Ratio (P Value) |
||||
|---|---|---|---|---|---|---|---|
| No ART (n = 51a) | EFV (n = 31) | LPV/r (n = 30b) | NVP (n = 30) | EFV/No ART | LPV/No ART | NVP/No ART | |
| Artemether | |||||||
| Cmax, ng/mL | 35.3 (28.0–44.5) | 15.3 (11.2–21.1) | 26.4 (19.1–36.4) | 10.2 (7.6–13.8) | 0.43 (.0006) | 0.75 (.23) | 0.29 (<.0001) |
| Tmax, h | 2.0 (.7–3.0) | 2.1 (1.1–4.0) | 3.0 (1.1–4.0) | 2.1 (1.1–3.1) | 1.1 (.22) | 1.5 (.18) | 1.1 (.29) |
| AUC0–8h, h × ng/mL | 120 (97.6–147) | 48.5 (37.2–63.1) | 89.7 (68.6–117) | 36.3 (27.8–47.4)c | 0.40 (<.0001) | 0.75 (.16) | 0.30 (<.0001) |
| C24h, ng/mL | 1.2 (.7–1.7) | BLQ (BLQ–.8) | 1.2 (.6–2.5) | BLQ (BLQ–1.1) | <1 (<.0001) | 0.98 (.96) | <1 (<.0001) |
| DHA | |||||||
| Cmax, ng/mL | 66.9 (53.6–83.5) | 24.0 (18.0–32.0) | 55.3 (41.3–74.1) | 45.5 (34.1–60.8) | 0.36 (<.0001) | 0.83 (.39) | 0.68 (.08) |
| Tmax, h | 2.9 (2.0–3.1) | 3.0 (1.1–4.0) | 3.0 (2.0–4.1) | 2.2 (2.0–4.0) | 1.0 (.75) | 1.0 (.99) | 0.80 (.31) |
| AUC0–8h, h × ng/mL | 212 (176–256) | 62.9 (49.2–80.3)c | 171 (133–219) | 137 (107–175) | 0.30 (<.0001) | 0.81 (.25) | 0.65 (.02) |
| C8h, ng/mL | 6.5 (4.6–8.0) | 1.2 (.6–3.4) | 9.3 (4.6–16.5) | 4.5 (2.9–8.8) | 0.19 (<.0001) | 1.42 (.16) | 0.681 (.41) |
| AUC ratio (DHA/AR) | 1.7 (1.3–2.5) | 1.6 (.8–2.2) | 1.9 (1.5–3.4) | 4.1 (2.2–6.9) | 0.94 (.07) | 1.12 (.74) | 2.41 (<.0001) |
Data are presented as geometric mean (90% confidence interval) unless otherwise specified. Significance level: α = .0083 (0.05/6); Tmax, C8h, and C24h reported as median (interquartile range); mixed-models repeated-measures analysis to compare between all groups using log-transformed geometric means.
Abbreviations: AR, artemether; ART, antiretroviral therapy; AUC, area under the concentration-time curve; BLQ, below the limit of quantitation; Cmax, maximal concentration; DHA, dihydroartemisinin; EFV, efavirenz; HIV, human immunodeficiency virus; LPV, lopinavir; NVP, nevirapine; r, ritonavir; Tmax, time to maximal concentration.
a n = 53 for C24h, n = 49 for C8h.
b n = 29 for C8h.
c One subject in the group did not have sufficient points for AUC8h due to being below the lower limit of quantification; AUC last was used.
Figure 1.

Plasma concentration-time profile of artemether (A), dihydroartemisinin (DHA) (B), and lumefantrine (C) in human immunodeficiency virus (HIV)-uninfected (no antiretroviral therapy [ART]) and HIV-infected children (stabilized on either an efavirenz [EFV]-, nevirapine [NVP]-, or lopinavair/ritonavir [LPV/r]–based regimen). Data are represented as median, and values below the limit of quantitation (BLQ) are shown.
Comparisons between ART groups (ratios not shown) revealed that EFV and NVP were associated with lower artemether AUC0–8h compared with LPV/r (reduced 1.9-fold [P = .0096]) and 2.5-fold [P = .0001], respectively). For DHA, AUC0–8h was reduced in children on EFV compared to children on either LPV/r- or NVP-based ART (reduced 2.7-fold [P < .0001] and 2.2-fold [P = .0003], respectively). The DHA/artemether AUC0–8h ratio was 2-fold higher in children on NVP compared with all other groups (P ≤ .0003 for all comparisons).
Pharmacokinetics of Lumefantrine
Pharmacokinetic parameters are summarized in Table 3 and Figure 1. Compared with HIV-uninfected children, children on ART demonstrated highly significant and contrasting changes in lumefantrine AUC0-∞: a 2.1-fold decrease with EFV and 2.1-fold increase with LPV/r, but no change with NVP. Half-life did not change with LPV/r, but was 61% shorter with EFV compared with HIV-uninfected children (P < .0001). Compared to HIV-uninfected children, median day 7 lumefantrine concentrations were 3.1-fold lower in EFV-treated and 3.4-fold higher in LPV/r-treated children (P < .0001 for both).
Table 3.
Lumefantrine Pharmacokinetics Following a 6-Dose Regimen of Artemether-Lumefantrine in Human Immunodeficiency Virus (HIV)-Uninfected and HIV-Infected Children
| Pharmacokinetic Parameter | HIV-Uninfected |
HIV-Infected |
Ratio (P Value) |
||||
|---|---|---|---|---|---|---|---|
| No ART | EFV | LPV/r | NVP | EFV/No ART | LPV/No ART | NVP/No ART | |
| Intensive PK arm | n = 54a | n = 31 | n = 30 | n = 30 | |||
| Cmax, ng/mL, geometric mean (90% CI) | 5782 (5025–6652) | 4725 (3921–5694) | 8905 (7366–11 000) | 6281 (5203–7582) | 0.82 (.15) | 1.54 (.003) | 1.09 (.56) |
| Tmax, h | 4.0 (0.0–8.0) | 4.2 (0.6–8.0) | 4.1 (0.0–8.1) | 8.0 (0.4–8.0) | 1.1 (.05) | 1.0 (.43) | 2.0 (.13) |
| T1/2, h | 64.3 (52.0–120.6) | 23.7 (21.8–46.0) | 98.7 (88.4–119.1) | 63.4 (46.8–111.1) | 0.369 (<.0001) | 1.54 (.13) | 0.988 (.51) |
| AUC0-∞, h × ug/mL, geometric mean (90% CI) | 270 (232–313) | 130 (107–157) | 579 (477–704) | 278 (228–339) | 0.48 (<.0001) | 2.14 (<.0001) | 1.03 (.84) |
| Intensive + sparse sampling PK arms | n = 186b | n = 48c | n = 70d | n = 62e | |||
| C7d, ng/mL | 340 (257–531) | 111 (63–192) | 1140 (515–2220) | 426 (282–733) | 0.30 (<.0001) | 3.4 (<.0001) | 1.3 (.13) |
| C14d, ng/mL | 86 (59–137) | BLQ (BLQ–BLQ) | 323 (174–584) | 115 (68–178) | <1 (<.0001) | 3.8 (<.0001) | 1.3 (.05) |
| C21d, ng/mL | BLQ (BLQ–63) | BLQ (BLQ–BLQ) | 159 (81–275) | BLQ (BLQ–83) | (.002) | >1 (<.0001) | (.28) |
Data are presented as median (interquartile range) unless otherwise specified. Significance level: α = .0083. All comparisons made using mixed-models repeated measures analysis of log-transformed values.
Abbreviations: ART, antiretroviral therapy; AUC, area under the concentration-time curve; BLQ, below the limit of quantitation; CI, confidence interval; Cmax, maximal concentration; EFV, efavirenz; HIV, human immunodeficiency virus; LPV, lopinavir; NVP, nevirapine; PK, pharmacokinetics; r, ritonavir; T1/2, elimination half-life; Tmax, time to maximal concentration.
a n = 52 for T1/2 and AUC.
b n = 183 (C14d) and 176 (C21d).
c n = 46 (C14d) and 44 (C21d).
d n = 65 (C14d and C21d).
e n = 60 (C14d) and 59 (C21d).
Comparing lumefantrine AUC0-∞ between ART groups, those on EFV and NVP had 4.1- and 2.0-fold lower exposure, respectively (P < .0001 for both), and day 7 concentrations were reduced 10.3- and 2.7-fold with EFV and NVP, respectively, compared with LPV/r (P < .0001 for both). Finally, for children on EFV, day 14 and 21 concentrations were markedly lower (76% and 80% were below detection limits, respectively) compared with children on NVP, LPV/r, and no ART (P ≤ .0018 for all comparisons).
Lumefantrine Exposure and Treatment Outcomes
Standardized WHO 28-day outcomes are in Table 4, and the 42-day cumulative risk of recurrent malaria for all groups is depicted in Figure 2 (42-day outcomes in Supplementary Data). Using GEEs adjusted for covariates, the odds of recurrent malaria in HIV-uninfected children was significantly higher than in children on all 3 ART regimens, differences which persisted through day 42 (Table 5; Supplementary Table 1).
Figure 3.
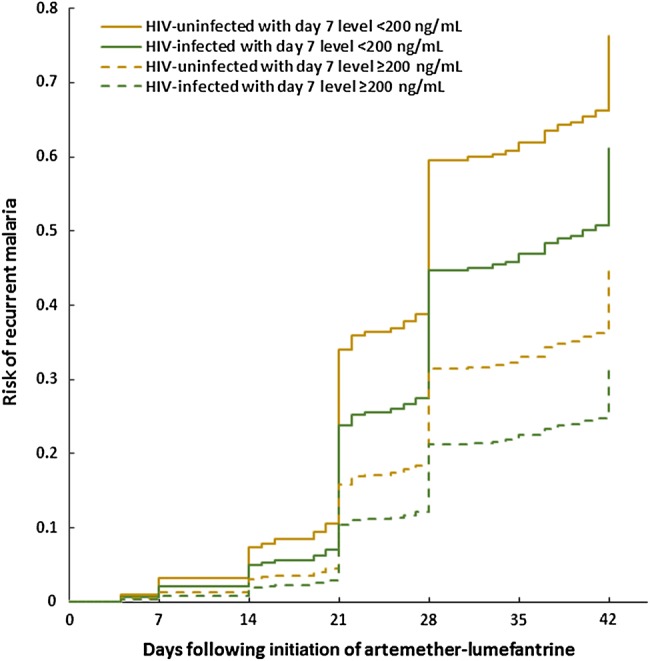
Cumulative risk of recurrent malaria by day 42 following treatment with artemether-lumefantrine stratified by human immunodeficiency virus (HIV) status and a day 7 lumefantrine concentration of 200 ng/mL. Includes children from intensive and sparse cohorts. Risk-adjusted for repeated measures, age, parasite density, and hemoglobin on day 0.
Figure 2.
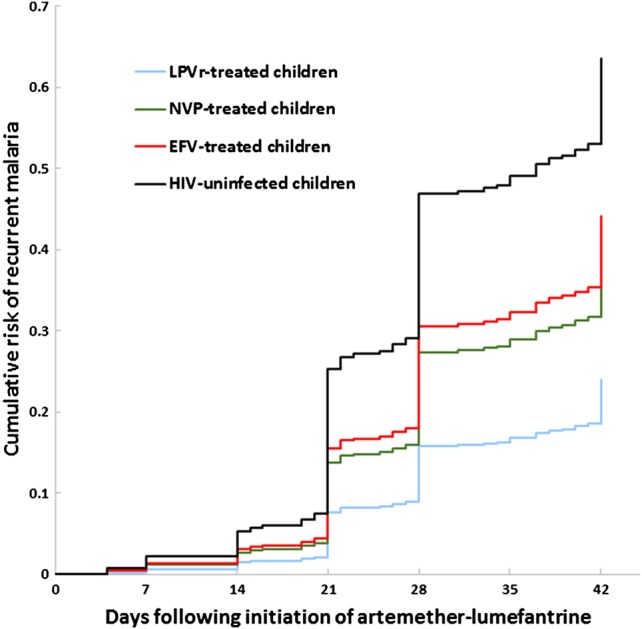
Cumulative risk of recurrent malaria by day 42 following treatment with artemether-lumefantrine stratified by human immunodeficiency virus (HIV) status and antiretroviral therapy regimen. Includes children from intensive and sparse cohorts. Risk-adjusted for repeated measures, age, parasite density, and hemoglobin on day 0. Abbreviations: EFV, efavirenz; LPV/r, lopinavir/ritonavir; NVP, nevirapine.
Table 5.
Generalized Estimating Equation Logistic Regression on Recurrent Malaria by Day 28
| Variables | Recurrent Malaria by Day 28 |
|||||
|---|---|---|---|---|---|---|
| Unadjusted OR (95% CI) | P Value | Adjusted OR Without LR Day 7 (95% CI) | P Value | Adjusted OR With LR Day 7 (95% CI) | P Value | |
| LPV/r | Reference | |||||
| EFV | 3.38 (1.01–11.35) | .049 | 3.74 (1.02–13.74) | .04 | 1.77 (.36–8.81) | .48 |
| NVP | 2.68 (.84–8.56) | .10 | 2.86 (.85–9.60) | .09 | 2.26 (.64–8.05) | .21 |
| HIV-uninfected | 6.32 (2.23–17.95) | .0005 | 6.48 (2.18–19.21) | .0008 | 5.03 (1.58–15.98) | .006 |
| NVP | Reference | |||||
| EFV | 1.26 (.5–3.17) | .62 | 1.31 (.49–3.51) | .59 | 0.78 (.32–2.37) | .66 |
| HIV-uninfected | 2.36 (1.21–4.58) | .01 | 2.27 (1.12–4.60) | .02 | 2.22 (1.10–4.48) | .03 |
| EFV | Reference | |||||
| HIV-uninfected | 1.87 (.86–4.06) | .11 | 1.73 (.73–4.11) | .21 | 2.84 (1.04–7.78) | .04 |
| Parasite density at baseline, log | 1.12 (1.01–1.24) | .03 | 1.04 (.92–1.18) | .53 | 1.03 (.91–1.18) | .61 |
| Hb at baseline, g/dL | 0.82 (.69–.97) | .02 | 0.82 (.68–1.00) | .05 | 0.82 (.67–.99) | .04 |
| Age, y | 0.81 (.7–.95) | .008 | 0.90 (.76–1.07) | .22 | 0.92 (.77–1.10) | .36 |
| LR level at day 7 in ng/mLa | 0.73 (.58–.92) | .008 | 0.75 (.58–.96) | .02 | ||
| LR day 7 ≥ 200 ng/mL | 0.56 (.34–.92) | .02 | ||||
| LR day 7 ≥ 175 ng/mL | 0.57 (.33–.97) | .04 | ||||
| LR day 7 ≥ 280 ng/mL | 0.65 (.41–1.02) | .06 | ||||
Abbreviations: CI, confidence interval; EFV, efavirenz; Hb, hemoglobin; HIV, human immunodeficiency virus; LPV/r, lopinavir/ritonavir; LR, lumefantrine; NVP, nevirapine; OR, odds ratio.
a Natural log-transformed lumefantrine concentration on day 7.
Associations between outcomes and previously defined day 7 protective “thresholds” are noted in Table 5. Day 7 concentrations ≥200 ng/mL or ≥175 ng/mL were associated with a 44% and 43% reduced 28-day odds of recurrence, respectively [23–25]. Neither threshold was predictive at 42 days (Supplementary Table 1).
Comparing outcomes between ART groups revealed that children on EFV-based ART had a 3.7 higher odds (P = .04) of 28-day recurrence than did children on LPV/r-based ART (Table 5). Inclusion of day 7 lumefantrine concentration into the model resulted in a reduction in odds ratio (OR) to 1.8 and loss of statistical significance. Formal mediation analysis indicated that 60% of the difference at day 28 between the EFV and LPV/r groups was attributable to differences in day 7 lumefantrine concentrations (median, 111 vs 1140 ng/mL; P < .0001). Similarly, 27% of the effect of LPV/r-based ART vs no ART was attributable to day 7 concentrations; both 28-day mediation effects were significant (P = .005, P < .0001, respectively). Similar 42-day mediation effects were not observed.
In the intensive cohort, lumefantrine day 7 concentrations were highly correlated with AUC (Pearson r = 0.87; P < .0001). In a GEE analysis for this smaller intensive cohort, the odds of 28-day recurrence were not associated with lumefantrine day 7 concentration (OR, 0.91; 95% confidence interval [CI], .70–1.17; P = .46), while the odds of recurrence was significantly associated with lumefantrine AUC (OR, 0.61; 95% CI, .38–.96; P = .03). Neither parameter was a significant predictor of 42-day outcomes in this smaller cohort.
Treatment Outcomes Adjusted for Genotyping
Ninety-six percent of recurrent malaria episodes were genotyped with 11 cases of recrudescence by day 28 (8.5%), but there was no difference in distribution based on age, HIV status, or ART (3 NVP, 2 LPV/r, 1 EFV, and 5 HIV-uninfected). Lower day 7 concentrations were seen in recrudescent infections (224 ng/mL) compared with adequate clinical and parasitological response (ACPR) (421 ng/mL) (P = .008). Lower day 14 lumefantrine concentrations were seen in recrudescent (62 ng/mL) compared with both new infections and ACPR (80 ng/mL [P = .02] and 115 ng/mL [P = .002], respectively).
Safety and Tolerability of Artemether-Lumefantrine
Artemether-lumefantrine was well tolerated throughout the study with <20 cases of grade 3 laboratory abnormalities in the context of 366 episodes of malaria (eg, reduced platelet or neutrophil count) that were independent of study arm. Abnormalities were followed until grade 2 or lower and were largely attributed to HIV, ART, or malarial infection.
DISCUSSION
For HIV-infected children requiring treatment of uncomplicated P. falciparum malaria with artemether-lumefantrine, selection of ART has a highly significant impact on antimalarial PK exposure and clinical outcomes. We compared PK exposure of artemether-lumefantrine in HIV-infected children on 3 first-line ART regimens to HIV-uninfected children, demonstrating that EFV-based ART results in 2.1- to 3.4-fold reductions in the AUC of artemether, DHA, and lumefantrine. In contrast, LPV/r-based ART had the opposite impact on lumefantrine exposure, increasing the AUC by 2.1-fold, and had no impact on the artemisinins, while the use of NVP-based ART significantly reduced exposure to artemether only. Comparisons of day 7 lumefantrine concentrations between ART groups revealed that concentrations were approximately 10-fold and 3-fold lower in children on EFV and NVP, respectively, compared with children on LPV/r. AUC, a more robust PK parameter of exposure, differed significantly between groups as well.
Differences in exposure to lumefantrine played a critical role in determining treatment outcomes. The risk of recurrent malaria differed significantly between EFV- and LPV/r-based ART, with those on EFV exhibiting a nearly 4-fold higher 28-day odds of recurrent malaria. Mediation analysis demonstrated that the increased 28-day risk was driven largely by lower lumefantrine exposure in those on EFV. This effect was not evident at 42 days, which may reflect that lumefantrine concentrations in all individuals are below a protective threshold by day 42, leaving them equally at risk of new infections in this high-transmission region. We confirmed that concentrations of >175 and >200 ng/mL were predictive of 28-day recurrent malaria; children exceeding these values had approximately 44% lower risk of recurrence. We also demonstrated that those with recurrence compared to ACPR had lower median lumefantrine concentrations [4, 23–25]. Interestingly, in the intensive cohort, AUC was a stronger predictor of outcomes than day 7 concentration. A 1-natural-log increase in AUC yielded a 40% decrease in the 28-day odds of recurrence (P = .03), while day 7 concentration in this smaller group of children was not a significant predictor, underscoring the important information generated by the intensive PK design.
ART also variably altered artemisinin PK. Compared with HIV-uninfected controls, EFV and NVP reduced the AUC0–8h of artemether and DHA up to 3.5-fold, LPV/r had no effect, and NVP increased artemether to DHA conversion by 2-fold; these are differences that may impact outcomes and/or selective pressure for artemisinin resistance, although our study did not assess these associations. Differences in how each ART impacts artemether or lumefantrine PK are attributed to varying effects on CYP3A4, whereas changes for DHA may be due to altered uridine glucuronosyltransferase activity [13, 26, 27].
EFV-based ART is first-line for all children >3 years of age, including adolescents, adults, and pregnant/breastfeeding women. Because EFV reduces exposure to all 3 antimalarial components and increases the risk of recurrent malaria, close clinical follow-up of artemether-lumefantrine–treated children on EFV is necessary. In addition, underexposure may increase the risk for the selection and spread of ACT-resistant parasites, such as was seen following the systematic underdosing of sulfadoxine-pyrimethamine in children [13, 28]. In our study, true recrudescence was documented in 9% of recurrent infections, and although there were no risk differences in recrudescence between ART groups, day 14 lumefantrine concentrations were significantly lower in children with recrudescence vs new infections. With the independent emergence and spread of ACT resistance in Asia, and the selection for resistance-mediating mutations in Africa, ensuring optimal ACT dosing for the large numbers of HIV/malaria-coinfected individuals on EFV-based ART is critical [13, 29–32]. The study of modified dosing regimens of artemether-lumefantrine, such as extension of dosing to 5 days in the setting of EFV, is urgently needed, sentiments supported by others [33, 34]. In contrast, LPV/r appears to be protective against recurrent malaria, and our group and others have demonstrated that LPV/r has in vitro and possible in vivo direct antimalarial effects [4, 35–40]. The use of LPV/r is predicted to avert 278–1043 annual incidences of malaria per 1000 children compared to NNRTI-based regimens in varied sub-Saharan settings [9, 41–43].
In addition to the effects of ART, the use of TMP-SMX reduces one's risk of malaria [11, 44]. Ninety-six percent of our HIV-infected participants were on TMP-SMX, as recommended for all HIV-infected children by the WHO [8]. Indeed, HIV-infected children had a lower overall risk of recurrent malaria compared with HIV-uninfected children in our study, a difference that was evident even when lumefantrine exposure was similar (ie, for NVP-treated and HIV-uninfected children). Overall, our results underscore the benefits of daily TMP-SMX prophylaxis and first-line ART in reducing malaria morbidity in HIV-infected individuals compared with that seen in the pre-ART and pre-TMP-SMX eras [8, 10].
Artemether-lumefantrine and ART interactions have previously been studied, but few studies linked intensive PK with malaria clinical outcomes, and none were in children [33, 45–48]. Day 7 concentrations with EFV, but not LPV/r, were collected in Tanzanian adults, and demonstrated a >19-fold higher risk of recurrence in EFV-treated compared with HIV-infected adults not on ART [45]. PK studies from HIV-infected Ugandan adults reported similar effects of ART on terminal lumefantrine concentrations, although sample sizes were substantially smaller, sampling duration was shorter, subjects were not malaria infected, and no PK/PD associations were possible [33, 46–48]. Our own group initially studied antimalarial–ART interactions in healthy adults; however, the impact of EFV on artemether-lumefantrine was underestimated by >50%, emphasizing the value of conducting studies directly in the most relevant populations, particularly children who exhibit PK distinctions and higher risks of malaria [49–54].
In summary, our results demonstrate that the selection of first-line ART has extensive effects on PK exposure of the most widely adopted ACT, artemether-lumefantrine. Children on EFV-based ART had highly significant reductions in exposure to all 3 antimalarial components, and, compared with children on LPV/r-based ART, experienced higher rates of recurrent malaria. In 2014, 86% of the 58 WHO focus countries adopted EFV-based ART as the preferred first-line regimen, which will be widely used to treat the estimated 25 million ART-eligible individuals in sub-Saharan Africa [7, 9, 55]. Use of artemether-lumefantrine at standard doses for treating malaria in the context of EFV may enhance the emergence and selection of ACT-resistant parasites and, therefore, study of modified dosing regimens such as an extended duration of artemether-lumefantrine in this setting is urgently needed. Until data on a modified artemether-lumefantrine dosing regimen is available, we advise close clinical follow-up for recurrent malaria following treatment with artemether-lumefantrine in the setting of EFV-based ART. In contrast, use of artemether-lumefantrine with LPV/r-based ART in this study led to pronounced increases in lumefantrine exposure with no change in artemisinin exposure, and therefore provides further evidence that LPV/r-based ART provides important benefits for treating malaria in endemic regions [43].
Supplementary Data
Supplementary materials are available at http://cid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. The investigators thank Dr Moses Kamya for his leadership of the Infectious Disease Research Collaboration (IDRC, based in Kampala, Uganda) and his expert consultation; staff of the IDRC including Abel Kakura, Francis Orukan, Catherine Tugaineyo, and Bridget Nzarbara; and Tamara Clark and Melissa Conrad at the University of California, San Francisco (UCSF). We also thank Dr Myaing M. Nyunt from the University of Maryland for her important contributions to the study. In addition, we thank the research staff of Dr Aweeka's laboratory at UCSF including Patricia Lizak, Florence Marzan, and David Gingrich for their excellent analytical work for all pharmacokinetics results. We thank Philip Rosenthal and Grant Dorsey for their comments on the manuscript during its preparation; Martina Wade in Dr Parikh's laboratory for her work on molecular genotyping; and Nitin Sukumar and Fangyong Li from the Yale Center for Analytic Studies for assistance with the statistical analysis. Most importantly, we are grateful to the families in Tororo who participated in all study procedures, without whose support this study would not have been possible.
Disclaimer. The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health (NIH).
Financial support. This work was supported by the NIH (award number R01 HD068174) and by the UCSF Center for AIDS Research (award number P30 AI022763).
Potential conflicts of interest. All authors: No potential conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization. World malaria report. Geneva, Switzerland: WHO, 2014. [Google Scholar]
- 2.Yeka A, Gasasira A, Mpimbaza A et al. Malaria in Uganda: challenges to control on the long road to elimination: I. Epidemiology and current control efforts. Acta Trop 2012; 121:184–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jagannathan P, Muhindo MK, Kakuru A et al. Increasing incidence of malaria in children despite insecticide-treated bed nets and prompt anti-malarial therapy in Tororo, Uganda. Malar J 2012; 11:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achan J, Kakuru A, Ikilezi G et al. Antiretroviral agents and prevention of malaria in HIV-infected Ugandan children. N Engl J Med 2012; 367:2110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. HIV/AIDS fact sheet. Geneva, Switzerland: WHO, 2014. [Google Scholar]
- 6.Joint United Nations Programme on HIV/AIDS. The gap report. Geneva, Switzerland: WHO, 2014. [Google Scholar]
- 7.World Health Organization. Guideline on when to start antiretroviral therapy and on pre-exposure prophylaxis for HIV. Geneva, Switzerland: WHO, 2015. [PubMed] [Google Scholar]
- 8.World Health Organization. Guidelines on post-exposure prophylaxis for HIV and the use of co-trimoxazole prophylaxis for HIV-related infections among adults, adolescents and children: recommendations for a public health approach. Geneva, Switzerland: WHO, 2014. [PubMed] [Google Scholar]
- 9.World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. Geneva, Switzerland: WHO, 2013. [PubMed] [Google Scholar]
- 10.Flateau C, Le Loup G, Pialoux G. Consequences of HIV infection on malaria and therapeutic implications: a systematic review. Lancet Infect Dis 2011; 11:541–56. [DOI] [PubMed] [Google Scholar]
- 11.Church JA, Fitzgerald F, Walker AS, Gibb DM, Prendergast AJ. The expanding role of co-trimoxazole in developing countries. Lancet Infect Dis 2015; 15:327–39. [DOI] [PubMed] [Google Scholar]
- 12.Khoo S, Back D, Winstanley P. The potential for interactions between antimalarial and antiretroviral drugs. AIDS 2005; 19:995–1005. [DOI] [PubMed] [Google Scholar]
- 13.Barnes KI, Watkins WM, White NJ. Antimalarial dosing regimens and drug resistance. Trends Parasitol 2008; 24:127–34. [DOI] [PubMed] [Google Scholar]
- 14.Ilett KF, Ethell BT, Maggs JL et al. Glucuronidation of dihydroartemisinin in vivo and by human liver microsomes and expressed UDP-glucuronosyltransferases. Drug Metab Dispos 2002; 30:1005–12. [DOI] [PubMed] [Google Scholar]
- 15.Wong RP, Salman S, Ilett KF, Siba PM, Mueller I, Davis TM. Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether-lumefantrine treatment outcome. Antimicrob Agents Chemother 2011; 55:1194–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenhouse B, Myrick A, Dokomajilar C et al. Validation of microsatellite markers for use in genotyping polyclonal Plasmodium falciparum infections. Am J Trop Med Hyg 2006; 75:836–42. [PMC free article] [PubMed] [Google Scholar]
- 17.Ashley EA, Stepniewska K, Lindegardh N et al. How much fat is necessary to optimize lumefantrine oral bioavailability? Trop Med Int Health 2007; 12:195–200. [DOI] [PubMed] [Google Scholar]
- 18.Huang L, Olson A, Gingrich D, Aweeka FT. Determination of artemether and dihydroartemisinin in human plasma with a new hydrogen peroxide stabilization method. Bioanalysis 2013; 5:1501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang L, Li X, Marzan F, Lizak PS, Aweeka FT. Determination of lumefantrine in small-volume human plasma by LC-MS/MS: using a deuterated lumefantrine to overcome matrix effect and ionization saturation. Bioanalysis 2012; 4:157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Institute of Allergy and Infectious Diseases—Division of AIDS. Division of AIDS table for grading the severity of adult and pediatric adverse events, 2004. Available at: www.niaid.nih.gov/labsandresources/resources/daidsclinrsrch/documents/daidsaegradingtable.pdf Accessed 23 October 2015.
- 21.World Health Organization. Methods for surveillance of antimalarial drug efficacy, 2009. Available at: http://www.who.int/malaria/publications/atoz/9789241597531/en/ Accessed 23 October 2015.
- 22.MacKinnon DP, Dwyer JH. Estimating mediated effects in prevention studies. Eval Rev 1993; 17:144–58. [Google Scholar]
- 23.Price RN, Uhlemann AC, van Vugt M et al. Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis 2006; 42:1570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White NJ, van Vugt M, Ezzet F. Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether-lumefantrine. Clin Pharmacokinet 1999; 37:105–25. [DOI] [PubMed] [Google Scholar]
- 25.WorldWide Antimalarial Resistance Network Lumefantrine PKPDSG. Artemether-lumefantrine treatment of uncomplicated Plasmodium falciparum malaria: a systematic review and meta-analysis of day 7 lumefantrine concentrations and therapeutic response using individual patient data. BMC Med 2015; 13:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riska P, Lamson M, MacGregor T et al. Disposition and biotransformation of the antiretroviral drug nevirapine in humans. Drug Metab Dispos 1999; 27:895–901. [PubMed] [Google Scholar]
- 27.Back D, Gibbons S, Khoo S. Pharmacokinetic drug interactions with nevirapine. J Acquir Immune Defic Syndr 2003; 34(suppl 1):S8–14. [DOI] [PubMed] [Google Scholar]
- 28.Barnes KI, Little F, Smith PJ, Evans A, Watkins WM, White NJ. Sulfadoxine-pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin Pharmacol Ther 2006; 80:582–96. [DOI] [PubMed] [Google Scholar]
- 29.Takala-Harrison S, Jacob CG, Arze C et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 2015; 211:670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashley EA, Dhorda M, Fairhurst RM et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014; 371:411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dondorp AM, Nosten F, Yi P et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009; 361:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conrad MD, LeClair N, Arinaitwe E et al. Comparative impacts over 5 years of artemisinin-based combination therapies on Plasmodium falciparum polymorphisms that modulate drug sensitivity in Ugandan children. J Infect Dis 2014; 210:344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoglund RM, Byakika-Kibwika P, Lamorde M et al. Artemether-lumefantrine co-administration with antiretrovirals: population pharmacokinetics and dosing implications. Br J Clin Pharmacol 2015; 79:636–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maganda BA, Ngaimisi E, Kamuhabwa AA, Aklillu E, Minzi OM. The influence of nevirapine and efavirenz-based anti-retroviral therapy on the pharmacokinetics of lumefantrine and anti-malarial dose recommendation in HIV-malaria co-treatment. Malar J 2015; 14:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobbs CV, Neal J, Conteh S et al. HIV treatments reduce malaria liver stage burden in a non-human primate model of malaria infection at clinically relevant concentrations in vivo. PLoS One 2014; 9:e100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hobbs CV, Voza T, Coppi A et al. HIV protease inhibitors inhibit the development of preerythrocytic-stage plasmodium parasites. J Infect Dis 2009; 199:134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Redmond AM, Skinner-Adams T, Andrews KT et al. Antimalarial activity of sera from subjects taking HIV protease inhibitors. AIDS 2007; 21:763–5. [DOI] [PubMed] [Google Scholar]
- 38.Porter KA, Cole SR, Eron JJ Jr et al. HIV-1 protease inhibitors and clinical malaria: a secondary analysis of the AIDS Clinical Trials Group A5208 study. Antimicrob Agents Chemother 2012; 56:995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White NJ, Stepniewska K, Barnes K, Price RN, Simpson J. Simplified antimalarial therapeutic monitoring: using the day-7 drug level? Trends Parasitol 2008; 24:159–63. [DOI] [PubMed] [Google Scholar]
- 40.Parikh S, Gut J, Istvan E, Goldberg DE, Havlir DV, Rosenthal PJ. Antimalarial activity of human immunodeficiency virus type 1 protease inhibitors. Antimicrob Agents Chemother 2005; 49:2983–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Violari A, Cotton MF, Gibb DM et al. Early antiretroviral therapy and mortality among HIV-infected infants. N Engl J Med 2008; 359:2233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruel TD, Kakuru A, Ikilezi G et al. Virologic and immunologic outcomes of HIV-infected Ugandan children randomized to lopinavir/ritonavir or nonnucleoside reverse transcriptase inhibitor therapy. J Acquir Immune Defic Syndr 2014; 65:535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenhalgh S, Ndeffo M, Galvani AP, Parikh S. The epidemiological impact of HIV antiretroviral therapy on malaria in children. AIDS 2015; 29:473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gasasira AF, Kamya MR, Ochong EO et al. Effect of trimethoprim-sulphamethoxazole on the risk of malaria in HIV-infected Ugandan children living in an area of widespread antifolate resistance. Malar J 2010; 9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maganda BA, Minzi OM, Kamuhabwa AA, Ngasala B, Sasi PG. Outcome of artemether-lumefantrine treatment for uncomplicated malaria in HIV-infected adult patients on anti-retroviral therapy. Malar J 2014; 13:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Byakika-Kibwika P, Lamorde M, Mayito J et al. Significant pharmacokinetic interactions between artemether/lumefantrine and efavirenz or nevirapine in HIV-infected Ugandan adults. J Antimicrob Chemother 2012; 67:2213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Byakika-Kibwika P, Lamorde M, Okaba-Kayom V et al. Lopinavir/ritonavir significantly influences pharmacokinetic exposure of artemether/lumefantrine in HIV-infected Ugandan adults. J Antimicrob Chemother 2012; 67:1217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kredo T, Mauff K, Workman L et al. The interaction between artemether-lumefantrine and lopinavir/ritonavir-based antiretroviral therapy in HIV-1 infected patients. BMC Infect Dis 2016; 16:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.German P, Greenhouse B, Coates C et al. Hepatotoxicity due to a drug interaction between amodiaquine plus artesunate and efavirenz. Clin Infect Dis 2007; 44:889–91. [DOI] [PubMed] [Google Scholar]
- 50.German P, Parikh S, Lawrence J et al. Lopinavir/ritonavir affects pharmacokinetic exposure of artemether/lumefantrine in HIV-uninfected healthy volunteers. J Acquir Immune Defic Syndr 2009; 51:424–9. [DOI] [PubMed] [Google Scholar]
- 51.Huang L, Parikh S, Rosenthal PJ et al. Concomitant efavirenz reduces pharmacokinetic exposure to the antimalarial drug artemether-lumefantrine in healthy volunteers. J Acquir Immune Defic Syndr 2012; 61:310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sambol NC, Yan L, Creek DJ et al. Population pharmacokinetics of piperaquine in young Ugandan children treated with dihydroartemisinin-piperaquine for uncomplicated malaria. Clin Pharmacol Ther 2015; 98:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mwesigwa J, Parikh S, McGee B et al. Pharmacokinetics of artemether-lumefantrine and artesunate-amodiaquine in children in Kampala, Uganda. Antimicrob Agents Chemother 2010; 54:52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parikh S, Fehintola F, Huang L et al. Artemether-lumefantrine exposure in HIV-infected Nigerian subjects on nevirapine-containing antiretroviral therapy. Antimicrob Agents Chemother 2015; 59:7852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doherty M, Beusenberg M, Asamoah-Odei E et al. Rapid uptake and adoption of the WHO 2013 consolidated ARV guideline recommendations: paving the way to achieving the 90/90/90 global target. J Int AIDS Soc 2015; 18:26–7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.