

Abstract
Background
DMBT1 is a gene that shows extensive copy number variation (CNV) that alters the number of bacteria-binding domains in the protein and has been shown to activate the complement pathway. It lies next to the ARMS2/HTRA1 genes in a region of chromosome 10q26, where single nucleotide variants have been strongly associated with age-related macular degeneration (AMD), the commonest cause of blindness in Western populations. Complement activation is thought to be a key factor in the pathogenesis of this condition. We sought to investigate whether DMBT1 CNV plays any role in the susceptibility to AMD.
Methods
We analysed long-range linkage disequilibrium of DMBT1 CNV1 and CNV2 with flanking single nucleotide polymorphisms (SNPs) using our previously published CNV and HapMap Phase 3 SNP data in the CEPH Europeans from Utah (CEU). We then typed a large cohort of 860 AMD patients and 419 examined age-matched controls for copy number at DMBT1 CNV1 and CNV2 and combined these data with copy numbers from a further 480 unexamined controls.
Results
We found weak linkage disequilibrium between DMBT1 CNV1 and CNV2 with the SNPs rs1474526 and rs714816 in the HTRA1/ARMS2 region. By directly analysing copy number variation, we found no evidence of association of CNV1 or CNV2 with AMD.
Conclusions
We have shown that copy number variation at DMBT1 does not affect risk of developing age-related macular degeneration and can therefore be ruled out from future studies investigating the association of structural variation at 10q26 with AMD.
Background
Age-related macular degeneration (AMD) is the leading cause of severe visual impairment in individuals over the age of 50, and affects the central region of the retina (the macula) containing the highest concentration of cone photoreceptors responsible for normal visual acuity [1–3]. Although the etiology and pathogenesis of AMD are not fully understood, numerous studies indicate that risk factors are both genetic and environmental include age, sex, ethnicity, smoking, hypertension and diet [4, 5]. Despite the multifactorial nature of AMD, and variable phenotype definitions, two genetic regions at 1q32 and 10q26 have been repeatedly implicated by linkage analysis and subsequently by genome-wide association studies [5–13]. The estimated effect sizes of the index SNPs at these two loci are also notable, with a fifty-fold increase in AMD risk in those individuals who are homozygous at both loci, with 65 % of AMD cases attributable to variation at these two SNPs [14].
The genetic signal at 1q32 has subsequently been shown to be due to variation involving genes of the Regulators of Complement Activation (RCA) alpha block, including complement factor H (CFH) and the complement factor H related genes (CHFR1-5). The Y402H polymorphism in CFH is strongly associated with AMD as well as several other SNPs in the CFH region [5, 15, 16]. Furthermore, an 84 kb deletion removing the CFHR3 and CHFR1 genes shows protection against AMD [17, 18]. This deletion is part of a spectrum of different copy number variants within the RCA region, with deletion and duplication mediated by different segmental duplications, and there is suggestive evidence that a rare deletion involving CFHR1 and CFHR4 is associated with bilateral geographic atrophy, one of the two main phenotypic variants of AMD [19, 20]. In addition to the identification of a strong genetic association between AMD and the RCA region, variation within several other complement genes such as complement factor B (CFB)/complement 2 (C2) [21], complement 3 (C3) [22] and complement factor I (CFI) [23] has been found to be associated with AMD. Taken together, these findings point to an important role of the complement innate immune response in the etiology of AMD [24].
The functional basis of the association at 10q26 remains unclear. Although there is some evidence that rs10490924 in the ARMS2 gene affects systemic complement activation [25], this remains controversial and the genes HTRA1 and PLEKHA1 do not have convincing links with the complement pathway. In contrast, the DMBT1 (Deleted in Malignant Brain Tumors 1) gene, 106 kb distal of rs10490924, encodes a glycoprotein which is known to bind complement C1Q [26] activate complement by the mannose-binding lectin pathway [27, 28], and promotes VEGF expression. DMBT1 (also known as gp340, salivary agglutinin, muclin or hensin) is present on the surfaces of the eye, being abundant in tears and in the lacrimal glands with a lesser amount detected in cornea and conjunctiva [29], and is expressed in the retina [30].
Sequence variation within and surrounding DMBT1 is poorly represented on SNP genotyping chips. However, given the distance between the rs10490924 risk allele and DMBT1, it is unlikely that a common single nucleotide polymorphism around DMBT1 is responsible for the association with AMD. We considered it possible, however, that the association may, at least in part, be due to a synthetic association with a copy number variant (CNV) of very strong effect size. This might be possible particularly if that CNV was rare or due to recurrent mutation that happened to occur on a rs10490924 risk allele background, and therefore be in LD with that allele [31]. Indeed, the observation that 10q26 had been identified in linkage studies of AMD is not inconsistent with a synthetic association with a rare or moderate frequency allele of very strong effect.
We and others have previously shown DMBT1 exhibits extensive copy number variation that affects the number of scavenger-receptor cysteine-rich domains (SRCR) within the protein [32–34] (Fig. 1). The copy number variation is confined to two loci within the gene, termed CNV1 and CNV2. Both show a high copy number mutation rate (of the order of 1–2 % per generation) and copy number at the two loci is not correlated at the population level. There is extensive variation at the population level, such that individuals are predicted to have between 7 and 20 SRCR repeats per DMBT1 molecule.
Fig. 1.
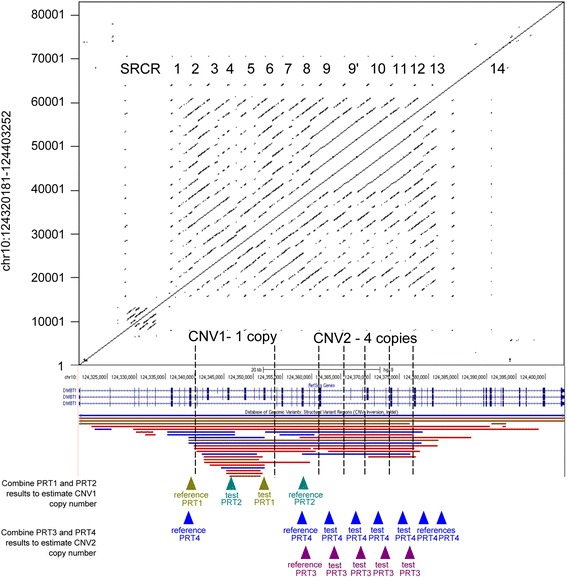
Overview of copy number variation within the DMBT1 gene. The top half shows a dot plot of the DMBT1 gene (shown below, in blue, from a screenshot from the UCSC genome browser) aligned against itself. Black dots indicate regions of sequence identity, with the diagonal lines showing the tandemly-repeated nature of the DMBT1 gene. The tandemly-arranged SRCR repeat regions are annotated as numbers on this dot plot, including SRCR14 which does not bind bacteria [44]. The bottom half shows the genome assembly of the DMBT1 gene, with one assembled copy of CNV1 and four assembled copies of CNV2. CNV regions, as recorded in the Database of Genomic Variants, are shown below the DMBT1 gene structure. Below these, location of reference and test amplicons of the four independent paralogue ratio tests (PRTs) that measure copy number of CNV1 and CNV2 are shown
Investigating the relationship between flanking SNP variation and CNV also casts light on the extent to which CNV can be tagged by flanking SNPs because of linkage disequilibrium (LD). Even partial tagging would allow CNV to be indirectly imputed rather than directly genotyped, and this remains a topic of considerable interest in the literature. This is primarily because even imperfect imputation could indicate a role in disease susceptibility for certain CNVs, particularly given extremely large sample sizes. It is generally established that, using current imputation panels, whether a CNV is taggable depends on its mutational history, with CNVs generated by a unique mutational event such as most simple deletions and duplications being more taggable than complex multiallelic CNVs generated by recurrent mutation [35, 36].
We sought to explore the role of DMBT1 CNV in AMD by investigating the linkage disequilibrium between copy number variations at DMBT1 and common SNP alleles surrounding the gene and the association of DMBT1 CNV with AMD in a large case–control cohort.
Methods
AMD case–control collection, DMBT1 CNV and SNP genotype data
The AMD case–control collection comprised cases with predominantly advanced AMD (either geographic atrophy or choroidal neovascularization) and spouse controls recruited from hospital ophthalmic clinics in London and the South East of England [22]. All subjects were examined by an ophthalmologist and had colour, stereoscopic fundus photography of the macular region. All the images were graded at the Reading Centre, Moorfields Eye Hospital, London using the International Classification of Age-related Maculopathy and Macular Degeneration [37]. All subjects described themselves as “white” on a recruitment questionnaire. DNA samples were obtained from a total of 1533 individuals. Each DNA plate contained both patients and controls and those undertaking the analysis were unaware of disease status of each sample.
DMBT1 CNV data from the HapMap Phase 1 CEU cohort and the UK Human Random Control (HRC) cohort was published previously [34]. SNP genotype data for the Phase 3 (release 27) HapMap 1 CEU cohort was downloaded from the International HapMap Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap/). SNP genotype data for the AMD case–control cohort was available from previous whole-genome SNP genotyping studies [5].
DMBT1 copy number typing
Typing of diploid copy number of DNA samples used paralogue ratio test methods previously described [34], which involves amplification of 10 ng of DNA using fluorescently labelled primers matching both test and reference loci. Briefly, CNV1 was typed using two distinct PRT assays, PRT1 and PRT2, with CNV2 typed using a further two distinct PRT assays, PRT3 and PRT4. Copy numbers for CNV1 and CNV2 were subsequently called using a Gaussian mixture model on the mean value of both PRTs, implemented using the CNVtools v1.42.3 package for the statistical software R v2.15.3 [38]. Previous analysis has shown that these assays have an error rate of 0.37 % for CNV1 and 0.33 % for CNV2 [34].
Linkage disequilibrium and association analysis
Linkage disequilibrium (LD) in HapMap trio data was analysed using Haploview [39]. Association analysis between CNV and flanking SNPs on HapMap CEU founder individuals was performed using PLINK v.1.0.7, using an additive model for the SNPs, treating CNV copy number as a quantitative trait, and visualised using LocusZoom [40]. Samples from the AMD dataset where DMBT1 CNV was called were matched to clinical data and case–control association analysis was carried out on late AMD cases and examined controls using logistic regression and Stata (version 13.1, StataCorp LP, College Station, TX).
Results and discussion
We began this study by using our previously published DMBT1 CNV data [34] and publically available SNP data on the HapMap CEU population, as a representative of a north-west European population, to investigate any evidence for long-range LD involving DMBT1 CNV1 and CNV2. There are several problems inherent in examining the LD between multiallelic copy number variants and SNPs. Firstly, multiallelic CNVs are often within regions of segmental duplication, where SNPs cannot be genotyped, and indeed may not exist as true diallelic polymorphisms. Secondly, most methods that measure copy number variation, such as qPCR, PRT, arrayCGH and sequence read-depth, rely on dosage information. This dosage information is for both alleles of the CNV, so that the resulting dosage is a sum of the two alleles at a diploid locus. Resolving this dose information into a true copy number genotype – for example determining whether a diploid copy number of 4 is a 2–2 genotype, 3–1 genotype or 4–0 genotype – requires observation of copy number in extended pedigrees so that the constituent alleles segregate as different combinations in different offspring.
Although the HapMap SNP data provides a densely SNP genotyped map of the human genome, the SNP density is low within and surrounding the DMBT1 gene (Fig. 2), such that only long-range LD can be examined. For CNV1 of DMBT1, we directly determined diploid genotypes of HapMap CEU trios using long PCR. Because most CNV1 variation can be regarded as presence or absence of a deletion allele, we recoded this variation as diallelic, equivalent to the DMBT1SR47- deletion reported previously [32], such that a copy number of 0 is regarded as a homozygous deletion, a copy number of 1 as a heterozygous deletion and copy numbers of 2 and higher as homozygous non-deleted. This allowed conventional determination of linkage disequilibrium. Pairwise LD of CNV1 with rs10490924, which has previously been shown to be most strongly associated with AMD, showed a D’ value of 1 and r2 value of 0.031, although the logarithm of odds (LOD) score, was only 0.75, reflecting weak confidence in the value of D’.
Fig. 2.
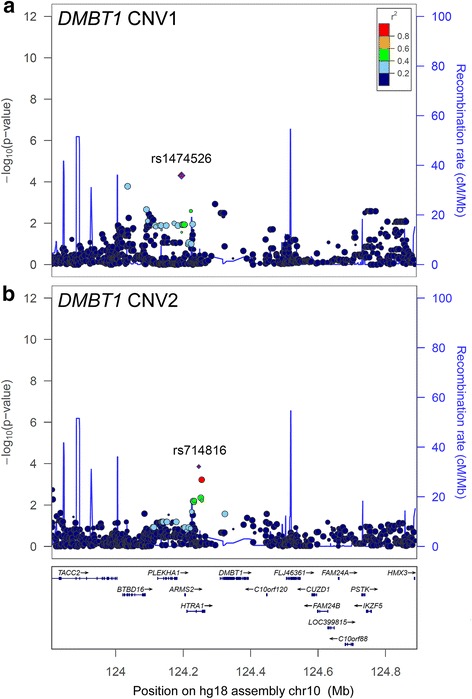
Regional association plots of DMBT1 copy number variation and flanking SNPs. Analysis of SNPs within the 1 Mb region flanking DMBT1 and association with diploid copy number of a CNV1 and b CNV2. The left-hand y-axis shows statistical support for an association, as measured by the negative log10 of the p-value of the linear regression test of association. The right-hand y-axis shows the recombination rate inferred from HapMap population data. The index SNP is shown as a diamond, and is annotated with its rs number. The size of each point reflects the number of genotypes for that particular SNP, and the colour of each point reflects the strength of association with the index SNP
We also treated copy number for the two independent CNVs within DMBT1 as a quantitative trait for association analysis in the unrelated CEU individuals. The lack of genotype information for CNVs is likely to result in a loss of power. Furthermore, an r2 value, reflecting the squared correlation coefficient of the SNP genotype and copy number, is used as a measure of allelic association, but is not directly comparable to r2 values between SNPs. Nevertheless, such information gives an indication of the relationship between a CNV and flanking SNPs. In general, as expected, there is weak association between copy number and flanking SNPs. Copy number at CNV1 is associated with rs1474526 (r2 = 0.14, p = 3.2 × 10−3), a PLEKHA1/ARMS2 intergenic SNP which itself is only in weak LD with flanking SNPs (Fig. 2a). For CNV2, strongest association was with rs714816 (r2 = 0.12, p = 6.6 × 10−3, Fig. 2b), which is within the intron of the HTRA1 gene and has been previously associated with AMD, although it is not the most strongly associated in the region [41].
Given the suggestive evidence at least some allelic association between CNVs and SNPs around the ARMS2 and HTRA1 genes, as well as the strong functional candidacy of DMBT1, we directly tested for an association of CNV1 or CNV2 copy number with AMD using a case–control design. Copy number typing of cases and controls showed raw values that clustered about integer copy numbers, and integer copy number was assigned to each individual using Gaussian mixture modelling (Fig. 3). Of 1533 DNA samples of cases and examined controls typed for this study, 5 had a posterior probability of a call less than 0.99 for CNV1, of which 2 were excluded following retesting, and for CNV2, 328 out of 1533 had a had a posterior probability of a call less than 0.99 but all had a posterior probability >0.5 for any given call. A small number of outliers (for example one DNA sample with CNV1 copy number 4 sample) were removed prior to Gaussian mixture model calling of copy number, and copy number called manually. The samples where copy number was called were matched to clinical data and the resulting 860 advanced AMD cases and 419 examined controls were used in the case–control association analysis. Copy number distributions ranged from 0 to 4 for CNV1, and from 2 to 13 for CNV2, consistent with previous studies (Tables 1, 2, [34]). We found no association of diploid copy number with disease status (Table 3). We also reclassified CNV1 as DMBT1SR47- deletion as previously [34], and found no association of CNV1 or CNV2 with disease status, either using matched examined controls or larger control cohort including 479 unexamined controls from the HRC cohort (Table 3). Furthermore, because the AMD cases and examined controls had been genotyped for rs10490924, we were able to analyse LD between this SNP and DMBT1SR47-. There was no evidence of LD between the two loci (r2 = 0.004, D’ = 0.25). As expected, rs10490924 genotype was strongly associated with AMD (Table 4).
Fig. 3.
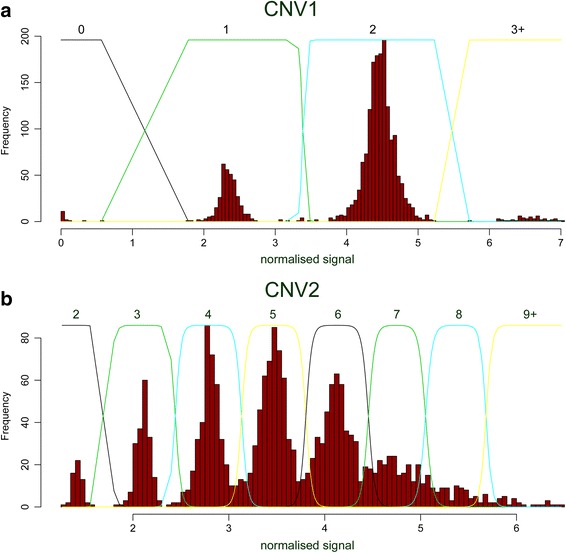
Raw copy number histograms and Gaussian mixture models. Raw copy number data, plotted as a histogram, for a CNV1 and b CNV2. The normalised signal, shown on the x-axis, reflects raw average PRT values normalised so that the overall standard deviation of the data is 1. Lines indicate the Gaussian mixture model fit, with numbers above each curve indicating the diploid copy number inferred
Table 1.
Frequency of CNV1 in advanced AMD cases and controls
| CNV1 | Cases | Controls (matched) | Controls (HRCa) | |||
|---|---|---|---|---|---|---|
| Count | Frequency | Count | Frequency | Count | Frequency | |
| 0 | 8 | <0.01 | 6 | 0.01 | 2 | <0.01 |
| 1 | 141 | 0.16 | 75 | 0.18 | 79 | 0.16 |
| 2 | 687 | 0.80 | 329 | 0.79 | 387 | 0.81 |
| 3 | 23 | 0.03 | 8 | 0.02 | 11 | 0.02 |
| 4 | 1 | <0.01 | 0 | 0 | 1 | <0.01 |
| total | 860 | 418 | 480 | |||
aHuman Random Control collection
Table 2.
Frequency of CNV2 in advanced AMD cases and controls
| CNV2 | Cases | Controls (matched) | Controls (HRC) | |||
|---|---|---|---|---|---|---|
| Count | Frequency | Count | Frequency | Count | Frequency | |
| 2 | 27 | 0.03 | 11 | 0.03 | 11 | 0.02 |
| 3 | 83 | 0.10 | 45 | 0.11 | 48 | 0.1 |
| 4 | 162 | 0.19 | 79 | 0.19 | 110 | 0.23 |
| 5 | 230 | 0.27 | 114 | 0.27 | 125 | 0.26 |
| 6 | 206 | 0.24 | 104 | 0.25 | 104 | 0.22 |
| 7 | 84 | 0.10 | 46 | 0.11 | 51 | 0.11 |
| 8 | 44 | 0.05 | 13 | 0.03 | 23 | 0.05 |
| 9 | 18 | 0.02 | 7 | 0.02 | 4 | <0.01 |
| 10 | 4 | <0.01 | 0 | 0 | 4 | <0.01 |
| 11 | 1 | <0.01 | 0 | 0 | 0 | 0 |
| 12 | 0 | 0 | 0 | 0 | 0 | 0 |
| 13 | 1 | <0.01 | 0 | 0 | 0 | 0 |
| total | 860 | 419 | 480 | |||
Table 3.
Association of DMBT1 CNV1 and CNV2 copy number with advanced AMD
| Variation | Control group | Odds ratio (95 % CI) | p-value |
|---|---|---|---|
| CNV1 | Matched controls (unadjusted) | 1.18 (0.92–1.51) | 0.20 |
| Matched controls (adjusted for age, sex and smoking status) | 1.15 (0.88–1.51) | 0.30 | |
| All Controls | 1.06 (0.86–1.30) | 0.57 | |
| CNV2 | Matched controls | 1.04 (0.96–1.12) | 0.32 |
| Matched controls (adjusted for age, sex and smoking status) | 1.01 (0.93–1.10) | 0.81 | |
| All Controls | 1.03 (0.98–1.11) | 0.22 | |
| DMBT1 SR47- | Matched controls | 0.87 (0.66–1.14) | 0.31 |
| Matched controls (adjusted for age, sex and smoking status) | 0.89 (0.66–1.19) | 0.43 | |
| All controls | 0.96 (0.77–1.21) | 0.73 |
Table 4.
Association of rs10490924 with advanced AMD
| Cases | Controls | |||
|---|---|---|---|---|
| Count | Frequency | Count | Frequency | |
| GG | 250 | 0.29 | 260 | 0.62 |
| GT | 423 | 0.50 | 140 | 0.34 |
| TT | 175 | 0.21 | 16 | 0.04 |
| Total | 848 | 1.00 | 416 | 1.00 |
| missing genotypes | 12 | - | 3 | - |
Unadjusted p value 7.8 × 10−30 OR = 3.25 (95 % CI: 2.65–3.98), n = 1264
Adjusted p value (for age, sex, smoking) 1.5 × 10−28 OR = 3.37 (95 % CI: 2.72–4.18), n = 1175
Sequence variation within and immediately surrounding the DMBT1 gene was not investigated for association with AMD in this study. It is poorly represented on genotyping chips, and uncertainties in aligning short sequence reads to a tandemly-repeated structure have prevented a full interpretation of variation of the gene. Nevertheless, analysis of low-coverage sequencing data outside the known CNV regions has led to the 5′ end of DMBT1 being identified as a region that has been subject to balancing selection [42]. The role of a rapidly-mutating CNV, putatively under selection pressure, on surrounding SNP variation remains to be explored. However, although such SNP variation may be effectively untagged by current SNP markers, it is very unlikely to account for the strong association signal with AMD at 10q26 because of the clear breakdown of LD between SNPs from rs10490924 towards DMBT1.
Could variation at rs10490924 be affecting expression levels of DMBT1? Initial examination of eQTL datasets suggests not, although such long range interactions are very likely; indeed, three SNPs within DMBT1 are eQTLs for HTRA1 in monocytes [43]. It should also be noted that no results from an eQTL analysis of retinal tissues, or indeed any tissues where DMBT1 is significantly expressed, have been published.
Conclusion
We have shown that copy number variation at DMBT1 does not affect risk of developing age-related macular degeneration, and can therefore be ruled out from future studies investigating the nature of association signal at 10q26.
Acknowledgements
This work was funded by a Government of India Ministry of Social Justice and Empowerment PhD studentship to SP and EJH. EJH was supported in part by an MRC New Investigator Grant (GO801123). The collection of patient and control samples was funded by a grant from the Medical Research Council to JRWY and ATM. VC is funded by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed in this article are those of the authors and not necessarily those of the Government of India, NHS, the NIHR or the Department of Health. We thank the clinicians who helped with recruitment, the Reading Centre at Moorfields Eye Hospital, London for grading fundus photographs and the subjects who participated in the research. We would like to thank Mark Jobling and Jenny Bowdrey for access and support in using an ABI 3130xl capillary electrophoresis machine.
Authors’ contributions
EJH, JRWY and VC designed the study. JCK, HS, ATM and JRWY provided samples. SP generated the data, and VC, SP and EJH analysed the data. EJH and VC wrote the paper with contributions from SP, ATM and JRWY. All authors have read and approved the final manuscript.
Competing interests
EJH has received grant funding from Pfizer Inc on a project unrelated to the project described in this paper.
Ethics approval and consent to participate
The study protocol followed the tenets of the Declaration of Helsinki and Multicenter Research Ethics Committee and Local Research Ethics Committee approvals were obtained. All participants gave informed written consent.
References
- 1.Wong T, Chakravarthy U, Klein R, Mitchell P, Zlateva G, Buggage R, Fahrbach K, Probst C, Sledge I. The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology. 2008;115(1):116–26. doi: 10.1016/j.ophtha.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. The Lancet. 2012;379(9827):1728–38. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 3.Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014;15:151. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, Buggage R, Pleil A, Mitchell P. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. 2010;10(1):31. doi: 10.1186/1471-2415-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45(4):433–9. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, Iyengar SK, Klein BE, Klein R, Lee KE, Majewski J, et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14(15):2257–64. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 8.Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, Weber BH. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14(21):3227–36. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 9.Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 11.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 12.Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77(3):389–407. doi: 10.1086/444437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeWan A, Liu M, Hartman S, Zhang SS-M, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314(5801):989–92. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 14.Schaumberg DA, Hankinson SE, Guo Q, Rimm E, Hunter DJ. A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol. 2007;125(1):55–62. doi: 10.1001/archopht.125.1.55. [DOI] [PubMed] [Google Scholar]
- 15.Sofat R, Casas JP, Webster AR, Bird AC, Mann SS, Yates JR, Moore AT, Sepp T, Cipriani V, Bunce C, et al. Complement factor H genetic variant and age-related macular degeneration: effect size, modifiers and relationship to disease subtype. Int J Epidemiol. 2012;41(1):250–62. doi: 10.1093/ije/dyr204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Atmaca-Sonmez P, Othman M, Branham KE, Khanna R, Wade MS, Li Y, Liang L, Zareparsi S, Swaroop A, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38(9):1049–54. doi: 10.1038/ng1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38(10):1173–7. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 18.Raychaudhuri S, Ripke S, Li M, Neale BM, Fagerness J, Reynolds R, Sobrin L, Swaroop A, Abecasis G, Seddon JM, et al. Associations of CFHR1-CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent. Nat Genet. 2010;42(7):553–5. doi: 10.1038/ng0710-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantsilieris S, White SJ, Richardson AJ, Guymer RH, Baird PN. Comprehensive analysis of copy number variation of genes at chromosome 1 and 10 loci associated with late age related macular degeneration. PLoS One. 2012;7(4):e35255. doi: 10.1371/journal.pone.0035255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmid-Kubista KE, Tosakulwong N, Wu Y, Ryu E, Hecker LA, Baratz KH, Brown WL, Edwards AO. Contribution of copy number variation in the regulation of complement activation locus to development of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(11):5070–9. doi: 10.1167/iovs.09-3975. [DOI] [PubMed] [Google Scholar]
- 21.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357(6):553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 23.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17(1):100–4. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khandhadia S, Cipriani V, Yates J, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217(2):127–46. doi: 10.1016/j.imbio.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 25.Smailhodzic D, Klaver CC, Klevering BJ, Boon CJ, Groenewoud JM, Kirchhof B, Daha MR, den Hollander AI, Hoyng CB. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology. 2012;119(2):339–46. doi: 10.1016/j.ophtha.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 26.Boackle RJ, Connor MH, Vesely J. High molecular weight non-immunoglobulin salivary agglutinins (NIA) bind C1Q globular heads and have the potential to activate the first complement component. Mol Immunol. 1993;30(3):309–19. doi: 10.1016/0161-5890(93)90059-K. [DOI] [PubMed] [Google Scholar]
- 27.Leito JT, Ligtenberg AJ, Van Houdt M, Van den Berg TK, Wouters D. The bacteria binding glycoprotein salivary agglutinin (SAG/gp340) activates complement via the lectin pathway. Mol Immunol. 2011;49(1):185–90. doi: 10.1016/j.molimm.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Reichhardt MP, Loimaranta V, Thiel S, Finne J, Meri S, Jarva H. The salivary scavenger and agglutinin binds MBL and regulates the lectin pathway of complement in solution and on surfaces. Front Immun. 2012;3:205. [DOI] [PMC free article] [PubMed]
- 29.Jumblatt MM, Imbert Y, Young WW, Foulks GN, Steele PS, Demuth DR. Glycoprotein 340 in normal human ocular surface tissues and tear film. Infect Immun. 2006;74(7):4058–63. doi: 10.1128/IAI.01951-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller H, Hu J, Popp R, Schmidt MH, Muller-Decker K, Mollenhauer J, Fisslthaler B, Eble JA, Fleming I. Deleted in malignant brain tumors 1 is present in the vascular extracellular matrix and promotes angiogenesis. Arterioscler Thromb Vasc Biol. 2012;32(2):442–8. doi: 10.1161/ATVBAHA.111.239830. [DOI] [PubMed] [Google Scholar]
- 31.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renner M, Bergmann G, Krebs I, End C, Lyer S, Hilberg F, Helmke B, Gassler N, Autschbach F, Bikker F, et al. DMBT1 confers mucosal protection in vivo and a deletion variant is associated with Crohn’s disease. Gastroenterology. 2007;133(5):1499–509. doi: 10.1053/j.gastro.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki H, Betensky RA, Cairncross JG, Louis DN. DMBT1 polymorphisms relationship to malignant glioma tumorigenesis. Cancer Res. 2002;62(6):1790–6. [PubMed] [Google Scholar]
- 34.Polley S, Louzada S, Forni D, Sironi M, Balsakas T, Hains D, Yang F, Hollox EJ. Evolution of the rapidly-mutating human salivary agglutinin gene (DMBT1) and population subsistence strategy. Proc Natl Acad Sci. 2015;112(15):5105–10. doi: 10.1073/pnas.1416531112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantsilieris S, White SJ. Correlating multiallelic copy number polymorphisms with disease susceptibility. Hum Mutat. 2013;34(1):1–13. doi: 10.1002/humu.22172. [DOI] [PubMed] [Google Scholar]
- 36.Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. Large multiallelic copy number variations in humans. Nat Genet. 2015;47(3):296–303. doi: 10.1038/ng.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bird A, Bressler N, Bressler S, Chisholm I, Coscas G, Davis M, De Jong P, Klaver C, Klein B, Klein R, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol. 1995;39(5):367–74. doi: 10.1016/S0039-6257(05)80092-X. [DOI] [PubMed] [Google Scholar]
- 38.Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, Clayton D, Hurles ME. A robust statistical method for case–control association testing with copy number variation. Nat Genet. 2008;40(10):1245–52. doi: 10.1038/ng.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 40.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibbs D, Yang Z, Constantine R, Ma X, Camp NJ, Yang X, Chen H, Jorgenson A, Hau V, DeWan A, et al. Further mapping of 10q26 supports strong association of HTRA1 polymorphisms with age-related macular degeneration. Vision Res. 2008;48(5):685–9. doi: 10.1016/j.visres.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 42.DeGiorgio M, Lohmueller KE, Nielsen R. A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet. 2014;10(8):e1004561. doi: 10.1371/journal.pgen.1004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, et al. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5(5):e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bikker FJ, Ligtenberg AJ, End C, Renner M, Blaich S, Lyer S, Wittig R, van’t Hof W, Veerman EC, Nazmi K, et al. Bacteria binding by DMBT1/SAG/gp-340 is confined to the VEVLXXXXW motif in its scavenger receptor cysteine-rich domains. J Biol Chem. 2004;279(46):47699–703. doi: 10.1074/jbc.M406095200. [DOI] [PubMed] [Google Scholar]