

Abstract
Photoinitiation of polymerizations based on the copper(i)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction enables spatio-temporal control and the formation of mechanically robust, highly glassy photopolymers. Here, we investigated several critical factors influencing photo-CuAAC polymerization kinetics via systematic variation of reaction conditions such as the physicochemical nature of the monomers; the copper salt and photoinitiator types and concentrations; light intensity; exposure time and solvent content. Real time Fourier transform infrared spectroscopy (FTIR) was used to monitor the polymerization kinetics in situ. Six different di-functional azide monomers and four different tri-functional alkyne monomers containing either aliphatic, aromatic, ether and/or carbamate substituents were synthesized and polymerized. Replacing carbamate structures with ether moieties in the monomers enabled an increase in conversion from 65% to 90% under similar irradiation conditions. The carbamate results in stiffer monomers and higher viscosity mixtures indicating that chain mobility and diffusion are key factors that determine the CuAAC network formation kinetics. Photoinitiation rates were manipulated by altering various aspects of the photo-reduction step; ultimately, a loading above 3 mol% per functional group for both the copper catalyst and the photoinitiator showed little or no rate dependence on concentration while a loading below 3 mol% exhibited 1st order rate dependence. Furthermore, a photoinitiating system consisting of camphorquinone resulted in 60% conversion in the dark after only 1 minute of 75 mW cm−2 light exposure at 400–500 nm, highlighting a unique characteristic of the CuAAC photopolymerization enabled by the combination of the copper(i)’s catalytic lifetime and the nature of the step-growth polymerization.
Introduction
Owing to the “click” nature of being a robust, orthogonal, and efficient reaction,1,2 the copper(i)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction has been widely utilized in bio-conjugation,3-6 surface functionalization,7-11 as a coupling chemistry,11-14 for labeling,15-18 and in polymer synthesis, particularly in the formation of complex polymer architectures.19-22 Mechanistic and kinetic investigations of the CuAAC reactions have generally been performed in highly dilute, solution-based systems, where small molecules bearing alkyne and azide functional groups were evaluated in an attempt to improve the efficiency, yield, and rate of the CuAAC reaction by varying an array of reaction conditions, including solvent types and catalyst concentrations.23-25 Vast arrays of ligands,25-29 solvents,28,30 copper salts,25,28,31 alkynes,30-33 and azide moieties30,31,33 have been screened at varying concentrations to optimize and understand the kinetics of the CuAAC reaction, to define a general rate law, or to assess a mechanistic aspect of the reaction.34,35 However, the conclusions from these experiments as well as the detailed kinetic constant measurements are highly sensitive to the reaction conditions that were used, making it difficult to draw conclusions about kinetic behavior, particularly for the significant extrapolation necessary for bulk CuAAC polymerizations in which the azide and alkyne concentrations are dramatically higher and where the solubility of copper, diffusion and mobility of reacting species, and the heat of reaction all play a crucial role in the reaction/polymerization kinetics.
Since the discovery of the CuAAC reaction in 2001 by Meldal and Sharpless,36,37 the direct addition of copper(i) salts and/or a 3 to 10 equivalent excess of a reducing agent such as sodium ascorbate with copper(ii) salts have been widely used to initiate the CuAAC reaction.38 The insolubility of sodium ascorbate in organic media38 and the lack of temporal control when using copper(i) salts has hindered the development of homogeneous bulk polymers using solvent-free CuAAC. Few studies have attempted the CuAAC polymerization in bulk or investigated kinetics and properties of the resulting polymers. In 2004 and 2007, Liu et al. performed CuAAC polymerizations using solutions of multi-functional alkyne and azide monomers on copper substrates in order to analyze adhesive properties of CuAAC polymers.39,40 Later in 2010, Sheng et al. made solvent-free CuAAC linear polymers by the direct addition of copper(i) salts with limited concentrations of copper used due to solubility issues.41 The discovery and implementation of the photo-reduction of copper(ii) upon light exposure from several research groups promoted CuAAC polymerization as a means to enhance the spatio-temporal control of the reaction.42 In 2006, Ritter and König presented the photogeneration of copper(i) by the reduction of copper(ii) using an excitation of the chromophore in the presence of an electron donor.43 In 2009, Poloukhtine et al. discovered the photo-mediated copper-free azide–alkyne reaction using light to decompose cyclopropenones into cyclooctynes, which then proceed via a cycloaddition reaction with azides.44 Adzima et al. implemented spatio-temporal control of photo-CuAAC reactions as well as photo-polymerizations of multi-functional monomers using a visible light photoinitiator to reduce copper(ii),45 and Tasdelen et al. introduced UV-initiated CuAAC reactions based on electron charge transfer from amine ligands to copper(ii).46,47 Building on these approaches to the photoinduced reduction of copper, bulk photoinitiated CuAAC polymerization of crosslinked networks was successfully demonstrated in homogeneous and stable resin mixtures that contained multi-functional alkynes and azides. Specifically, Gong et al. reported kinetic profiles of bulk CuAAC polymerizations initiated by light in the presence of visible light photoinitiators,48 and Sandmann et al. presented photo-reduction of copper(ii) acetates via light without the presence of photo-initiators on the CuAAC resins containing at least 15 weight% methanol.49
Scheme 1 presents the photo-CuAAC reaction scheme that occurs when using a radical generating photoinitiator along with several plausible side reactions in four distinct stages: initiation, reduction of copper, cycloaddition, and termination. Initiation, in this case, involves the cleavage of photo-responsive compounds to generate radicals upon UV or visible light irradiation. Subsequently, the reduction of the copper(ii) species into catalytically active copper(i) occurs45 parallel to other competing reactions, such as re-oxidation of copper(i) to copper(ii), further reduction of copper(i) to copper(0), and disproportionation of copper(i) to copper(ii) and copper(0).23 The cycloaddition step itself is a complex, multi-step mechanism involving copper diffusion, σ- and π-coordination with alkynes, six-membered ring formation between copper-acetylides and azides, and the ultimate release of copper.50 Termination takes place when copper(i) loses its catalytic activity by oxidation or disproportionation. Previously, other mechanistic studies dealing with either experimental or computational modeling confirmed that the CuAAC reaction rate had a second order dependence on copper concentration,50 the formation of six-member rings during cycloaddition was a rate determining step,51 and other plausible side reactions such as alkyne coupling hindered the reaction rate by forming inactive species23,52 though all of these conclusions depend at least somewhat on the reaction conditions used.
Scheme 1.
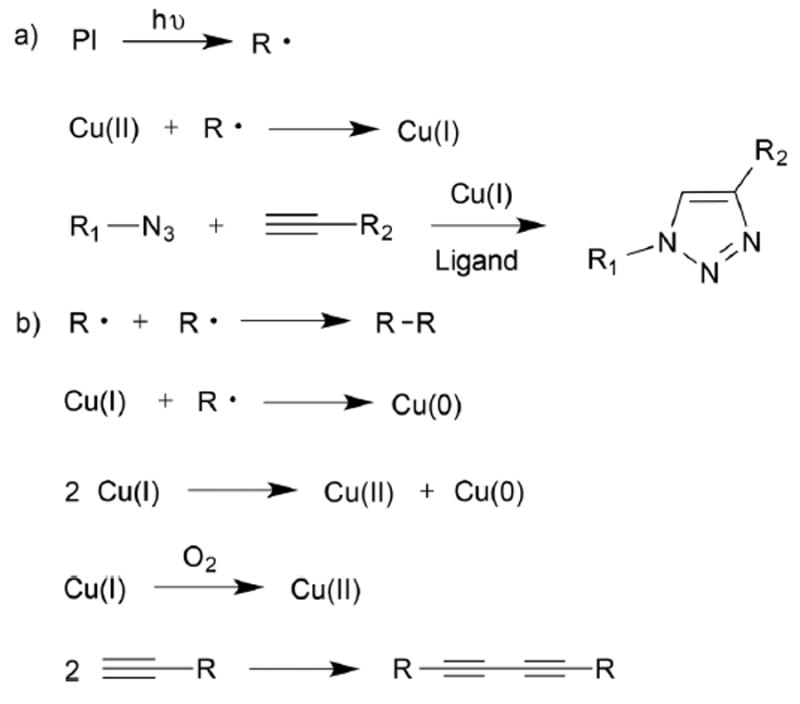
Proposed reaction diagram of one approach to photo-initiated CuAAC-based polymerizations: (a) photoinitiation, copper reduction to form Cu(i), and cycloaddition between azides and alkynes. (b) Side reactions that can potentially occur during the course of the reaction: radical coupling, copper disproportionation, copper oxidation, and alkyne coupling reactions.
The nature of step-growth polymerizations enables the CuAAC polymerization to form relatively homogeneous polymer networks,53 where the rigid-aromatic triazole adducts formed throughout the network as a product of the CuAAC reactions exhibit excellent thermal and chemical stability, while also increasing the polymer stiffness and glass transition temperature.1,48 However, the azide moieties can be explosive when sufficiently concentrated; therefore, designing higher molecular weight azide monomers is essential to enable bulk polymerizations to be performed safely and efficiently.54 In addition, the solubility of copper in organic substrates is often insufficient, either requiring an addition of chelating ligands to increase solubility or only allowing for minimal concentrations of copper to be incorporated into the resin mixtures.31 Due to the aforementioned challenges, previous investigations of the CuAAC polymerization kinetics in bulk are limited. Herein, we explore the effects of monomer structure, copper and photoinitiator concentrations, light exposure conditions, temperature, solvent, light intensity, and irradiation times on the rate of bulk CuAAC polymerization to understand this complex polymerization and enable the determination of optimal polymerization conditions for spatially and temporally controlled formation of photopolymerized CuAAC thermosets.
Experimental section
Materials
1,3-Bis(isocyanatomethyl)cyclohexane, 4,4-methylenebis(cyclohexyl isocyanate), 1,3-bis(2-isocyanatopropan-2-yl)benzene, 4,4′-methylenebis(phenyl isocyanate), bis(4-hydroxyphenyl)-methane, 6-chloro-1-hexanol, dibutyltin dilaurate, sodium azide, 1,1,1-tris(hydroxymethyl)propane, pentaerythritol, 1,3,5-tris(bromomethyl)benzene, phloroglucinol, propargyl alcohol, sodium hydride, diethyl azodicarboxylate, tetrabutylammonium iodide, N,N,N′,N′,N″ -pentamethyldiethyl-enetriamine (PMDETA), copper(ii) chloride, triphenyl-phosphine, 2,2-dimethoxy-2-phenylacetophenone (DMPA), propargyl bromide, camphorquinone (CQ), tetrahydrofuran, and acetonitrile were used as received from Sigma Aldrich. 2,2,4,4-tetramethyl-1,3-cyclobutanediol, 5-hexyn-1-ol, hexyl isocyanate, 6-chloro-1-hexyne, 1-phenyl-1,2-propanedione (PPD), 2,2-bis-(bromomethyl)-1,3-propanediol, sodium hydroxide, potassium carbonate, potassium hydroxide, hydrochloric acid, methanol, acetone, methylene chloride, and dimethylformamide were used as received from Fisher Scientific. Diphenyl(2,4,6-trimethylbenzoyl)-phosphine oxide (Lucirin-TPO) was used as received from VWR International. Bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide (I819) was used as received from BASF. All azides were synthesized according to the azide safety rules and handled with appropriate care and precaution, and generally working with the monomers, resins and polymers in small quantities.54 Three facile reaction schemes, alcoholysis of isocyanates, Mitsunobu, and Williamson ether, were used to synthesize di-functional azides and multi-functional alkynes as indicated in Fig. 1. All NMR measurements and yields of monomers are presented in the ESI.†
Fig. 1.

Monomer libraries with systematic structural variations for difunctional azides 2a–2f and multifunctional alkynes 3–8, photoinitiators, and copper catalysts used in the bulk photo-CuAAC photopolymerizations studied here.
Synthesis of dicarbamate halide intermediates (1a–1d)
A solution of diisocyanate – 1,3-bis(isocyanatomethyl)cyclohexane (a), 4,4-methylenebis(cyclohexyl isocyanate) (b), 1,3-bis-(2-isocyanatopropan-2-yl)benzene (c), or 4,4′-methylenebis (phenyl isocyanate) (d) (4.09 mmol) – and dibutyltin dilaurate (5 drops) in THF (3 mL) was added in a round bottom flask and purged under nitrogen. The reaction mixture was cooled to 0 °C in an ice bath, followed by dropwise addition of 6-chloro-1-hexanol (8.60 mmol, 1.17 g). Removal of the ice bath allowed the reaction mixture to stir at room temperature for 12 h. The reaction mixture was then flowed through a silica plug with excess THF and purified by column chromatography if necessary. The product – dicarbamate chlorides – was dried in vacuo as a colorless oil (1a–1c) or white solid (1d).
Synthesis of dicarbamate halide intermediates (1f)
A solution of hexyl isocyanate (34.3 mmol, 5 ml) and dibutyltin dilaurate (5 drops) in THF (20 mL) was added in a round bottom flask and purged under nitrogen. The reaction mixture was cooled to 0 °C in an ice bath, followed by dropwise addition of a solution of 2,2-bis(bromomethyl)-1,3-propanediol (f) (17.2 mmol, 4.5 g) in THF (10 ml). Removal of the ice bath allowed the reaction mixture to stir at room temperature for 12 h. The reaction mixture was then flowed through a silica plug with excess THF. The product was recrystallized in ethyl acetate as a white solid (1f).
Synthesis of carbamate diazides (2a–2d, 2f)
A solution of dicarbamate chlorides (1a–1d, 1f) (4.15 mmol) and sodium azides (16.6 mmol, 1.08 g) in DMF (30 ml) was added to a round bottom flask connected with a reflux condenser. The reaction mixtures containing (1a–1d) were stirred at 80 °C for 12 h and for (1f) for 30 h. The product was extracted with ethyl acetate and water, dried with Na2SO4, purified by column chromatography if necessary, and dried in vacuo as a colorless oil (2a–2c) or a white solid (2d,2f).
Synthesis of ether diazides, (2e)
A solution of bis(4-hydroxy-phenyl)methane (4.99 mmol, 1 g), 6-chloro-1-hexanol (14.9 mmol, 2.05 g), and triphenylphosphine (14.9 mmol, 3.93 g) in THF (5 ml) was added to a round bottom flask and placed in an ice bath inside a sonicator. After dropwise addition of diethyl azodicarboxylate (14.9 mmol, 5.88 ml of a 40% solution in toluene) at 0 °C, the reaction mixture was sonicated for 2 h and then stirred for 12 h at room temperature. Triphenylphosphine was removed from the reaction mixture by crystallization in ethyl acetate. The product –diether chlorides – was dried in vacuo as a colorless oil, and sodium azides (20.6 mmol) in DMF (60 ml) was added to a round bottom flask connected with a reflux condenser. The reaction mixture was stirred at 80 °C for 12 h. The product was extracted with ethyl acetate, water, and 1 M NaOH, dried with Na2SO4, purified by column chromatography if necessary, and dried in vacuo as a colorless oil.
Synthesis of trialkynes, (3)
A solution of 1,1,1-tris(hydroxymethyl)propane (14.7 mmol, 1.97 g) and 40 w/w% NaOH/water in DMSO (15 ml) was added in a round bottom flask and stirred for 1 h at room temperature. After dropwise addition of propargyl bromide (94 mmol, 8.9 ml of 80% solution in toluene), the reaction mixture was stirred for 5 days. The product was extracted with diethyl ether and water, dried with Na2SO4, purified by column chromatography if necessary, and dried in vacuo as a colorless oil.
Synthesis of tetraalkynes, (4)
A solution of pentaerythritol (73.45 mmol, 10 g), KOH (1016 mmol, 57 g), and TBAI (0.95 mmol, 0.35 g) in THF (250 ml) was added to a round bottom flask connected with a reflux condenser under a nitrogen purge. After dropwise addition of propargyl bromide (691 mmol, 65.45 ml of 80% solution in toluene), the reaction mixture was stirred for 3.5 h at 70 °C. The reaction mixture was extracted with ethyl acetate, water, and 1 M NaOH, dried with Na2SO4. The product was recrystallized in ethyl acetate at 0 °C as a yellow solid.
Synthesis of trialkynes, (5)
A solution of propargyl alcohol (4.51 mmol, 0.25 g) and 60% NaH (4.51 mmol, 0.18 g) oil dispersion in DMF (15 ml) was added in a round bottom flask under a nitrogen purge at 0 °C in an ice bath. After 10 min of stirring, 1,3,5-tris(bromomethyl)benzene (1.40 mmol, 0.50 g) was added to the reaction mixture and stirred for 24 h at room temperature. The reaction mixture was neutralized with HCl. The product was extracted with ethyl acetate and water, dried with Na2SO4, purified by column chromatography if necessary, and dried in vacuo as a yellow oil.
Synthesis of trialkynes, (6)
A solution of phloroglucinol (79.3 mmol, 10.0 g) and K2CO3 (476 mmol, 65.8 g) in DMF (500 ml) was added in a round bottom flask connected with a reflux condenser under a nitrogen purge. After dropwise addition of propargyl bromide (560 mmol, 53 ml of an 80% solution in toluene), the reaction mixture was stirred for 24 h at 80 °C. The reaction mixture was extracted with ethyl acetate, water, and 1 M NaOH, dried with Na2SO4. The product was recrystallized in methanol as a white solid.
Synthesis of trialkynes, (7)
A solution of phloroglucinol (7.93 mmol, 1.0 g) and K2CO3 (47.6 mmol, 6.58 g) in DMF (50 ml) was added in a round bottom flask connected with a reflux condenser under a nitrogen purge. After dropwise addition of 6-chloro-1-hexyne (55.50 mmol, 6.8 ml), the reaction mixture was stirred for 24 h at 100 °C. The reaction mixture was extracted with ethyl acetate, water, and 1 M NaOH, dried with Na2SO4, purified by column chromatography, and dried in vacuo as a white solid.
Synthesis of dialkynes, (8)
A solution of 2,2,4,4-tetramethyl-1,3-cyclobutanediol (6.93 mmol, 1 g), KOH (47.15 mmol, 2.65 g), and TBAI (0.048 mmol, 17.5 mg) in THF (20 ml) was added to a round bottom flask connected with a reflux condenser under a nitrogen purge. After dropwise addition of propargyl bromide (27.74 mmol, 3 ml of 80% solution in toluene), the reaction mixture was stirred for 3.5 h at 70 °C. The reaction mixture was extracted with ethyl acetate, water, and 1 M NaOH, and dried with Na2SO4. The product was flowed through a silica plug with hexane/ethyl acetate solvent system (9 : 1) and recrystallized from methanol as a white solid.
Preparation of CuCl2[PMDETA] complex
1 : 1 molar mixture of CuCl2 and PMDETA (N,N,N′,N′,N″-pentamethyldiethyl-enetriamine) in acetonitrile was stirred overnight at room temperature and dried in vacuo to a blue-green solid.
Methods
Sample preparation
Stoichiometric mixtures of a diazide, trialkyne (1 : 1 N3 : alkyne), and various mole percentages of CuCl2[PMDETA] and photoinitiator per functionality were prepared. Methanol, DCM, or acetone was used to homogenize the mixture, depending on the solubility of the resin mixtures with copper, and was later removed in vacuo. The solvent content of each resin was verified by 1H-NMR using a Bruker Avance-III 400 MHz spectrometer with 16 scans s−1 and 1 s of relaxation time prior to any polymerization.
Fourier transform infrared spectroscopy
An FTIR spectrometer (Nicolet 8700, Fisher Scientific) incorporated with a heating stage was used to monitor the real-time polymerization kinetics of the functional group conversion in transmission mode. Irradiation was performed using a light guide connected to a mercury lamp (Acticure 4000, EXFO) with either a 365 nm or 400–500 nm bandgap filter, depending on the photoinitiator used. Samples were placed between NaCl plates, and the azide peak was monitored in the absorption range between 2300–2000 cm−1 having the alkane C–H stretching bonds as a reference peak between 2980–2840 cm−1 with 12 scans s−1 and 2 cm−1 resolution.
Results and discussion
Monomer structural variations
Combinations of azide and alkyne monomers containing monomers of various structure and functionality were examined while maintaining all other reaction conditions the same, including the copper loading, photoinitiator concentration, light exposure time and intensity, and temperature. The resulting CuAAC polymerization kinetics are shown in Fig. 2 and 3. Generally, despite being initiated by relatively low light intensities (only 10 mW cm−2), these polymerizations are largely complete after only 2–5 minutes of irradiation though persistent polymerization does occur well after exposure is complete, indicative of the long lifetime of the catalytic Cu(i) species formed during the exposure period.
Fig. 2.
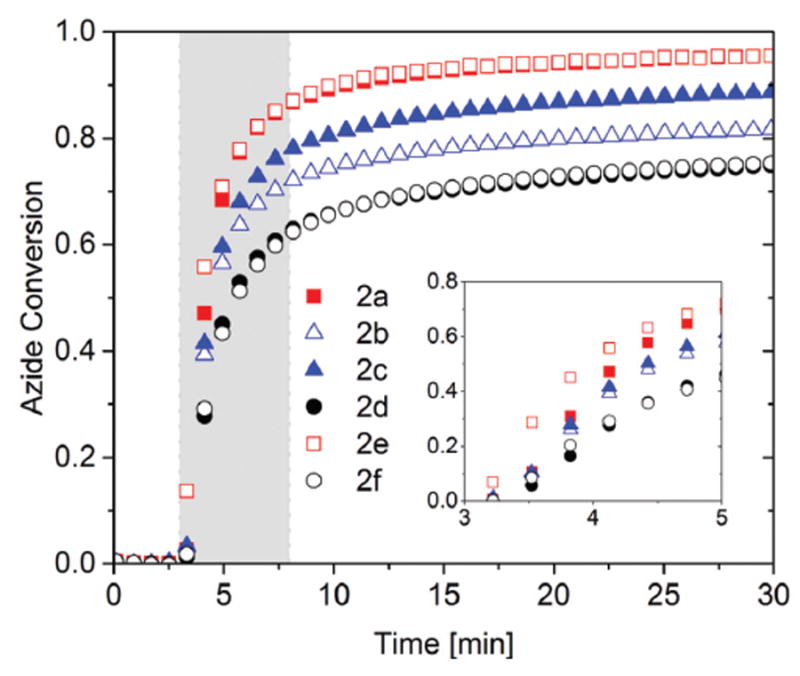
Bulk photo-CuAAC polymerization kinetics as measured by FTIR. 1 : 1 azide : alkyne mixture with varying azide structures, including 2a (closed square), 2b (open triangle), 2c (closed triangle), 2d (closed circle), 2e (open square), 2f (open circle). Each azide was polymerized stoichiometrically with alkyne 3 in the presence of 2 mol% CuCl2[PMDETA], 4 mol% DMPA per azide functional group, and <0.5 wt% methanol (<0.5 wt% acetone for 2e). Each mixture was irradiated for 5 min (gray shaded area) at 50 °C with 10 mW cm−2 of 365 nm light following 3 minutes in the dark as a baseline measuring period.
Fig. 3.
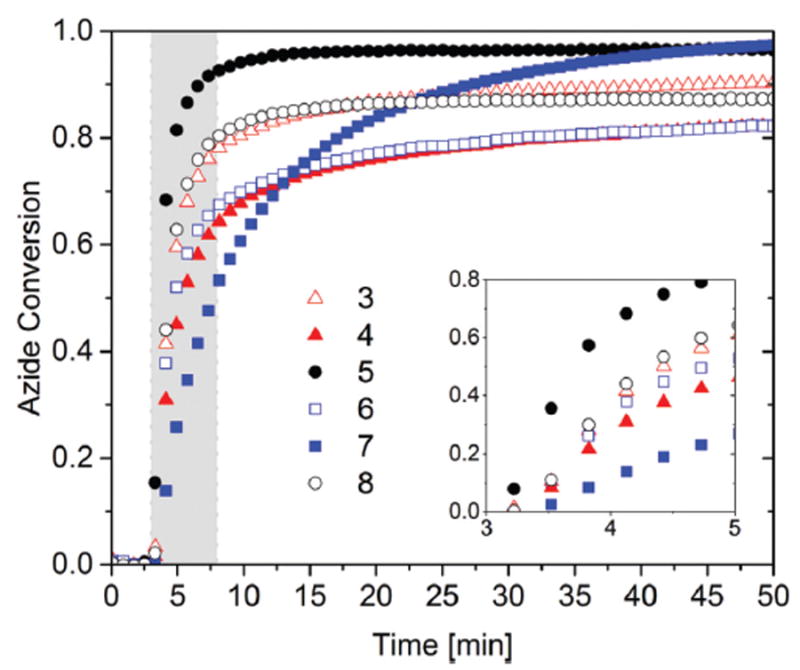
Bulk photo-CuAAC polymerization kinetics using FTIR. 1 : 1 azide : alkyne mixture with varying alkyne structures, including 3 (open triangle), 4 (closed triangle), 5 (closed circle), 6 (open square), 7 (closed square), 8 (open circle). Each alkyne was polymerized stoichiometrically with azide 2c in the presence of 2 mol% CuCl2[PMDETA], 4 mol% DMPA per azide functional group, and <0.5 wt% methanol. Each mixture was irradiated for 5 min (gray shaded area) at 50 °C with 10 mW cm−2 of 365 nm light following 3 minutes in the dark as a baseline measuring period.
As shown in Fig. 2, structural elements within the azide monomers such as the presence of either aromatic or non-aromatic cores and either the carbamate or ether linkages in the monomer backbones affect both the initial polymerization rate and final conversion significantly. Azides having cyclohexane cores, 2a and 2b, resulted in slightly more rapid initial polymerization rates and approximately 6.5% higher final conversion after 30 minutes, as compared to azides bearing aromatic cores, 2c and 2d. Similarly, an azide monomer 2e containing an ether linkage in contrast to a carbamate linkage as in monomer 2d, showed a two-fold increase in the initial polymerization rate during the first 2 minutes of irradiation and resulted in approximately 20% higher maximum conversion after 30 minutes. From the viscosity measurement for the pure azides at 50 °C via rheometry, the viscosity of the azide monomers with a single ring core, 2a and 2c, was 0.18 Pa s, while the viscosity of the azide monomers having two ring cores, 2b and 2d, was 5.8 Pa s at a shear rate of 20 s−1 (see Table S1†). Similarly, the azide monomer 2e containing an ether linkage had a viscosity of 0.05 Pa s, approximately 100 times less viscous than the azide monomer 2d with a carbamate linkage (Table S1†). By correlating the viscosity of the monomers with the kinetic profiles from Fig. 2, it is clear that increasing the monomer viscosity results in a reduction of both the initial rate of polymerization and the maximum conversion. The effect of viscosity on the polymerization rate is likely caused by diffusional limitations, either of the initiation reaction or of the CuAAC reaction itself. A similar effect of increasing viscosity was also observed in resins with azides 2f and 2d as shown in Fig. 2.
Azide 2f contains two azide functional groups held in close proximity by sterically hindered carbamate side groups. This short distance between the two azides has previously been reported to accelerate the CuAAC reaction, proposing that the formation of a first triazole works as a ligand to aid copper coordination for the very proximate neighboring azide.55 However, no significant difference in kinetics was observed between azides 2d and 2f, suggesting that this proximal effect is of minimal importance in these bulk, highly concentrated reaction environments.
As observed in Fig. 3, structural variations in the alkyne monomers have a pronounced effect on the initial polymerization rate and the final conversion. Alkynes 3, 4, 5, 6, and 8 showed a noticeable increase in the average initial polymerization rate, as compared to alkyne 7, during the first 2 minutes of irradiation, mainly due to the higher reactivity of an alkyne functional group next to an ether linkage compared to a hydrocarbon linkage.32,34 The alkyne reactivity was also confirmed through a study of small molecule model compound reactivity in solution by FTIR, using a 2 M solution in DMF of propargyl alcohol or 5-hexyn-1-ol, difunctional azides 2c, 2% CuCl2-[PMDETA], and 4% DMPA, irradiated at ambient temperature (Fig. S1†). The average initial reaction rate using propargyl alcohol was 4.5 mol (L min)−1 while the average initial rate using 5-hexyn-1-ol was only 1.7 mol (L min)−1, suggesting approximately 2.6 times higher reactivity of propargyl alcohol towards the CuAAC reaction under these conditions. However, despite a slower average initial polymerization rate as compared with alkyne 6, a resin containing alkyne 7 reached 97% conversion after 50 minutes, as compared to alkyne 6 which reached only 82% conversion after 50 minutes. Longer hydrocarbon linkages between the alkyne functional groups in 7 in contrast to 6 provide flexibility to the resin, which aids in increasing the final conversion, as long as the copper(i) species are able to remain active. Furthermore, by increasing the alkyne functionality from 3 to 4, alkyne 4 yielded only 82% conversion after 50 minutes while alkyne 3 achieved 90% conversion after the same time. However, when the alkyne functionality was further reduced to 2, alkyne 8 only exhibited 87% conversion after 50 minutes, where the number average degree of polymerization is predicted to be 8 repeat units using the Carothers equation, which corresponds to a number average molecular weight of 6000 (PDI = 1.9). Clearly, the final conversion is strongly affected by vitrification and structural elements of the monomer that impact the final glass transition temperature of the polymer will have a significant effect on the final conversion as well. Specifically, the correlation between conversion and the structural rigidity of monomers, rather than the reactivity of functional groups, suggests that the kinetics of CuAAC bulk polymerizations are highly diffusion-limited, especially at the later stages of polymerization.
Fig. 4 illustrates the effect of the presence of solvent on the final conversion of the CuAAC polymerization via variations in methanol concentration within a single resin mixture. With increasing methanol content, one expects the initial viscosity and final glass transition temperature for the polymer to be reduced. Here, the primary effect was found to be the plasticization of the methanol which increases chain mobility.56 The conversion after 10 minutes reaction time was increased dramatically by the presence of the methanol with a negligible influence on the initial polymerization rate. In addition, the effect of methanol content on polymerization conversion was only significant at ambient temperature, while negligible differences were observed at elevated temperature, 50 °C.
Fig. 4.
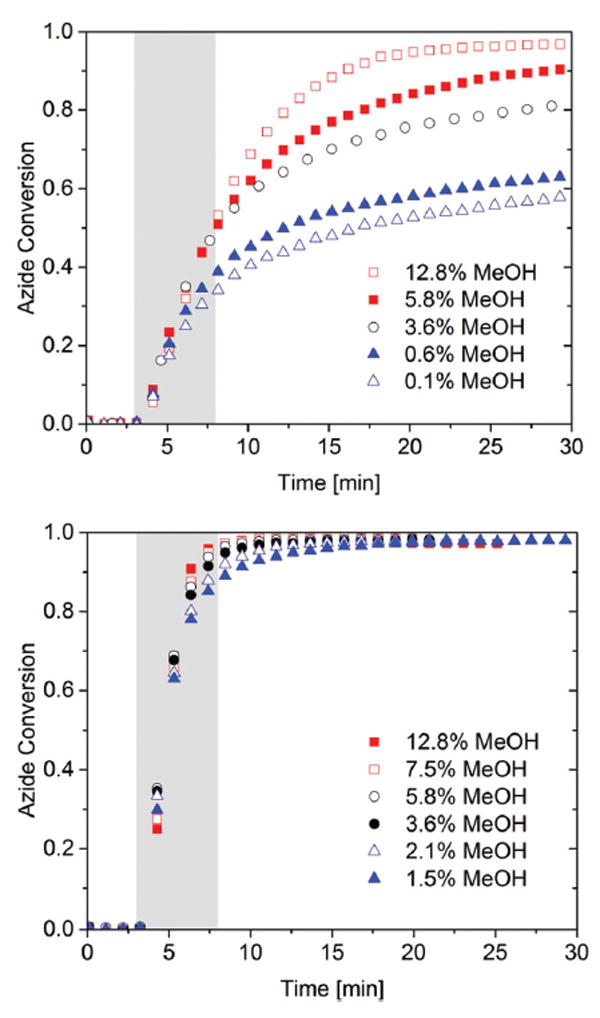
Bulk photo-CuAAC polymerization kinetics using FTIR. 1 : 1.2 azide : alkyne mixture with varying methanol concentration ranging from 0.1 to 12.8% by weight. Azide 2c was polymerized with alkyne 3 in the presence of 2 mol% CuCl2[PMDETA], 4 mol% DMPA per azide functional group, and methanol. Each mixture was irradiated for 5 min (gray shaded area) at (top) ambient temperature and (bottom) 50 °C with 10 mW cm−2 of 365 nm light following 3 minutes in the dark as a baseline measuring period.
Visible light photoinitiators
In Fig. 5, a variety of visible light photoinitiators, including CQ (camphorquinone), PPD (1-phenyl-1,2-propanedione), I819 (bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide), and Lucirin TPO (2,4,6-trimethylbenzoyl-diphenylphosphine oxide) – were tested to examine their effectiveness in initiating the CuAAC polymerization. A 1 : 1 stoichiometric mixture of azide 2c and alkyne 3 with 2 mol% CuCl2[PMDETA] and 2 mol% visible light photoinitiators was polymerized using 10 mW cm−2 of 400–500 nm light. For all cases, over 60% conversion was achieved with substantially different initial polymerization rates under the same condition tested. The most obvious behavior observed was that rapid initial polymerization rates were only achieved for photoinitiators with higher molar extinction coefficient such as TPO and I819;57 however, relatively high conversions were obtained using the CQ initiation system over an extended time period, indicative of the versatility of copper reduction by nearly any radical initiator and the longevity of the copper(i) catalyst that is formed.
Fig. 5.
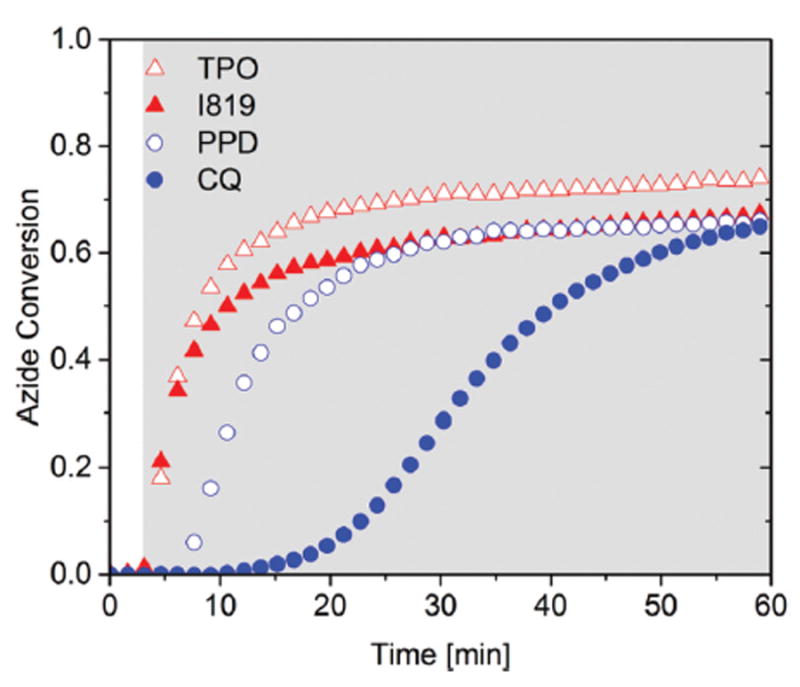
Bulk photo-CuAAC polymerization kinetics using FTIR. 1 : 1 azide : alkyne mixture with varying photoinitiators, including TPO (open triangle), I819 (closed triangle), PPD (open circle), CQ (closed circle). Azide 2c was polymerized stoichiometrically with alkyne 3 in the presence of 2 mol% CuCl2[PMDETA], 2 mol% photoinitiators per azide functional group, and <1 wt% DCM. Each mixture was irradiated continuously (gray shaded area) at 35 °C with 10 mW cm−2 of 400–500 nm light following 3 minutes in the dark as a baseline measuring period.
Copper & photoinitiator concentration
Fig. 6 presents the effects of the photoinitiator and copper concentration on the average initial polymerization rate, as measured by the time required to react from 10% to 30% conversion. In order to minimize the number of variables affecting the polymerization rate other than the concentration of copper and photoinitiator, one specific reaction condition was chosen as follows: azide 2c and alkyne 3 were polymerized stoichiometrically with CuCl2[PMDETA] as a catalyst, and CQ as a visible light photoinitiator, with continuous irradiation at a light intensity of 75 mW cm−2, wavelength range of 400–500 nm, and a temperature of 35 °C. It should be noted that the absorption efficiency of the photoinitiator, molar extinction coefficient of the photoinitiator, the solubility of copper(ii), the stability of copper(i) and several other factors are all also important factors controlling initiation and copper reduction that are intended to be preserved by using a single polymerization condition.
Fig. 6.

The average initial polymerization rate taken between 10% to 30% conversion from FTIR as a function of the copper concentration. A 1 : 1 azide : alkyne mixture with varying copper and photoinitiator concentration. Azides 2c was polymerized stoichiometrically with alkynes 3 in the presence of different molar ratio of CuCl2[PMDETA] and CQ per azide functional group, and <1 wt% methanol. Each mixture was irradiated for continuous at 35 °C with 75 mW cm−2 of 400–500 nm light following 3 minutes in the dark as a baseline measuring period. [PI]/[Cu] = 1 (closed square) and [PI]/[Cu] = 2 (closed circle) indicate the molar ratio of the photoinitiator to copper (PI : Cu) is fixed at 1 and 2 while [PI] = 1 (closed triangle) represents when the photoinitiator concentration is fixed at 1 mol% with varying copper concentration.
For both PI : Cu ratios cases, the average initial polymerization rates increased linearly as the copper concentration varied from 0 mol% to 3 mol%, indicating that the initial rate is first order in copper concentration under these circumstances. However, as the concentration of copper increased from 3 mol% to 5 mol%, either a subtle increase or a plateau in the average polymerization rate was observed. It is worth noting that precipitation of copper catalysts started to appear for resin formulations containing higher copper loadings above 4 mol% several hours after mixing. This slight increase or plateau in the rate regardless of higher copper and photo-initiator loadings is possibly due to phenomena associated with various CuAAC reactions. First, at higher copper concentrations, disproportionation of copper(i), aggregation and/or precipitation of insoluble copper species can occur rapidly, and each one promotes the formation of inactive copper species. Furthermore, at higher photoinitiator concentrations, a higher density of radicals generated from the photoinitiator increases radical–radical recombination/termination reactions which then eliminates radicals that would otherwise be available to reduce copper.
For the case when the PI concentration is 1 mol%, the average initial polymerization rates remained constant as the copper concentration varied from 1 mol% to 5 mol%. This constant rate over a wide range of copper loadings indicates that copper reduction which strongly dictates the average initial polymerization rates is highly restricted by insufficient amounts of photoinitiator relative to the copper.
Light exposure
To probe the influence of light intensity and exposure dose on the polymerization rate, a single resin formulation consisting of a 1 : 1 stoichiometric mixture of azide 2c and alkyne 3 with 2 mol% CuCl2[PMDETA] and 2 mol% CQ was polymerized using different light intensities and exposure times. Fig. 7 demonstrates the effects of light intensity on the polymerization kinetics. The initial polymerization rate was significantly improved by increasing light intensity using CQ, mainly because the lower molar extinction coefficient of CQ requires higher light intensity to effectively generate radicals without the presence of excess amine as a co-initiator.58 It must be noted that at 75 mW cm−2, a 5 °C temperature increase was observed, due to the heat generated at the higher light intensity. The small increase in temperature also serves to accelerate the CuAAC polymerization, though not enough to significantly increase conversion. Interestingly, the lag time between the start of irradiation and the maximum rate of polymerization increased monotonically with decreasing light intensity, ranging from 3 minute to 26 minutes under the conditions tested. After 60 minutes of irradiation, all samples with different light intensities resulted in a conversion over 60%.
Fig. 7.

Bulk photo-CuAAC polymerization kinetics using FTIR. 1 : 1 azide : alkyne mixture with varying light intensity, including 75 mW cm−2 (closed triangle), 50 mW cm−2 (open triangle), 20 mW cm−2 (closed square), 10 mW cm−2 (open square). Azide 2c was polymerized stoichiometrically with alkyne 3 in the presence of 2 mol% CuCl2[PMDETA], 2 mol% CQ per azide functional group, and <1 wt% DCM. Each mixture was irradiated continuously (gray shaded area) at 35 °C with different light intensities of 400–500 nm light following 3 minutes in the dark as a baseline measuring period.
As shown in Fig. 8, different exposure times at a fixed light intensity, 75 mW cm−2, were screened to define an optimal exposure dose required to yield the maximum polymerization rate. Exposure times greater than 3 minutes were found to have a limited benefit, as essentially the same kinetic profiles were observed as with continuous irradiation. In order to determine the concentration of CQ after 3 minutes of light exposure, a solution containing 0.044 M CQ with 0.044 M PMDETA as a coinitiator in methanol was irradiated using 75 mW cm−2 of light irradiation at 400–500 nm (Fig. S2–3†) and the absorption spectra was measured at different exposure times. Approximately 70% of the CQ was bleached following 3 minutes of light exposure in the presence of PMDETA. Given that outcome in an optically thick sample, one would reasonably expect that at elevated temperatures and in the presence of monomers also bearing carbamate functionality which provide an additional coinitiator source for CQ,59 CQ is largely decomposed during a 3 minute irradiation period used in Fig. 8. Thus, the lack of any benefit associated with continuous exposure after this period likely results from the lack of generation of any additional radicals that would be capable of reducing Cu(ii) to Cu(i). Further, from the emission spectra of the mercury arc lamp with 400–500 nm band pass filter and using the absorbance spectra of the CQ photoinitiator as measured by the UV/Vis spectrometer, approximately 2 moles of photons are absorbed per mole of CQ during 3 minutes of irradiation (Fig. S4†). With only 3 minutes of exposure time, 82% azide conversion was achieved after 60 minutes, but approximately 55% of this reaction conversion occurred after the light was turned off. Interestingly, this extended dark polymerization was also observed after as little as 1 minute of irradiation; less than 2% conversion was obtained during the first minute of irradiation, but an additional 60% conversion was achieved during 56 minutes of time in the dark. Furthermore, the cases of 1, 2, and 3 minutes of irradiation all achieved more than 50% conversion in the dark despite the differences in the initial polymerization rate and the final conversion at 60 minutes. This extent of dark polymerization highlights the longevity of the catalytically active Cu(i) species as compared to conventional radical processes, where radical termination events cease the polymerization rapidly after initiation is halted.
Fig. 8.

Bulk photo-CuAAC polymerization kinetics using FTIR. 1 : 1 azide : alkyne mixture with varying exposure times, including 0 minute (closed circle), 0.5 minute (open circle), 1 minute (closed triangle), 2 minutes (open triangle), 3 minutes (closed square), 60 minutes (open square). Azide 2c was polymerized stoichiometrically with alkyne 3 in the presence of 2 mol% CuCl2[PMDETA], 2 mol% CQ per azide functional group, and <1 wt% methanol. Each mixture was irradiated at 35 °C with 75 mW cm−2 of 400–500 nm light following 3 minutes in the dark as a baseline measuring period. Only the start of light irradiation is highlighted in gray dotted line.
Conclusions
The kinetics of CuAAC photopolymerizations are highly influenced by the resin viscosity and the ultimate glass transition temperature of the polymer, both being strongly dictated by the monomer structure. CuAAC photopolymerizations exhibit a rapid initial rate followed by the attainment of a maximum conversion that is limited by vitrification and therefore increases dramatically with the addition of plasticizers. The most efficient photo-reduction of copper occurred here when 3 mol% of the copper per functional group were present. Of significant practical benefit, it was demonstrated that the copper(i) catalyst persists long after irradiation is ceased causing polymerization to continue without additional light exposure. Although this extent of dark polymerization limits the temporal control of the photo-induced CuAAC polymerization, controlled initiation of the reaction is readily achieved. For numerous applications, the persistence of the polymerization long after exposure is a significant benefit, enabling dark polymerization for an extended time following only a short exposure. For example, in one instance, approximately 50% to 60% conversion occurred in the dark after only one minute of irradiation.
Supplementary Material
Acknowledgments
The authors acknowledge financial support from the National Institutes of Health (NIH:5UO1DE023774) and the National Science Foundation (NSF:CHE1214109).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01655j
References
- 1.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Barner-Kowollik C, Du Prez FE, Espeel P, Hawker CJ, Junkers T, Schlaad H, Van Camp W. Angew Chem, Int Ed. 2011;50:60–62. doi: 10.1002/anie.201003707. [DOI] [PubMed] [Google Scholar]
- 3.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem, Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Presolski SI, Hong VP, Finn MG. Curr Protoc Chem Biol. 2011;3:153–162. doi: 10.1002/9780470559277.ch110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mckay CS, Finn MG. Chem Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Díaz Velázquez H, Ruiz García Y, Vandichel M, Madder A, Verpoort F. Org Biomol Chem. 2014;12:9350–9356. doi: 10.1039/c4ob01350f. [DOI] [PubMed] [Google Scholar]
- 7.Vutti S, Buch-Månson N, Schoffelen S, Bovet N, Martinez KL, Meldal M. ChemBioChem. 2015;16:782–791. doi: 10.1002/cbic.201402629. [DOI] [PubMed] [Google Scholar]
- 8.Smyth T, Petrova K, Payton NM, Persaud I, Redzic JS, Graner MW, Smith-jones P, Anchordoquy TJ. Bio-conjugate Chem. 2014;25:1777–1784. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beauvilliers EE, Topka MR, Dinolfo PH. RSC Adv. 2014;4:32866. [Google Scholar]
- 10.Wang C, Podgórski M, Bowman CN. Mater Horiz. 2014;1:535–539. [Google Scholar]
- 11.Iha RK, Wooley KL, Nyström AM, Burked DJ, Kade MJ, Hawker CJ. Chem Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang C, Wu B, Chen Y, Zhang K. Macromol Rapid Commun. 2015;36:750–754. doi: 10.1002/marc.201400717. [DOI] [PubMed] [Google Scholar]
- 13.Macdonell A, Johnson NAB, Surman AJ, Cronin L. J Am Chem Soc. 2015;137:5662–5665. doi: 10.1021/jacs.5b02466. [DOI] [PubMed] [Google Scholar]
- 14.Reuther JF, Siriwardane DA, Kulikov OV, Batchelor BL, Campos R, Novak BM. Macromolecules. 2015;48:3207–3216. [Google Scholar]
- 15.Sibbersen C, Lykke L, Gregersen N, Jørgensen KA, Johannsen M. Chem Commun. 2014;50:12098–12100. doi: 10.1039/c4cc05246c. [DOI] [PubMed] [Google Scholar]
- 16.Hu Q, Deng X, Kong J, Dong Y, Liu Q, Zhang X. Analyst. 2015;140:4154–4161. doi: 10.1039/c5an00566c. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Gu Z, Liu L, Wang S, Xing G. Chem Commun. 2015;51:8606–8609. doi: 10.1039/c5cc01907a. [DOI] [PubMed] [Google Scholar]
- 18.Hong V, Steinmetz NF, Manchester M, Finn MG. Bioconjugate Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doran S, Yagci Y. Polym Chem. 2015;6:946–952. [Google Scholar]
- 20.Lu D, Hossain MD, Jia Z, Monteiro MJ. Macromolecules. 2015;48:1688–1702. [Google Scholar]
- 21.Iskin B, Yilmaz G, Yagci Y. Polym Chem. 2011;2:2865. [Google Scholar]
- 22.Liang L, Astruc D. Coord Chem Rev. 2011;255:2933–2945. [Google Scholar]
- 23.Berg R, Straub BF. Beilstein J Org Chem. 2013;9:2715–2750. doi: 10.3762/bjoc.9.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hein JE, Fokin VV. Chem Soc Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng JC, Fokin VV, Finn MG. Tetrahedron Lett. 2005;46:4543–4546. [Google Scholar]
- 26.Rodionov VO, Presolski SI, Díaz DD, Fokin VV, Finn MG. J Am Chem Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 27.Presolski SI, Hong V, Cho SH, Finn MG. J Am Chem Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamura T, Asano K, Matsubara S. Org Lett. 2010;12:4988–4991. doi: 10.1021/ol101990d. [DOI] [PubMed] [Google Scholar]
- 29.Lewis WG, Magallon FG, Fokin VV, Finn MG. J Am Chem Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 30.Shao C, Zhu R, Luo S, Zhang Q, Wang X, Hu Y. Tetrahedron Lett. 2011;52:3782–3785. [Google Scholar]
- 31.Gonda Z, Novák Z. Dalton Trans. 2010;39:726–729. doi: 10.1039/b920790m. [DOI] [PubMed] [Google Scholar]
- 32.Kislukhin AA, Hong VP, Breitenkamp KE, Finn MG. Bioconjugate Chem. 2013;24:684–689. doi: 10.1021/bc300672b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell-Verduyn LS, Mirfeizi L, Dierckx RA, Elsinga PH, Feringa BL. Chem Commun. 2009:2139–2141. doi: 10.1039/b822994e. [DOI] [PubMed] [Google Scholar]
- 34.Michaels HA, Zhu L. Chem – Asian J. 2011;6:2825–2834. doi: 10.1002/asia.201100426. [DOI] [PubMed] [Google Scholar]
- 35.Meldal M, Tornøe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 36.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 38.Binder WH, Sachsenhofer R. Macromol Rapid Commun. 2008;29:952–981. [Google Scholar]
- 39.Díaz DD, Punna S, Holzer P, Mcpherson AK, Sharpless KB, Fokin VV, Finn MG. J Polym Sci, Part A: Polym Chem. 2004;42:4392–4403. [Google Scholar]
- 40.Liu Y, Díaz DD, Accurso AA, Sharpless KB, Fokin VV, Finn MG. J Polym Sci, Part A Polym Chem. 2007;45:5182–5189. [Google Scholar]
- 41.Sheng X, Rock DM, Mauldin TC, Kessler MR. Polymer. 2011;52:4435–4441. [Google Scholar]
- 42.Chatani S, Kloxin CJ, Bowman CN. Polym Chem. 2014;5:2187–2201. [Google Scholar]
- 43.Ritter SC, König B. Chem Commun. 2006:4694–4696. doi: 10.1039/b610696j. [DOI] [PubMed] [Google Scholar]
- 44.Poloukhtine AA, Mbua NE, Wolfert MA, Boons GJ, Popik VV. J Am Chem Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adzima BJ, Tao Y, Kloxin CJ, DeForest CA, Anseth KS, Bowman CN. Nat Chem. 2011;3:256–259. doi: 10.1038/nchem.980. [DOI] [PubMed] [Google Scholar]
- 46.Tasdelen MA, Yagci Y. Tetrahedron Lett. 2010;51:6945–6947. [Google Scholar]
- 47.Alzahrani AA, Erbse AH, Bowman CN. Polym Chem. 2014;5:1874–1882. [Google Scholar]
- 48.Gong T, Adzima BJ, Baker NH, Bowman CN. Adv Mater. 2013;25:2024–2028. doi: 10.1002/adma.201203815. [DOI] [PubMed] [Google Scholar]
- 49.Sandmann B, Happ B, Vitz J, Hager MD, Burtscher P, Moszner N, Schubert US. Polym Chem. 2013;4:3938–3942. [Google Scholar]
- 50.Worrell BT, Malik JA, Fokin VV. Science. 2013;340:457–460. doi: 10.1126/science.1229506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantillo D, Ávalos M, Babiano R, Cintas P, Jiménez JL, Palacios JC. Org Biomol Chem. 2011;9:2952–2958. doi: 10.1039/c0ob01001d. [DOI] [PubMed] [Google Scholar]
- 52.Fokin VV. In: Organic Chemistry - Breakthroughs and Perspectives. Ding K, Dai L-X, editors. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2012. pp. 247–277. [Google Scholar]
- 53.Ye S, Cramer NB, Bowman CN. Macromolecules. 2011;44:490–494. doi: 10.1021/ma200098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bräse S, Gil C, Knepper K, Zimmermann V. Angew Chem, Int Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]
- 55.Rodionov VO, Fokin VV, Finn MG. Angew Chem, Int Ed. 2005;44:2210–2215. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 56.Immergut EH, Mark HF. Plasticization and Plasticizer Processes. Vol. 48. Polytechnic Institute of Brooklyn; Brooklyn, N Y: 1965. pp. 1–26. [Google Scholar]
- 57.Neumann MG, Schmitt CC, Ferreira GC, Corrêa IC. Dent Mater. 2006;22:576–584. doi: 10.1016/j.dental.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Moon HJ, Shin DH. Restor Dent Endod. 2012;37:96–102. doi: 10.5395/rde.2012.37.4.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asmusen S, Arenas G, Cook WD, Vallo C. Dent Mater. 2009;25:1603–1611. doi: 10.1016/j.dental.2009.08.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.