

Abstract
Chromosomes in all organisms are highly organized and divided into multiple chromosomal interaction domains, or topological domains. Regions of active, high transcription help establish and maintain domain boundaries, but precisely how this occurs remains unclear. Here, using fluorescence microscopy and chromosome conformation capture in conjunction with deep sequencing (Hi‐C), we show that in Caulobacter crescentus, both transcription rate and transcript length, independent of concurrent translation, drive the formation of domain boundaries. We find that long, highly expressed genes do not form topological boundaries simply through the inhibition of supercoil diffusion. Instead, our results support a model in which long, active regions of transcription drive local decompaction of the chromosome, with these more open regions of the chromosome forming spatial gaps in vivo that diminish contacts between DNA in neighboring domains. These insights into the molecular forces responsible for domain formation in Caulobacter likely generalize to other bacteria and possibly eukaryotes.
Keywords: chromosomal domains, transcription, chromosome organization, Hi‐C, supercoil diffusion
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination; Microbiology, Virology & Host Pathogen Interaction
Introduction
In all living organisms, chromosomes are not haphazardly packed inside the cells but rather are compacted into highly organized structures, presumably to support the execution and fidelity of DNA‐based processes such as replication, transcription, repair, recombination, and chromosome segregation. The three‐dimensional organization of chromosomes has recently emerged through the microscopic analysis of fluorescently tagged DNA loci and by chromosome conformation capture in conjunction with deep sequencing (Hi‐C) (Umbarger et al, 2011; Le et al, 2013; Wang et al, 2013; Badrinarayanan et al, 2015). Hi‐C provides quantitative information about the spatial proximity of genomic loci in vivo by measuring the frequency with which different regions of the chromosome can be cross‐linked together (Dekker et al, 2002; Lieberman‐Aiden et al, 2009). The first application of Hi‐C to bacteria examined the chromosome of Caulobacter crescentus (Le et al, 2013). Caulobacter cells can be easily synchronized with respect to the cell cycle and DNA replication, enabling Hi‐C analysis of large, nearly homogeneous populations of cells.
Hi‐C analysis of wild‐type Caulobacter cells revealed a pattern of DNA–DNA interactions consistent with the origin of replication being located at one cell pole, the terminus near the opposite pole, and the two chromosomal arms running in parallel and colinearly down the long axis of the cell (Le et al, 2013). Additionally, Hi‐C and recombination‐based assays demonstrated that the Caulobacter chromosome is subdivided into a series of chromosomal interaction domains (CIDs), averaging ~120 kb in length, where loci within a domain interact more frequently with other loci in the same domain. Chromosomal domains have also recently been reported in B. subtilis using Hi‐C (Marbouty et al, 2015; Wang et al, 2015). CIDs represent just one level of chromosome organization in bacteria. Multiple CIDs are often nested, with several consecutive CIDs comprising a larger entity that may correspond to the so‐called macrodomains first documented in E. coli (Valens et al, 2004). The genomic DNA within each CID is also further organized, likely forming plectonemic loops of supercoiled DNA (Deng et al, 2005). Individual supercoil domains have been estimated to be ~10 kb on average meaning there may be several supercoil domains within a given CID (Higgins et al, 1996). Such supercoil domains are difficult to see by Hi‐C given their size, which is close to or below the resolution of Hi‐C. Additionally, supercoils can diffuse, so the genomic distribution of supercoil domains may not be consistent across a population of cells.
Domains similar to CIDs, called topologically associated domains (TADs), have been documented in a range of eukaryotic cells (reviewed in Dekker & Heard, 2015). In both bacteria and eukaryotes, domain boundaries are frequently associated with highly transcribed genes. TAD boundaries have also been correlated with several other factors, including CTCF and cohesin (Dekker & Heard, 2015), but the factors ultimately responsible for TAD formation are still unclear. However, studies of C. crescentus indicate that chromosomal interaction domains in bacteria (hereafter referred to as chromosomal domains or just domains for brevity) are frequently created directly by highly transcribed genes (Le et al, 2013). In addition to a strong correlation between domain boundaries and the locations of very highly expressed genes, inhibiting transcription in Caulobacter with rifampicin almost completely eliminates domain boundaries (Le et al, 2013). Strikingly, the insertion of a highly expressed gene, rsaA, was also sufficient to produce a new domain boundary at the site of insertion (Le et al, 2013). Nevertheless, the precise relationship between gene expression and chromosomal domains is uncertain. Not all highly expressed genes form domain boundaries, and for those that do, the molecular or physical mechanisms responsible remain unknown.
Here, we use Hi‐C along with microscopy‐based analyses of individual loci, to characterize domain boundary formation in Caulobacter. We show that highly expressed genes must be of a sufficient length to create barriers in vivo that prevent long‐range DNA interactions. We provide evidence that the transcription of long, highly expressed genes, and not their translation, which can occur concurrently with transcription in bacteria, is critical to domain boundary formation. We show that longer genes are slightly more efficient than shorter genes in blocking supercoil diffusion, helping to establish boundaries, although our results indicate that long, highly expressed genes can establish boundaries primarily by creating decompacted, extensible regions of the chromosome that physically separate and insulate the flanking domains. In sum, our findings reveal a primary mechanism driving the formation of chromosomal domains in Caulobacter and are likely applicable to all bacterial chromosomes and possibly eukaryotes as well.
Results
Starvation‐induced changes in global transcription redistribute domain boundaries
Prior studies indicating that regions of high gene expression in Caulobacter often correspond to chromosomal domain boundaries were done exclusively in a rich growth medium. To confirm that gene expression globally affects chromosome organization in Caulobacter, we first generated and compared Hi‐C contact maps for cells growing in exponential phase or following nutrient starvation when gene expression patterns change dramatically (Fig 1). Caulobacter cells were starved of carbon and nitrogen by resuspending a population of synchronized swarmer cells in M2 salts for 90 min before fixing with 1% formaldehyde and performing Hi‐C. To analyze the Hi‐C data, we divided the genome into 10‐kb bins and assigned to the appropriate bins the interaction frequencies for all informative ligation products that were sequenced. Interaction frequencies were visualized as a contact map with each matrix position, m ij, reflecting the relative interaction frequency of loci in bin i with those in bin j. The contact map contains two prominent diagonals (Fig 1A and Appendix Fig S1A). The main diagonal represents high‐frequency interactions between DNA loci on the same arm of the chromosome, i.e. intra‐arm interactions. The less prominent diagonal reflects lower‐frequency interactions between loci on one arm of the chromosome and those on the opposite arm, i.e. inter‐arm interactions (Appendix Fig S1A).
Figure 1. Starvation induces a global change in the locations of domain boundaries in Caulobacter crescentus .
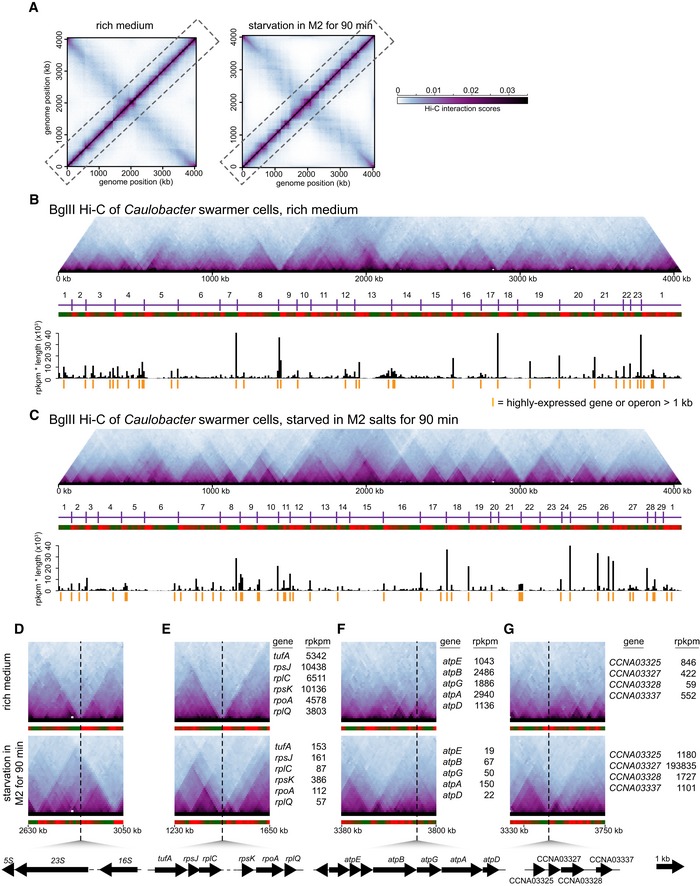
-
ANormalized Hi‐C contact maps showing contact frequencies between 10‐kb bins across the Caulobacter genome during exponential growth (Le et al, 2013) and following nutrient starvation. Axes indicate genome position of each bin.
-
B, CHi‐C contact maps for intra‐chromosomal arm interactions (dashed box in panel A) rotated 45° clockwise. Images were resized in each dimension after rotation, but otherwise unprocessed relative to (A). CID boundaries are marked with vertical purple lines and numbered. Directional preference plots were used to assign domain boundaries (see Materials and Methods and Appendix Fig S1B and C). Leftward and rightward preferences are shown as green and red bars, respectively. Abrupt transitions from leftward to rightward preference correspond to chromosomal interaction domain boundaries. Plots of gene expression “rpkpm * transcript length” for each 10‐kb bin along the chromosome are shown beneath the directional preference plots. Genomic locations of long, highly expressed genes or operons (see Dataset EV2) are indicated by orange vertical lines.
-
D–GComparison of normalized Hi‐C contact maps for regions of the Caulobacter chromosome during exponential growth and following starvation. The black dashed line shows the genomic location of (D) an rRNA locus, (E) a ribosomal protein gene cluster, (F) an ATP synthase gene cluster, and (G) starvation‐induced genes CCNA03325, CCNA03327, CCNA03328, and CCNA03337.
Both Hi‐C contact maps showed regions of high self‐interaction or chromosomal interaction domains. These domains appear as squares along the main diagonal, or as triangles if the contact map is rotated (Fig 1B and C, and Appendix Fig S1B and C). The existence of domains was independently verified previously using a recombination assay, and they are not artefacts of restriction site distribution (Le et al, 2013). Additionally, we verified here that domains do not arise because of differences in the cross‐linking efficiency of certain regions of the genome. If cross‐linking were substantially higher for certain regions of the genome, those regions would have significantly higher numbers of reads. However, the distribution of sequencing reads was relatively uniform across the entire genome with no correlation between reads and domain boundary location (Appendix Fig S1D).
We called the locations of domain boundaries by analyzing patterns of directional preference for individual bins (see Materials and Methods). Briefly, domain boundaries correspond to locations in which the directional preference of individual bins switches abruptly from negative to positive (Figs 1B and C, and EV1 and Dataset EV1). Although the Hi‐C maps for growing and starved cells were broadly similar, a careful inspection of the main diagonals revealed clear differences in domain organization (Fig 1B–G). For the starved cells shown in Fig 1C, there were 29 domains, whereas exponentially growing cells had 23, with 11 domain boundaries in common (Dataset EV1). Using RNA‐Seq to quantify mRNA abundances in starved and exponentially growing cells, we found a clear correspondence between the changes in gene expression and changes in domain boundary locations (Fig 1B–G). For instance, domain boundaries associated with ribosomal RNA (rRNA) clusters and genes encoding ribosomal proteins in growing cells became significantly less prominent in starved cells (Figs 1D and E, and EV1), and the transcription of these genes decreased significantly upon starvation, with the rpkpm values for ribosomal protein genes typically being greater than 3,800 in growing cells but less than 400 in starved cells (Dataset EV2). Similarly, domain boundaries found exclusively in starved cells (Figs 1B and C, and EV1) were typically associated with starvation‐induced genes. For instance, the genes CCNA02866, CCNA03169, and CCNA03327 (Fig 1G and Dataset EV2) exhibited dramatic increases in expression from 131, 365, and 422 rpkpm to 12,652, 184,180, and 193,835 rpkpm, respectively, when shifted from rich medium to starvation conditions, and each gene was located near a starvation‐specific domain boundary. We also found that the domain boundaries in starved cells were generally sharper and more well defined than in exponentially growing cells (Fig 1B–G). The reason for this increase is unclear, but likely not due to ATP depletion in starvation conditions reducing DNA movement as DNP‐induced dissipation of ATP alone did not result in more well‐defined boundaries (data not shown).
Figure EV1. Starvation induces a global change in locations of domain boundaries in Caulobacter crescentus .
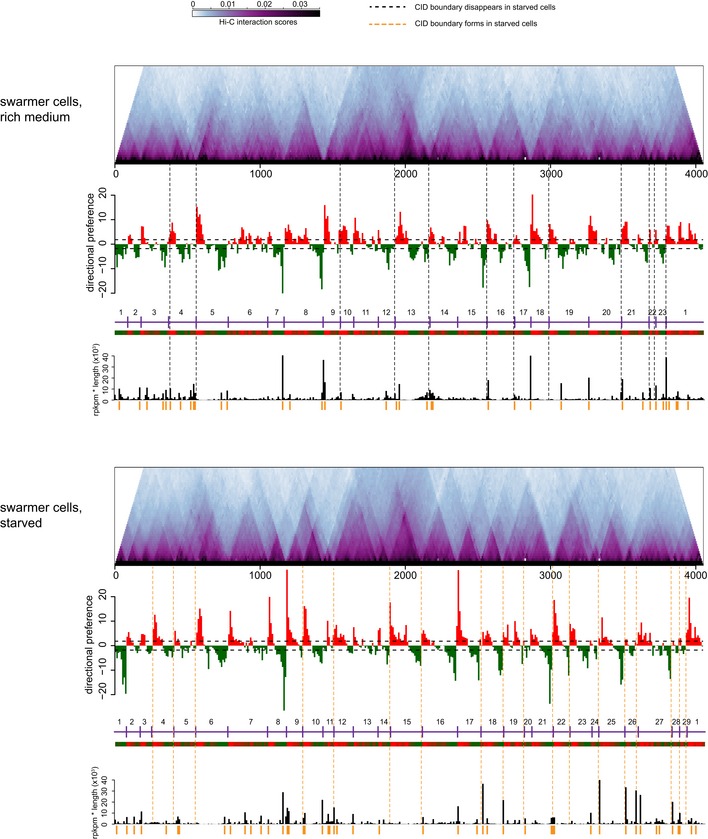
Hi‐C contact maps for intra‐chromosomal arm interactions rotated 45o clockwise. Directional preference plots were used to assign domain boundaries (see Appendix Supplementary Materials and Methods). Leftward and rightward preferences are shown as green and red bars, respectively. Abrupt transitions from leftward to rightward preference correspond to chromosomal interaction domain boundaries. Vertical black and orange dashed lines indicate boundaries specific to growing or starved cells, respectively. The directional preference plot and “rpkm * transcript length” plot corresponding to each condition are shown beneath the Hi‐C plots. For a complete tabulation of CID boundary locations, transcript lengths, and expression levels, see Dataset EV2.
The differences in domain boundaries between starved and exponentially growing cells were reproducible (Dataset EV1). A second, independent replicate of the Hi‐C for starved cells had 26 domain boundaries (Fig EV1), with 25 domain boundaries in common between the two replicates (Fig 1C), and again with 11 boundaries in common with those of exponentially growing cells. The correlation between the directional preference values derived from each replicate was 0.82 (P < 10−15). In contrast, the correlation between directional preference values for starved cells and exponentially growing cells was 0.34 and 0.4, respectively, for the two independent replicates of starved cells (Dataset EV1).
Length of a highly expressed transcript determines the strength of a domain boundary
Although high gene expression and domain boundaries are clearly correlated, not all highly transcribed genes were associated with domain boundaries, in both growing and starved cells. For example, genes encoding tRNAs are very highly expressed in fast‐growing cells, but were rarely associated with domain boundaries. Notably, tRNAs (~76–90 bp) are much shorter than an average bacterial gene (~1,000 bp). Moreover, we observed that the highly expressed genes associated with domain boundaries were often particularly long genes or operons, such as rsaA (3 kb), the ATP synthase gene cluster (~5.8 kb), the large ribosomal protein gene cluster (~14.5 kb), and rRNA loci (~5 kb). Thus, we hypothesized that transcript length, not just abundance, affects domain boundary formation.
To test this hypothesis, we first calculated “rpkpm * transcript length” for each gene or operon in the Caulobacter genome and plotted these values as a function of genome position (Figs 1B and C, and EV1 and Dataset EV2). For growing cells, 15 of the 18 highest (and 23 of the 40 highest) “rpkpm * transcript length” values were for genes or operons positioned at domain boundaries (P < 10−8 in each case, binomial test). For starved cells, the set of genes with the highest “rpkpm * transcript length” values changed substantially, but again, 15 of the 18 highest (and 24 of the 40 highest) values were for genes or operons located at domain boundaries (P < 10−8 in each case, binomial test). We found that there were 12 chromosomal domain boundaries specific to cells grown in a rich medium (Fig EV1 and Dataset EV2). Of these 12 boundaries, 11 coincided with long (> 1 kb) genes that were highly expressed in rich media but not in starved cells. Conversely, there were 18 boundaries specific to starved cells; 14 of these 18 coincided with long genes that were highly expressed in starved, but not nutrient‐replete cells. Taken together, these analyses support the hypothesis that domain boundaries frequently arise at the positions of long, highly expressed genes.
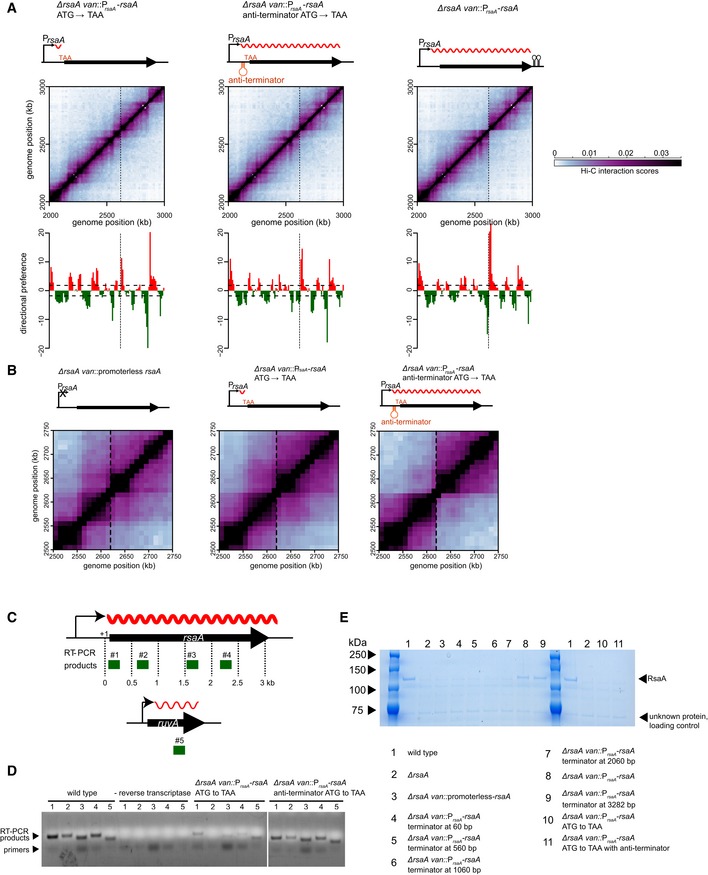
To further test this hypothesis, we ectopically inserted the ~3‐kb coding region for rsaA, a highly expressed gene encoding the Caulobacter S‐layer, together with its promoter at the vanillate utilization (van) locus. The van locus is normally transcriptionally inactive and resides within a chromosomal domain, but the insertion of PrsaA‐rsaA was sufficient to generate a sharp new domain boundary at this location (Fig 2) (Le et al, 2013). To alter the length of the transcribed region, we inserted a cassette containing two TAA stop codons and two transcriptional terminators 60‐, 560‐, 1,060‐, or 2,060‐bp downstream from the rsaA transcription start site (Fig 2). RT–PCR analysis of these strains confirmed that (i) transcription was efficiently terminated at the site of the inserted transcriptional terminator and (ii) levels of the truncated rsaA transcripts were comparable to the wild‐type rsaA in each case (Fig EV2A and B). We also used Western blotting with an anti‐FLAG antibody to confirm that truncated RsaA variants harboring an N‐terminal FLAG tag had the predicted molecular weights and were produced at roughly comparable levels (Fig EV2C).
Figure 2. Longer transcripts are more effective in establishing chromosomal domain boundaries.
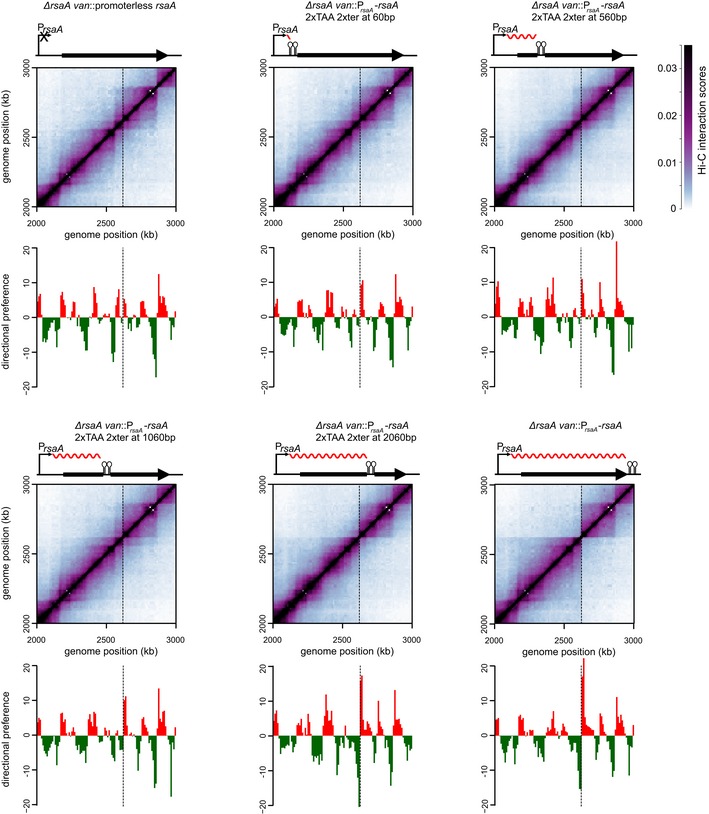
Normalized Hi‐C contact maps for ΔrsaA van::promoterless rsaA and derivatives of ΔrsaA van::PrsaA‐rsaA with transcription terminators inserted 60, 560, 1,060, or 2,060 bp from the transcription start site. Only the region of the genome containing the van locus (dashed line) is shown. Above each Hi‐C contact map is a schematic of the corresponding rsaA construct with the transcription terminator shown as a black hairpin and transcript as a wavy red line. Below each map is the corresponding directional preference plot.
Figure EV2. Expression levels and transcript lengths of wild‐type rsaA and its derivatives.
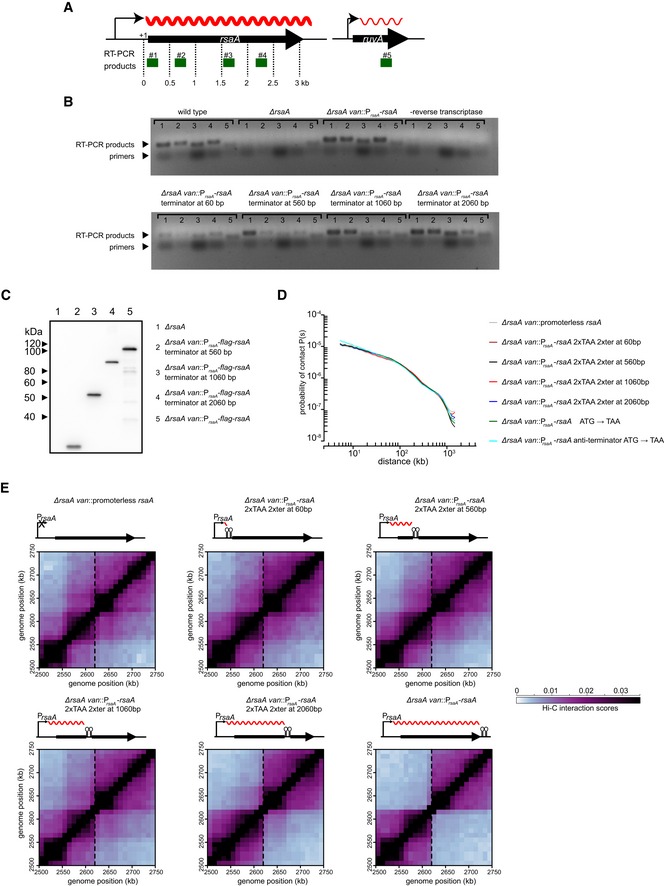
- A schematic showing the location of RT–PCR products (green) examined for rsaA and the ruvA control. Transcripts are denoted as wavy red lines.
- RT–PCR products from RNA extracted from strains harboring the wild‐type rsaA or the derivative indicated. Samples where no reverse transcriptase was added were included to control for genomic DNA contamination.
- Western blot of FLAG‐tagged RsaA and its various derivatives indicated. Anti‐FLAG primary antibody and anti‐mouse secondary antibody were used.
- Expanded view of the region surrounding the site of insertion (dashed line) for each strain examined in Fig 2.
We then examined domain boundary induction by each rsaA variant using Hi‐C analysis of swarmer cells from each strain grown to exponential phase. The Hi‐C maps were very similar (Fig EV2D) except at the van locus, with a sharp domain boundary present when the terminator cassette was at least 2,060 bp from the transcriptional start site (Figs 2 and EV2E). There was no domain boundary or a very weak boundary when the transcript length was engineered to be < 2,060 bp. To quantify the sharpness of the boundary formed, we calculated the difference in directional preference between the two bins flanking the site of insertion for each strain constructed. This value increased from 6.9 in the case of a promoterless rsaA to 12.0, 12.9, 14.2, 27.9, and 32.3 in the strains producing rsaA transcripts of length 60, 560, 1,060, 2,060, or 3,060 bp, respectively. Taken together, our results indicate that the length of a highly expressed gene plays a critical role in determining whether it creates a chromosomal domain boundary.
Translating ribosomes do not contribute to domain boundary formation
Long, highly expressed genes could, in principle, form domain boundaries if multiple ribosomes, which can begin translating even before transcription finishes in bacteria, sterically block or diminish the interactions between flanking loci. To test this possibility, we sought to eliminate ribosome binding to the mRNA of an ectopically inserted rsaA gene by mutating the ATG start codon to a TAA stop codon (Figs 3 and EV3A and B). This mutation strongly reduced the intensity of the domain boundary at the van locus; however, transcription of this mutant rsaA (ATG→TAA) was severely diminished, likely due to Rho‐dependent transcription termination (Figs 3 and EV3B–D). Thus, to retain high levels of transcription while preventing translation, we inserted a Caulobacter anti‐termination sequence (Arnvig et al, 2008) into the leader sequence of rsaA. RT–PCR analysis confirmed that rsaA transcription in this strain (anti‐termination + ATG→TAA) was restored to near wild‐type levels (Fig EV3B–D), and S‐layer protein extraction confirmed the absence of rsaA translation (Fig EV3E). Cells were then synchronized and subsequently analyzed by Hi‐C. The resulting Hi‐C contact map indicated that a sharp chromosomal domain boundary was clearly formed at the site harboring this untranslated rsaA construct. As before, we calculated the difference in directional preference for the two genomic bins flanking the insertion site. For the anti‐termination + ATG→TAA construct, this difference was 25.6, whereas for the ATG→TAA construct lacking the anti‐terminator, this value was only 12.6, with the promoterless rsaA construct producing a value of 6.9. These analyses demonstrate that ribosome binding does not contribute significantly to domain boundary formation in Caulobacter (Fig 3).
Figure 3. The recruitment of ribosomes to highly expressed genes does not contribute to domain boundary formation.
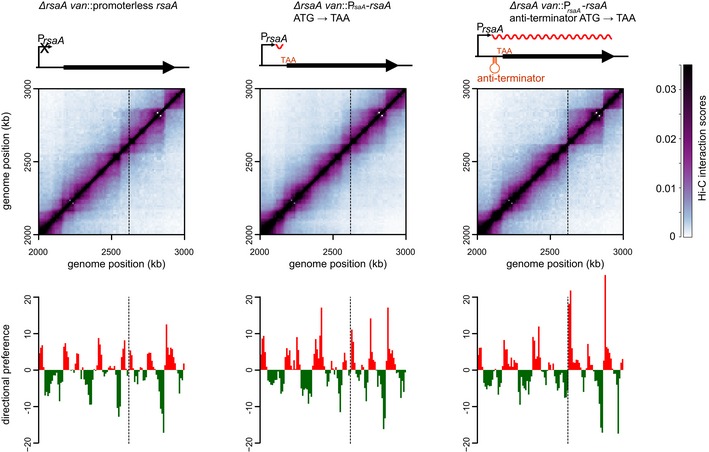
Normalized Hi‐C contact maps for ΔrsaA van::promoterless rsaA, ΔrsaA van::PrsaA‐rsaA (ATG→TAA), and ΔrsaA van::PrsaA‐rsaA (anti‐termination + ATG→TAA) cells. Only the region of the genome containing the van locus (dashed line) is shown. Above each Hi‐C contact map is a schematic of the corresponding rsaA construct. The transcription anti‐terminator is shown as a red hairpin, the start codon mutated to TAA is in red font, and the transcript produced is shown as a wavy red line. Below each map is the corresponding directional preference plot.
Figure EV3. Expression levels and transcript lengths of rsaA variants.
- Expanded view of the region surrounding the site of insertion (dashed line) for each strain examined in Fig 3.
- A schematic showing the location of RT–PCR products (green) examined for rsaA and a ruvA control locus. mRNA transcripts are denoted with wavy red lines.
- RT–PCR products from RNA extracted from a strain expressing wild‐type rsaA or the derivative indicated. Wild‐type samples where no reverse transcriptase was added were included to control for genomic DNA contamination.
- SDS–PAGE showing protein level after S‐layer extraction by low pH for the strains indicated below. The position of RsaA on the gel is denoted by a black arrow.
We also tested the role of ribosomes and translation in chromosome domain formation using Hi‐C on cells treated with chloramphenicol, an inhibitor of peptide chain elongation. Caulobacter cells growing in exponential phase were synchronized and then treated with 50 μg/ml chloramphenicol for 30 min before fixation. The contact map of chlor‐treated cells had the same global, X‐shaped pattern seen with untreated cells (Fig 4A). Most domain boundaries seen in the control cells were retained in the chlor‐treated sample although their strengths were substantially reduced and most boundaries were considerably less well defined (Fig 4B). This effect likely stems from Rho‐dependent termination, which attenuates transcription when translation is blocked (Richardson et al, 1975). The two domain boundaries that remained prominent, and were in fact slightly sharper, in the chlor‐treated cells correspond to the 10‐kb bins harboring the two rRNA clusters (Fig 4B), which are, of course, not translated. Consistent with sharper boundaries near these rRNA loci, RT–qPCR analysis indicated that rRNA levels increased ~2–3‐fold in chlor‐treated cells relative to the untreated control cells (data not shown). Prior work in E. coli has also found that translation‐inhibited cells increase rRNA promoter activity, likely as an attempt to overcome inhibition (Shen & Bremer, 1977; Schneider et al, 2002). Collectively, our results support the conclusion that long, highly active genes induce chromosomal domain boundaries and that the translation of such genes is not necessary for boundary formation or maintenance.
Figure 4. The effect of inhibiting translation on chromosomal domain boundaries.
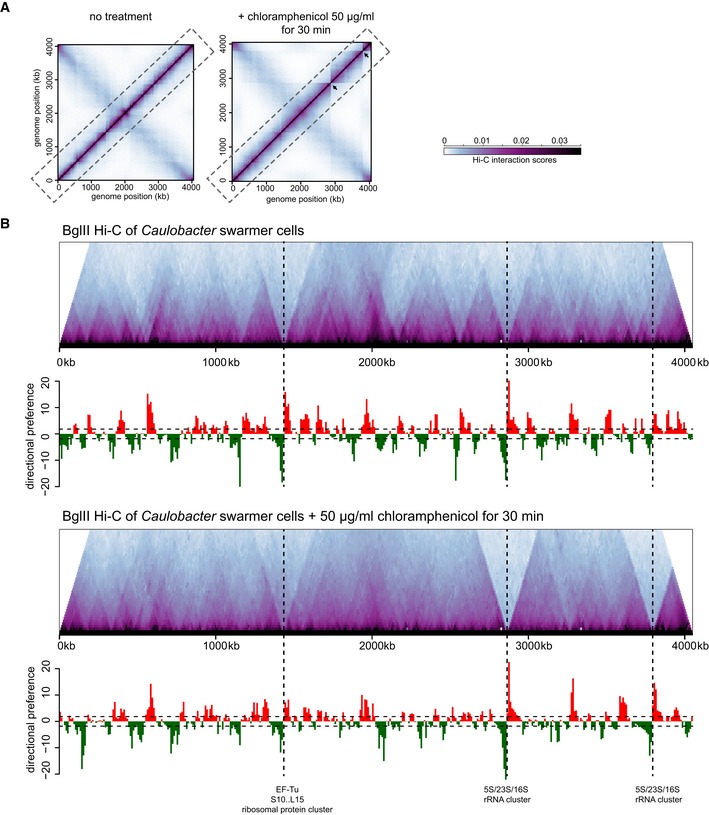
- Normalized Hi‐C contact maps for untreated (left) and chloramphenicol‐treated cells (right).
- Hi‐C contact maps for intra‐chromosomal arm interactions (dashed box in panel A) rotated 45° clockwise with directional preference plots below. Images were resized in each dimension after rotation, but otherwise unprocessed relative to (A). Leftward and rightward preferences are shown as green and red bars, respectively. Vertical dashed black lines indicate the positions of the largest ribosomal protein cluster and the two rRNA loci in the Caulobacter chromosome.
Transcription rate and transcript length contribute to an inhibition of supercoil diffusion
The diffusion of supercoils can drive DNA–DNA contacts by bringing together within a plectoneme two loci that are relatively distant in the primary sequence. As gene expression can interfere with supercoil diffusion (Deng et al, 2005; Booker et al, 2010), we sought to determine whether such a mechanism contributes to the formation of domains seen by Hi‐C. To quantify supercoil diffusion, we developed a recombination assay based on the γδ resolvase TnpR (Higgins et al, 1996; Deng et al, 2005) (Fig 5A). Briefly, two resolution (res) sites were inserted in the same orientation flanking a tetracycline resistance (tet R) cassette with TnpR expressed from a cumate‐inducible promoter. TnpR, which depends on supercoil diffusion to align the two res sites (Wells & Grindley, 1984; Benjamin et al, 1985), can drive site‐specific recombination, leading to excision of the tet R cassette, rendering cells tetracycline sensitive. We inserted the res‐tet R ‐res construct at the van locus and expressed TnpR from a plasmid for 2 h. Cells were then spotted on plates +/− tetracycline to determine the recombination frequency, as a proxy for the efficiency of supercoil diffusion. Recombination was highly efficient, with TnpR induction driving a 4‐log drop in plating viability in the presence of tetracycline (Fig 5B). Recombination was dependent on res‐binding as the induction of a TnpR variant lacking the DNA‐binding domain (TnpRΔDBD) did not reduce plating viability on tetracycline (Appendix Fig S2A). We also confirmed that recombination is promoted by negative supercoiling as treatment with novobiocin, an inhibitor of DNA gyrase, diminished the viability drop and thus recombination associated with TnpR induction (Appendix Fig S2B).
Figure 5. The ability of different rsaA constructs to inhibit supercoil diffusion does not correlate with their ability to establish a chromosomal domain boundary.
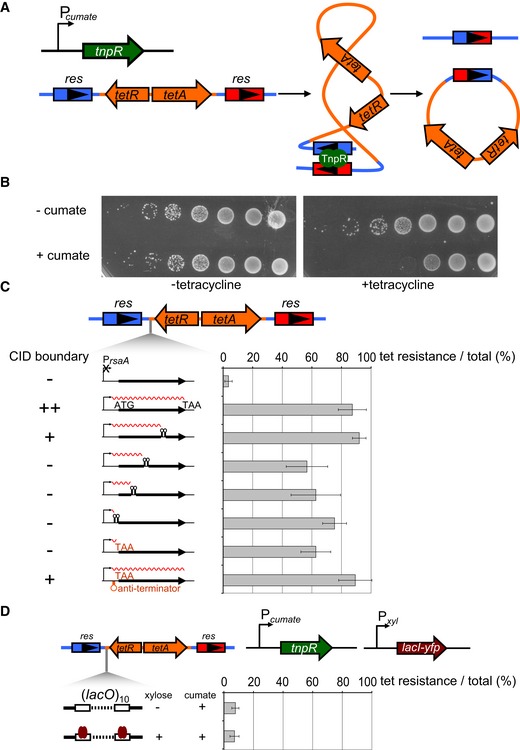
- A γδ TnpR recombinase (green) was induced from a cumate‐inducible promoter. TnpR recombines two res sites (blue and red arrows) that flank a tet resistance cassette (orange arrows) to excise the intervening DNA. Recombination requires supercoiling‐dependent juxtaposition of res sites.
- Induction of TnpR from Pcumate results in a loss of cell viability on tetracycline plates. Each spot is a 10‐fold dilution.
- Percentage of tetracycline‐resistant colonies represent a proxy for the extent of supercoil diffusion inhibition by the various PrsaA‐rsaA constructs indicated, each inserted between one res site and the tet R cassette. Error bars represent SD, n = 3. The ability of each construct to form a CID boundary is indicated on the left (see Figs 2 and 3).
- LacI binding to 10 consecutive lacO sites in between two res sites does not significantly inhibit supercoil diffusion. TnpR and LacI‐YFP (brown arrow) were induced from Pcumate and Pxyl, respectively. Error bars represent SD, n = 3.
We then measured how the various rsaA constructs characterized above (Figs 2 and 3) affected supercoil diffusion using our recombination assay. Each rsaA construct was inserted between the two res sites, adjacent to the tet R cassette with the efficiency of supercoil diffusion inhibition quantified as the percentage of tetR colonies (Fig 5C). A promoterless rsaA variant failed to block supercoil diffusion yielding only ~4% tetR colonies, whereas full‐length rsaA was highly efficient, producing ~87% tetR colonies, indicating that a high rate of transcription prevents supercoil diffusion and loss of the tet R cassette. Consistent with this conclusion, we found that inhibiting transcription by treating cells with rifampicin, an inhibitor of RNA polymerase elongation, also resulted in higher rates of recombination (Appendix Fig S2C). Transcription‐based inhibition of supercoil diffusion is likely not simply a consequence of DNA binding by RNA polymerase between two res sites. Cells harboring 10 consecutive lacO sites in between the res sites and expressing LacI‐YFP did not affect recombination (Fig 5D). We conclude that it is likely the DNA unwinding associated with active transcription, rather than protein binding, that drives supercoil diffusion inhibition, as also suggested previously from studies of Salmonella (Booker et al, 2010).
The rsaA variant producing a truncated, ~2‐kb transcript was as efficient as full‐length rsaA in preventing supercoil diffusion. The rsaA variants producing truncated transcripts of 1,060, 560, or 60 nucleotides were only slightly less efficient in blocking supercoil diffusion with 55, 62, and 75% tetR colonies, respectively (Fig 5C), and none of the differences relative to full‐length rsaA were statistically significant. Importantly, these results suggest that long transcripts likely do not create chromosomal domain boundaries simply by establishing more effective supercoil diffusion barriers. For instance, the truncated rsaA variant producing a 60 nucleotide transcript was nearly as effective as full‐length rsaA in blocking supercoil diffusion, but failed to produce a chromosomal domain boundary by Hi‐C (Fig 2). Similarly, we found that an rsaA variant with the start codon converted to a stop codon, which does not form a Hi‐C boundary, was also nearly as efficient in blocking supercoil diffusion as a variant with the start to stop codon change and an anti‐terminator, which does form a Hi‐C boundary (Figs 3 and 5C).
Long transcripts decompact DNA near domain boundaries, separating flanking loci
Taken all together, our results suggest that transcript length influences whether a gene or operon will produce a chromosomal domain boundary, but that longer transcripts are not substantially better at blocking supercoil diffusion, suggesting that chromosomal domains are not identical to supercoil diffusion barriers. Instead, because transcription requires the unwinding and local decompaction of DNA, long genes or polycistronic operons likely lead to greater physical separation of flanking loci, thereby preventing collisions and producing domain boundaries. Such a model predicts that if the chromosome were allowed to expand, as occurs in elongated cells, the DNA in regions of high gene expression would be more extensible than the compacted DNA within a chromosomal interaction domain. In other words, the chromosome should expand non‐uniformly, with greater extension occurring between domains compared to within domains.
To test this model, we fluorescently labeled two loci either within the same chromosomal domain or in adjacent domains using orthogonal ParB/parS systems and then measured inter‐focus distances in individual cells. The loci in each case were ~200 kb apart, at chromosomal positions +600 and +800 kb for examining intra‐domain interactions or +400 and +600 kb for examining inter‐domain interactions (Fig 6A and B). We measured these inter‐focus distances in cells where the only copy of dnaA is driven by a vanillate‐regulated promoter. Growth in medium lacking vanillate produced a population of cells that (i) contained only one chromosome and (ii) continued to grow without dividing, leading to elongated cells in which the chromosome continues to occupy nearly the entire cell volume, as judged by visualization of a fluorescent protein fused to the nucleoid‐associated protein HU (Appendix Fig S3A).
Figure 6. Intra‐ and inter‐domain DNA expand differentially in elongated Caulobacter cells.
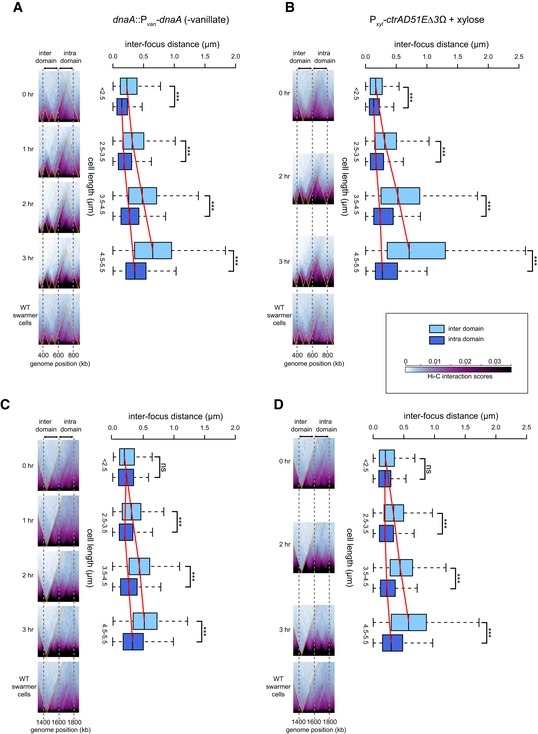
-
APairs of loci were labeled with YFP‐ParBp MT 1/parS p MT 1 and mCherry‐ParBP 1/par SP 1 at genomic positions 400 and 600 kb (inter‐domain) or at 600 and 800 kb (intra‐domain).
-
BSame as (A) but for a strain producing CtrA(D51E)Δ3Ω.
-
C, DSame as (A and B), but with pairs of loci labeled with YFP‐ParBp MT 1/parS p MT 1 and mCherry‐ParBP 1/par SP 1 at genomic positions 1,400 and 1,600 kb (inter‐domain) or at 1,600 and 1,800 kb (intra‐domain). Note that differences in Hi‐C chromosomal domain sharpness reflect differences in interaction patterns as the color scale and image contrast settings are identical across each time series.
Before initiating DnaA depletion, cells were, on average, approximately wild‐type length (~2.5 μm), with the distance between loci in different domains slightly greater than that between loci in the same domain (mean ± standard error: 0.267 ± 0.013 μm versus 0.176 ± 0.011 μm, P = 2 × 10−7, Student's t‐test) (Fig 6A and Appendix Fig S3B). As cells were depleted of DnaA and grew longer, this difference became more pronounced. After 3 h, cells were, on average, 4.2 μm long, with an average interfocus distance of 0.77 μm for loci in different domains and 0.46 μm for loci in the same domain. Additionally, the variance and range in inter‐focus distances was much greater for the loci in different domains than for those in the same domain (Fig 6A). Similar observations were made in a strain engineered to produce elongated cells via a different mechanism, namely by expressing ctrA (D51E)Δ3Ω, a constitutively active allele of ctrA (Domian et al, 1997) (Fig 6B and Appendix Fig S3C). We also performed the same experiment but with another set of loci (at +1,400, +1,600, and +1,800 kb); again, the average inter‐focus distance and especially the variance increased more substantially for the inter‐domain locus pair than the intra‐domain pair (Fig 6C and D, and Appendix Fig S3D and E).
In parallel with these microscopy analyses, we performed Hi‐C on the elongated cells resulting from DnaA depletion or overexpression of ctrA(D51E)Δ3Ω. In both cases, the locations of the chromosomal domains were unchanged, but the domain boundaries became progressively sharper and more well defined as cells elongated (Figs 6A–D and EV4A–C). The sharpness of domain boundaries indicates diminished interactions between domains and, consequently, reduced long‐range interactions in Hi‐C. Consistent with this conclusion, the contact probability graphs clearly indicated an increase in short‐range (< 100 kb) interactions and a corresponding decrease in long‐range (100–1,000 kb) interactions over a 3‐h time course (Fig EV4D and E), with the increased short‐range interactions arising through an increase in interaction frequencies within domains compared to between domains (Fig EV4F and G). Thus, the sharpness in Hi‐C boundaries indicates that, as cells elongate, the interactions within domains are increasingly favored relative to the interactions between domains, consistent with a preferential expansion of chromosomal DNA associated with domain boundaries. The difference in intra‐ and inter‐domain interactions seen likely reflects the decreased confinement of the chromosome as cells elongate; in addition, the synchronization procedure could cause global chromosome compaction that cells recover from over time. Whatever the case, our microscopy and Hi‐C analyses together indicate that nucleoid expansion does not occur uniformly across the chromosome. The DNA at chromosomal domain boundaries is likely less compacted and therefore stretches more than the DNA inside a domain. These results further support our conclusion that domain boundaries produced by highly expressed and long genes produce a spatial barrier in vivo that diminishes contacts between loci in adjacent domains.
Figure EV4. Hi‐C analysis of elongated Caulobacter cells.
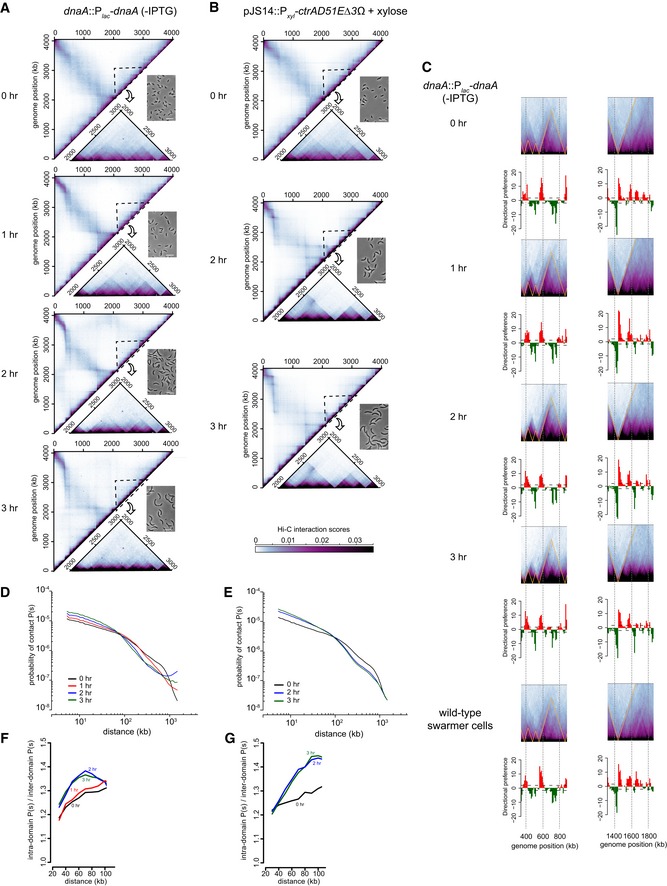
-
AHi‐C maps for cells depleted of DnaA for the times indicated. Only the top left half of each symmetric Hi‐C map is shown with a region outlined shown as an inset along with a representative phase contrast micrograph of cells from each time point.
-
BSame as (A) but for cells overproducing CtrA(D51E)Δ3Ω.
-
CSame portion of the Hi‐C maps shown in Fig 6A and B corresponding to the locations of loci examined by microscopy, but with directional preference plots below each panel.
-
D, EDNA–DNA contact probability P(s), plotted against genomic distance for (D) DnaA‐depleted cells, or (E) cells overproducing CtrA(D51E)Δ3Ω at different time points after synchronization.
-
F, GPlots show the ratio of P(s) values at length scales up to 100 kb for locus–locus interactions within and between domains for (F) DnaA‐depleted cells or (G) cells overproducing CtrA(D51E)Δ3Ω at different time points after synchronization.
Discussion
Hi‐C studies in a range of organisms, from bacteria to humans, have demonstrated that chromosomes are comprised of discrete domains with individual loci interacting preferentially with other loci in the same domain (Dixon et al, 2012; Hou et al, 2012; Sexton et al, 2012; Le et al, 2013; Naumova et al, 2013; Hsieh et al, 2015). Although a number of factors and chromosomal features correlate with domain boundaries in eukaryotes, including highly expressed genes (Hsieh et al, 2015; Ulianov et al, 2016), a cause and effect relationship has not been defined or characterized. For bacteria, we previously showed that high rates of transcription are both necessary and sufficient to establish chromosomal domain boundaries (Le et al, 2013), but the precise mechanisms responsible were unclear. Here, we demonstrated that regions of high gene expression in Caulobacter can create domain boundaries primarily by locally decompacting the chromosome and creating a spatial gap or separation between flanking regions.
Boundary formation and the decrease in contacts between loci in neighboring domains do not depend on the recruitment of ribosomes. Because translation in bacteria can occur even before a full‐length transcript is produced, ribosomes could, in principle, sterically hinder interactions between adjacent domains. However, highly expressed genes that are naturally not translated (e.g. rRNAs) or are engineered not to be translated (Fig 3) were still fully capable of establishing a domain boundary. Nevertheless, not all highly expressed genes form domain boundaries, and we demonstrated that, at least for rsaA, only a relatively long (> ~2 kb) transcript could produce a clear domain boundary in Hi‐C. This observation likely explains why some highly expressed genes, such as tRNAs, are rarely associated with domain boundaries. More generally, these results suggest that a combination of promoter activity and transcript length enables domain boundary formation in Caulobacter. The presence of at least one gene or operon with a high “rpkpm * transcript length” value was strongly correlated with the presence of a chromosomal domain boundary. Additionally, in both growing and starved cells, the highest “rpkpm * transcript length” values were generally observed for genes or operons associated with the sharpest domain boundaries. Systematic assessments of promoter activity and transcript length may ultimately enable the prediction of boundary locations in other growth conditions or in other organisms. Whether expression level or transcript length should be weighted most heavily is not yet clear and the development of a predictive metric for boundary formation remains a challenge for the future.
Long, highly expressed genes may create domain boundaries in part by efficiently inhibiting the diffusion of supercoiled plectonemes, a phenomenon originally characterized in S. typhimurium (Deng et al, 2005; Booker et al, 2010). The slithering of DNA within a plectoneme can promote the spatial juxtaposition of loci separated in the primary genome sequence; such interactions would be blocked by regions of high gene expression and the concomitant unwinding of the DNA (Fig 7). However, we found that both short and long transcripts can block supercoil diffusion (Fig 5), but only the latter create clear domain boundaries by Hi‐C, indicating that Hi‐C and supercoiling domains are not equivalent. Moreover, most plectonemes are ~10–20 kb (Higgins et al, 1996), so the ability of highly expressed genes to diminish interactions at longer length scales, as is seen in Hi‐C, presumably arises primarily from a different mechanism.
Figure 7. Model of chromosomal domain boundary formation in the Caulobacter chromosome.
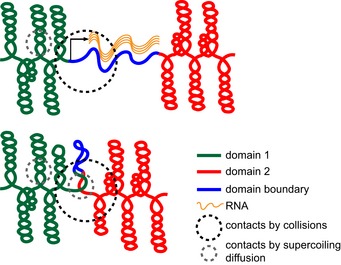
Compacted chromosomal interaction domains (red and green) are connected by a domain boundary denoted as a blue line. (Top) High rates of transcription that produce a relatively long (> 2 kb) transcript (wavy orange lines) locally decompact the DNA, thereby creating a spatial gap between flanking loci. This spatial separation diminishes the long‐range contacts that can occur within a fixed sphere (black dotted circle), leading to the domain boundaries visible by Hi‐C analysis. High rates of transcription can also prevent supercoil diffusion, helping to limit DNA–DNA interactions on a shorter length scale (gray dotted circle). (Bottom) In the absence of transcription, DNA at a domain boundary (blue) will become more compacted, reducing the spatial separation between adjacent domains, and enabling supercoil diffusion, thereby promoting long‐ and short‐range contacts, respectively.
Collectively, our results suggest that long, highly expressed genes create a spatial gap or barrier between flanking loci that effectively blocks, or at least substantially diminishes, interactions (Fig 7). The local decompaction and unwinding of DNA necessary for transcription likely physically separates neighboring loci; indeed, if fully stretched out, a 3‐kb segment of the genome would be ~1 μm. In Streptomyces coelicolor, DNase I sensitivity correlates positively with transcription levels, further implying that transcribed DNA has a more “open” structure (McArthur & Bibb, 2006). We found that, as cells elongate, domain boundaries in Hi‐C maps became sharper (Figs 6 and EV4), indicating that loci continue to interact strongly with other loci in the same domain but even less frequently with loci in other domains, consistent with a model in which domain boundaries create spatial barriers. Additionally, our microscopy analysis demonstrated that the distance between loci in different domains increased more in elongated cells and became significantly more variable, compared to loci in the same domain but at an equivalent distance in the primary sequence.
We propose that chromosomes have a “domains‐on‐a‐string” organization with domain boundaries created by long, highly expressed genes serving as flexible tethers between domains. Chromosomal interaction domains ~100 kb in length have also been seen in Bacillus subtilis (Marbouty et al, 2015; Wang et al, 2015) and in Vibrio cholerae, E. coli and Pseudomonas aeruginosa (Val et al, 2016; R. Koszul and F. Boccard, personal communication), suggesting that they are a conserved unit of chromosome organization in bacteria. In B. subtilis, domain boundaries also frequently coincide with highly expressed genes. The binding of certain nucleoid‐associated proteins was also suggested, though not yet validated, as an alternative mechanism for domain boundary formation (Marbouty et al, 2015), and could explain some of the boundaries in Caulobacter not associated with very long, highly expressed genes.
In sum, chromosomal interaction domains have emerged recently as a fundamental unit of chromosome structure (Dixon et al, 2012; Hou et al, 2012; Sexton et al, 2012; Le et al, 2013; Naumova et al, 2013; Hsieh et al, 2015) and these domains, in turn, likely influence a wide range of DNA‐based transactions inside cells, both prokaryotic and eukaryotic. The open, decompacted nature of domain boundaries might allow easier access of transcription factors and RNA polymerases to certain stretches of DNA. Thus, the high rates of transcription at domain boundaries may enable yet more transcription, potentially forming a positive feedback loop. If open DNA is not immediately compacted following starvation or other major changes in the environment, it could also help direct transcription to certain genes upon recovery. Domains may also impact the expression of less highly expressed genes. The chromosomal position of a gene can dramatically affect its expression level (Bryant et al, 2014), and we speculate that position relative to domain boundaries may contribute to these effects, although this idea remains to be explored. Finally, domain boundaries may help promote the segregation and decatenation of sister chromosomes, both during the cell cycle and possibly during periods of DNA damage and repair. In sum, the identification of chromosomal interaction domains and the elucidation of a primary mechanism of their formation now lay the foundation for future studies of chromosome organization and its relationship to a variety of other essential cellular processes.
Materials and Methods
Strains, media, and growth conditions
Escherichia coli and C. crescentus were grown in LB and PYE, respectively. When appropriate, media were supplemented with antibiotics at the following concentrations (liquid/solid media for C. crescentus; liquid/solid media for E. coli [μg/ml]): chloramphenicol (1/2; 20/30), kanamycin (5/25; 30/50), oxytetracycline (1/2; 12/12), spectinomycin (25/100; 50/50 and gentamycin (0.5/5; 15/20).
Synchronizations were performed on mid‐exponential phase cells using Percoll (GE Healthcare) and density gradient centrifugation. A detailed procedure is described in the Appendix. After synchronization, swarmer cells were released into PYE + 1% formaldehyde for fixation for chromosome conformation capture and Hi‐C analysis. For antibiotic treatment, swarmer cells were incubated with 50 μg/ml chloramphenicol (final concentration) for 30 min before fixing with formaldehyde. For starvation experiments, swarmer cells were released into an 1× M2 salts solution for 90 min before fixation for Hi‐C.
All plasmids, strains, and primers used are listed in Appendix Tables S1 and S2.
Chromosome conformation capture with deep sequencing (Hi‐C)
Hi‐C experiments were performed exactly as described previously (Le et al, 2013). Restriction enzymes (BglII or NcoI) used for Hi‐C and the number of valid reads are listed for each sample in Appendix Table S3. More details on generating Hi‐C contact maps and directional preference plots are described in the Appendix Supplementary Materials and Methods.
RNA‐seq
Exponentially growing Caulobacter cells were collected by centrifugation, released into 1× M2 salts for 90 min before being pelleted and snap‐frozen in liquid nitrogen in preparation for RNA extraction. Total RNA was extracted using a combination of hot Trizol (Thermo Scientific) and RNA extraction spin columns (Zymo Research). Subsequently, contaminated genomic DNA was removed from this RNA prep by Turbo DNaseI (Ambion) treatment. A detailed procedure is described in the Appendix. The integrity of the RNA was checked by Bioanalyzer before being submitted for Illumina library prep and deep sequencing (BioMicroCenter, MIT).
For analysis of RNA‐seq data, Hiseq 2500 Illumina short reads (40 bp) were mapped back to the Caulobacter NA1000 reference genome using Bowtie 1. The sequencing coverage was computed using BEDTools (Quinlan & Hall, 2010). The normalized value of reads per kb per million mapped reads (rpkpm) was calculated for each gene by a custom R script to enable comparison of gene expression within and between RNA‐seq datasets (Dataset EV2). To identify genes that are potentially responsible for the differences in CID boundaries in exponentially growing versus starved cells (Dataset EV2), we considered highly expressed genes with RPKPM values > 1,000 and gene length > 1 kb. Highly expressed genes < 1 kb in length were also considered if they are in an operon and the total length of the operon was > 1 kb or if there were multiple highly expressed genes in a 10‐kb bin near a given CID boundary and the total length of these genes was > 1 kb.
Determination of subcellular positions of chromosomal loci by orthogonal ParB/parS systems
Strains were grown to OD600 = 0.4 in the presence of appropriate antibiotics, vanillate, and glucose before the cells were collected by centrifugation and washed of residual vanillate, antibiotics and glucose twice with fresh PYE. Cells were then resuspended in PYE plus xylose (0.3% final concentration) (vanillate omitted) to deplete DnaA for 90 min before synchronization, thereby inducing cell elongation. Cells were imaged at 0, 1, 2 and 3 h post‐synchronization. Phase contrast (150 ms exposure) and fluorescence images (2,000 ms exposure) were collected. MicrobeTracker (http://microbetracker.org/) was used to detect cell outlines and SpotFinderZ to detect fluorescent foci positions. The number of cells used for construction of boxplots in Fig 6 (cell length μm/loci measured kb/number of cells) was as follows: (< 2.5/400–600/206), (< 2.5/600–800/254), (2.5–3.5/400–600/1,438), (2.5–3.5/600–800/998), (3.5–4.5/400–600/803), (3.5–4.5/600–800/651), (4.5–5.5/400–600/422), (4.5–5.5/600–800/458), (< 2.5/1,400–1,600/90), (< 2.5/1,600–1,800/152), (2.5–3.5/1,400–1,600/131), (2.5–3.5/1,600–1,800/714), (3.5–4.5/1,400–1,600/401), (3.5–4.5/1,600–1,800/346), (4.5–5.5/1,400–1,600/308), (4.5–5.5/1,600–1,800/520). The procedure for CtrA(D51E)Δ3Ω overexpression strains is described in the Appendix.
γδ TnpR recombination assay
Caulobacter strains used for the recombination assay were first grown to early exponential phase in PYE supplemented with tetracycline and kanamycin. Just before inducing TnpR, cells were collected by centrifugation, washed of tetracycline, and resuspended in fresh PYE + kanamycin to an OD600 of ~0.2. Cumate was then added to a final concentration of 100 μM, and cultures were incubated with shaking at 30°C for 2 h. Subsequently, cultures were 10‐fold serially diluted, and 5 μl of each dilution was spotted on both tetracycline + kanamycin and kanamycin PYE plates. To measure colony‐forming units (CFU), Caulobacter cultures were serially (20‐fold) diluted with 50 μl of appropriately diluted culture spread on tetracycline + kanamycin or kanamycin PYE plates. Plates were incubated at 30°C for 2 days before counting CFUs. A more detailed procedure is described in the Appendix.
Data availability
Hi‐C data are available in GEO, GSE74364.
Author contributions
TBKL and MTL conceived of the project. TBKL performed experiments. TBKL and MTL analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
We thank A. Badrinarayanan, D. Haakonsen, and M. Guo for comments on the manuscript and acknowledge A. Triassi for help with initial experiments; J. Nomellini, M. Imakaev, and L. Mirny for helpful discussion. This work was supported by a Gordon and Betty Moore Foundation Postdoctoral Fellowship from the Life Sciences Research Foundation (GBMF2550.02), a Royal Society University Research Fellowship (UF140053) to T.B.K.L. and a NIH grant (5R01GM082899) to M.T.L. who is also an Investigator of the Howard Hughes Medical Institute.
The EMBO Journal (2016) 35: 1582–1595
References
- Arnvig KB, Zeng S, Quan S, Papageorge A, Zhang N, Villapakkam AC, Squires CL (2008) Evolutionary comparison of ribosomal operon antitermination function. J Bacteriol 190: 7251–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badrinarayanan A, Le TBK, Laub MT (2015) Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol 31: 171–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin HW, Matzuk MM, Krasnow MA, Cozzarelli NR (1985) Recombination site selection by Tn3 resolvase: topological tests of a tracking mechanism. Cell 40: 147–158 [DOI] [PubMed] [Google Scholar]
- Booker BM, Deng S, Higgins NP (2010) DNA topology of highly transcribed operons in Salmonella enterica serovar typhimurium . Mol Microbiol 78: 1348–1364 [DOI] [PubMed] [Google Scholar]
- Bryant JA, Sellars LE, Busby SJW, Lee DJ (2014) Chromosome position effects on gene expression in Escherichia coli K‐12. Nucleic Acids Res 42: 11383–11392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295: 1306–1311 [DOI] [PubMed] [Google Scholar]
- Dekker J, Heard E (2015) Structural and functional diversity of topologically associating domains. FEBS Lett 589: 2877–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Stein RA, Higgins NP (2005) Organization of supercoil domains and their reorganization by transcription. Mol Microbiol 57: 1511–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domian IJ, Quon KC, Shapiro L (1997) Cell type‐specific phosphorylation and proteolysis of a transcriptional regulator controls the G1‐to‐S transition in a bacterial cell cycle. Cell 90: 415–424 [DOI] [PubMed] [Google Scholar]
- Higgins NP, Yang X, Fu Q, Roth JR (1996) Surveying a supercoil domain by using the gamma delta resolution system in Salmonella typhimurium . J Bacteriol 178: 2825–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Li L, Qin ZS, Corces VG (2012) Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell 48: 471–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ (2015) Mapping nucleosome resolution chromosome folding in yeast by Micro‐C. Cell 162: 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TB, Imakaev MV, Mirny LA, Laub MT (2013) High‐resolution mapping of the spatial organization of a bacterial chromosome. Science 342: 731–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbouty M, Le Gall A, Cattoni DI, Cournac A, Koh A, Fiche J‐B, Mozziconacci J, Murray H, Koszul R, Nollmann M (2015) Condensin‐ and replication‐mediated bacterial chromosome folding and origin condensation revealed by Hi‐C and super‐resolution imaging. Mol Cell 59: 588–602 [DOI] [PubMed] [Google Scholar]
- McArthur M, Bibb M (2006) In vivo DNase I sensitivity of the Streptomyces coelicolor chromosome correlates with gene expression: implications for bacterial chromosome structure. Nucleic Acids Res 34: 5395–5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, Dekker J (2013) Organization of the mitotic chromosome. Science 342: 948–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JP, Grimley C, Lowery C (1975) Transcription termination factor rho activity is altered in Escherichia coli with suA gene mutations. Proc Natl Acad Sci USA 72: 1725–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DA, Gaal T, Gourse RL (2002) NTP‐sensing by rRNA promoters in Escherichia coli is direct. Proc Natl Acad Sci USA 99: 8602–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G (2012) Three‐dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472 [DOI] [PubMed] [Google Scholar]
- Shen V, Bremer H (1977) Rate of ribosomal ribonucleic acid chain elongation in Escherichia coli B/r during chloramphenicol treatment. J Bacteriol 130: 1109–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A, Gelfand MS, Shevelyov YY, Razin SV (2016) Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26: 70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger MA, Toro E, Wright MA, Porreca GJ, Bau D, Hong SH, Fero MJ, Zhu LJ, Marti‐Renom MA, McAdams HH, Shapiro L, Dekker J, Church GM (2011) The three‐dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Mol Cell 44: 252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val ME, Marbouty M, de Lemos Martins F, Kennedy SP, Kemble H, Bland MJ, Possoz C, Koszul R, Skovgaard O, Mazel D (2016) A checkpoint control orchestrates the replication of the two chromosomes of Vibrio cholerae . Sci Adv 2: e1501914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valens M, Penaud S, Rossignol M, Cornet F, Boccard F (2004) Macrodomain organization of the Escherichia coli chromosome. EMBO J 23: 4330–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Montero Llopis P, Rudner DZ (2013) Organization and segregation of bacterial chromosomes. Nat Rev Genet 14: 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Le TBK, Lajoie BR, Dekker J, Laub MT, Rudner DZ (2015) Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis . Genes Dev 29: 1661–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RG, Grindley NDF (1984) Analysis of the γδ res site: sites required for site‐specific recombination and gene expression. J Mol Biol 179: 667–687 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Data Availability Statement
Hi‐C data are available in GEO, GSE74364.